




ABSTRACT
High-efficiency electrocatalysis could serve as the bridge that connects renewable energy technologies, hydrogen economy and carbon capture/utilization, promising a sustainable future for humankind. It is therefore of paramount significance to explore feasible strategies to modulate the relevant electrocatalytic reactions and optimize device performances so as to promote their large-scale practical applications. Microenvironment regulation at the catalytic interface has been demonstrated to be capable of effectively enhancing the reaction rates and improving the selectivities for specific products. In this review we summarize the latest advances in microenvironment regulation in typical electrocatalytic processes (including water electrolysis, hydrogen–oxygen fuel cells, and carbon dioxide reduction) and the related in situ/operando characterization techniques and theoretical simulation methods. At the end of this article, we present an outlook on development trends and possible future directions.
INTRODUCTION
The pressing issue of continuously elevating CO2 concentrations in the Earth's atmosphere urgently calls for a fundamental paradigm shift in energy-related technologies and the recycling of this greenhouse gas, and electrocatalysis powered by sustainable energies is expected to play a substantial role in this grand picture [1]. Hydrogen economy and electrochemical CO2 reduction reaction (CO2RR) have been extensively acknowledged as two promising solutions. Green hydrogen produced via water electrolysis, in combination with hydrogen–oxygen fuel cells, could potentially lead to a sustainable energy infrastructure with net-zero carbon emissions [2]. On the other hand, CO2RR could recycle CO2 and convert it into value-added chemical feedstocks and fuels, representing an attractive alternative for closing the carbon cycle [3]. At the current stage, water electrolyzers, hydrogen–oxygen fuel cells, and CO2 electrolyzers have demonstrated considerable prospects for commercialization. However, there still exist quite a few obstacles on the way towards their large-scale application. For instance, water electrolyzers and hydrogen–oxygen fuel cells are facing the issues of sluggish kinetics of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), unsatisfactory catalyst stability, and high costs due to inclusion of noble metal components [2,4,5]; for CO2RR, it is still rather challenging to achieve high selectivity for specific target high-value products [6]. Therefore, it is of great significance, both scientific and practical, to explore feasible strategies to boost the related electrocatalytic reactions.
Electrocatalytic reactions take place at the catalyst/electrolyte interface, where the electrical double layer (EDL) structure plays a crucial role in influencing reaction kinetics. Optimizing the catalytic structures and regulating the microenvironment at the interface are two effective options for promoting electrocatalytic processes [7]. The former aims to optimize the adsorption energies and activation barriers for the involved chemical species (including reactants, intermediates, and products) at the catalytically active sites in pursuit of high-performance catalysts, which has been one of the hottest topics in electrocatalysis during the past few decades [8]. Several review articles have already summarized research progress in the field of electrocatalysis, providing important guidance for the modulation of the microenvironment of electrocatalyst structures [9–11]. The latter, on the other hand, resorts to tactics such as controlling the electrolyte compositions (including pH, [12,13] anions [14–16] and cations [17,18]), altering the interfacial hydrophilicity/hydrophobicity [19–21], and constructing the organic-compound/electrode interface [22,23], and has also demonstrated its capabilities of elevating reaction rates, tuning reaction selectivities, and enhancing reaction stability. In addition, investigating the effect of microenvironment on the overall catalytic process could help to unveil the underlying catalytic mechanisms, and thus shed light on the development of next-generation electrocatalytic materials and devices [24]. For example, it has been reported that adding cyclohexanol could effectively block the direct adsorption of Nafion onto the Pt surface [25], and that employing chemically modified carbon supports with tailored porosity enabled the uniform dispersion of ionomer [26]. These strategies have successfully optimized the interface microenvironment of ionomers/catalysts, thereby improving the operational performance and robustness of fuel cell devices. In addition, modulating the cation concentrations [17], local pH [27] and local CO2/H2O ratio [28] at the catalytic interface could enable production of multi-carbon products via CO2RR. These pioneering works showcase the importance of microenvironment regulation for promoting practical applications. On the other hand, there are still some issues that need to be noted: most of the research works on microenvironment were based on the three-electrode system rather than practical devices. Similar to the challenges faced in catalyst development, a number of extraordinary results have been obtained using the rotating disk electrode (RDE) setup, and yet they could hardly be reproduced in the membrane electrode assembly (MEA) configuration [29]. This is probably because when the catalytic reactions are transferred from the planar electrodes in the conventional three-electrode setup to the triple-phase interface in MEA, the distinct differences in their operational conditions and microenvironments could lead to significant discrepancies [30]. Therefore, it is necessary to conduct cross validation between the three-electrode system and MEA for the research on microenvironment in the future. To sum up, the approach of starting from the electrode/electrolyte interface could help to establish a more comprehensive and in-depth understanding on the fundamentals of electrocatalysis, to explore feasible strategies for regulating the microenvironment, and to develop advanced electrochemical devices with superior performances.
Here in this paper, we present a brief review on the recent advances in microenvironment regulation for typical energy-related electrochemical processes (such as HOR, HER, ORR, OER, CO2RR) associated with water electrolyzers, hydrogen–fuel cells, and CO2 electrolyzers (Fig. 1). In the following sections, we first give an introduction on the establishment and development of EDL theory. Then we overview the latest advances in the research on microenvironment regulation, with particular emphasis on the effects of pH, anions, cations and organic compounds, and on the underlying principles for different regulation strategies. Next we summarize the most recent progress in the design and fabrication of membrane electrodes and corresponding electrocatalytic systems based on microenvironment regulation. An extra section is given for the in situ/operando characterization techniques (such as in situ visualization, surface-enhanced infrared/Raman spectroscopies) and theoretical simulation methods (such as ab initio molecular dynamics and continuum modeling). At the end of this review, we present a brief summary on the challenges currently facing this research area, and an outlook on possible future directions. We believe this comprehensive review of recent advancements in microenvironment regulation across a broad spectrum of electrochemical processes, coupled with practical insights into device-level optimization and in situ/operando characterization, can help to deepen understanding and facilitate the advance in microenvironment engineering.
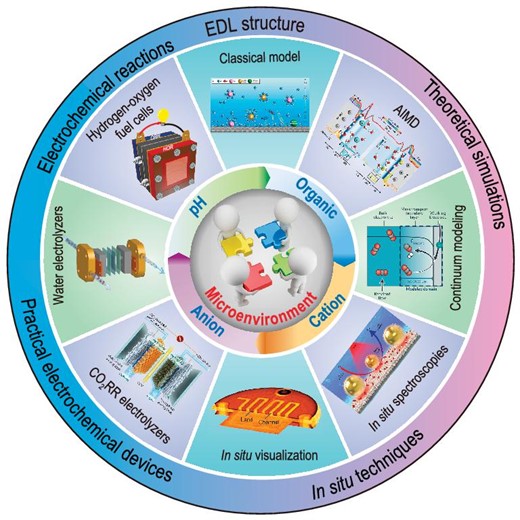
Schematic illustration of microenvironment regulation for electrocatalysis.
ELECTRODE–ELECTROLYTE INTERFACES: ELECTRICAL DOUBLE LAYER (EDL) STRUCTURE
The microenvironment of electrode–electrolyte interface (EEI) could have a significant impact on the thermodynamics, kinetics, reaction pathways and thus product selectivity. On the other hand, the electrocatalytic reactions themselves could also alter the interfacial microenvironment, such as the change of electric field, dynamic adsorption/desorption of reactants, intermediates and products at the electrode surface; in particular, when the reaction involves the generation/consumption of H+/OH−, the local pH at the electrode surface could deviate substantially from the bulk electrolyte. Understanding the structure of EEI at the atomic/molecular scale is a major challenge owing to the dynamic and complex interactions between electrocatalytic reactions and the interfacial environment [31]. Electrical double layer (EDL) theory lays the foundation of theoretical research on the EEI, and during nearly two centuries of development, the EDL model has undergone significant modifications, currently leading to the well-known Gouy–Chapman–Stern (GCS) model. After further incorporating the solvated ions and adsorption [32], the schematic illustration of the classic EDL model is shown in Fig. 2, which has been widely applied in the field of electrocatalysis.
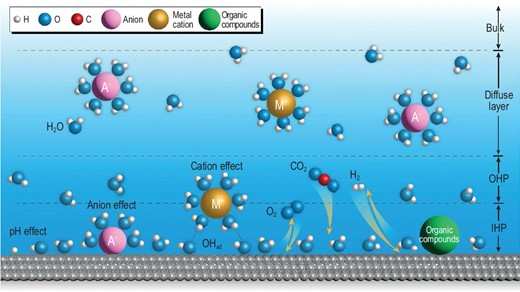
Schematic illustration of the EDL structure and the typical effects of microenvironment in electrocatalysis. The off-white, blue, red and silver-white spheres represent the H, O, C and the metal (electrode material) atoms, respectively. The golden, pink and green spheres represent the cations, the anions and organic species, respectively.
The EDL extends from the electrode towards the electrolyte, with the layer nearest to the electrode surface composed of solvent molecule dipoles arranged in an ordered manner, forming the inner Helmholtz plane (IHP). Certain specific adsorbed anions may lose a portion of their solvation shells and directly adhere to the electrode surface, which might compete with the adsorption of the reactant/intermediate species. On top is the outer Helmholtz plane (OHP) formed through the solvation of cations. On account of the high solvation enthalpies of cations, it is generally believed that cations are not in direct contact with the electrode. Nonetheless, the electrode interface would still be affected by the quasi-specific adsorption of cations coupling with other surface-adsorbed species (such as *OH) [18]. The electric potential in the vicinity of the electrode interface region (including IHP and OHP) is associated with the surface charge of the electrode, and follows a quasi-linear distribution. Farther out from OHP is the diffuse layer, where the distribution of cations/anions in this layer is still controlled partially by the electric potential, which varies almost exponentially with respect to the distance from the electrode.
The classic EDL model described above is the foundation for understanding the interfacial microenvironment, but it still has significant limitations. For example, the structure of interfacial water molecules, the degree of ion hydration and the position of cations at the interface are still rather vague. Recently, Bonn et al. discussed the formation of EDL on different types of charged interfaces and its impact on the interfacial water structure, emphasizing the limitations of the classic mean-field description of EDL in depicting the real systems involving charged surfaces, water, and ions at the molecular level [33]. Taking into consideration more microscopic details (such as interfacial water properties and specific ion interactions) as well as developing density functional theory (DFT)-based molecular dynamics simulations has positive implications for understanding interfacial water structures. In situ spectroscopies are crucial for probing the microscopic details of interfacial water structures. Tian et al. observed the interfacial water structure on electrified Au single-crystal electrode surfaces via in situ Raman spectroscopy [34]. They discovered that the O–H stretching vibrational mode of interfacial water underwent two transitions, accompanied by two changes in the number of hydrogen bonds when sweeping to negative potentials. In combination with ab initio molecular dynamics (AIMD) simulations, it was demonstrated that these transitions are due to the evolution of interfacial water molecules from the ‘parallel’ configuration to ‘one-H-down’ and then to ‘two-H-down’ configurations with the variation in interfacial electric potential.
Furthermore, the determination of actual parameters for the EDL, such as the potential drop (PD) and the potential of zero charge (PZC), is crucial for validating models and making necessary adjustments. Crumlin et al. directly probed the electric potential distribution in the EDL at the interface between electrified gold polycrystalline electrode and liquid electrolyte under polarization conditions via ambient-pressure X-ray photoelectron spectroscopy [35]. This not only discerned the shape of the EDL profile, but also helped to further understand the influence of electrolyte concentration and applied potential on the EDL structure. Koper et al. determined the PZC as 0.3 V vs. normal hydrogen electrode (NHE) on Pt(111) by measuring the Gouy–Chapman capacitance minimum at the interface between Pt(111) and aqueous perchlorate electrolyte [36]. Owing to the stronger ‘effective screening’ than predicted by purely electrostatic mean-field Poisson–Boltzmann theory, the actual diffuse double layer structure of Pt(111) deviates more from the theoretical values. Recently, Suntivich et al. reported a novel optical method for determining the PZC, which directly probes the electric field at the electrochemical interface through field-induced second-harmonic generation (SHG) effects [37]. Using this method, the team determined the PZC value of the polycrystalline Pt/water interface to be 231 ± 76 mV vs. standard hydrogen electrode (SHE). This phase-sensitive SHG method, which does not require probe molecules, also provides a promising approach to determining the PZC of other metal electrodes under different electrochemical conditions.
With the advance of in situ testing techniques and theoretical simulation methods, new impetus has been added to the development of EDL models. Understanding of the EDL structure at the atomic scale is of important significance for better utilizing the microenvironment to regulate electrocatalytic performances.
MICROENVIRONMENTAL REGULATION FOR HYDROGEN ELECTROCATALYSIS
Electrochemical hydrogen evolution and oxidation reactions (HER/HOR) are not only the foundation of a sustainable hydrogen economy, but also the typical processes for electrocatalytic reactions, serving as models for studying electrode kinetics and the fundamental principles of electrocatalysis. The non-Nernstian pH dependency of the HER/HOR kinetics leads to notably lower reaction rates in alkaline environments than under acidic conditions, and the origins are still unclear [38]. Yan et al. assessed the HER/HOR activities of Pt catalysts and the oxidation peak for the underpotentially deposited hydrogen (Hupd) in different buffer solutions over a wide pH range (0–13). It was found that as the pH increases, the HER/HOR activities gradually decrease whereas the Hupd peak potential (Epeak) shifts anodically [39]. Nørskov et al. established a direct correlation between Epeak and the hydrogen binding energy (HBE) of Pt based on DFT calculations [40], and thus the pH effect on Epeak seems to suggest that HBE is the sole descriptor of the HER/HOR activities on Pt. As is shown in Fig. 3a, a universal dependence between pH and the measured HBE has also been found on Pt-group metals other than Pt (including Ir, Pd and Rh) [12]. This provides an explanation for the suppressed HER/HOR activities in alkaline solutions, that the OH species would lead to the overbinding of Had owing to the higher HBE.
![Microenvironmental regulation for hydrogen electrocatalysis. (a) The exchange current density of HOR/HER on Pt/C, Ir/C, Pd/C and Rh/C as a function of Epeak. Reproduced with permission from Ref. [12]. Copyright 2016 American Association for the Advancement of Science. (b) The O–H stretching wavenumber of interfacial water as a function of potential. Reproduced with permission from Ref. [43]. Copyright 2020 American Chemical Society. (c) KIE of HER/HOR on Pt(111) and Pt(110) in HClO4/DClO4 and KOH/KOD. Reproduced with permission from Ref. [44]. Copyright 2020 American Chemical Society. (d) Schematic of operando ETS measurements and (e) the normalized ETS conductance of the PtNW device in 0.1 M MOH (M = Li, Na and K) electrolyte solutions as a function of potential. Reproduced with permission from Ref. [47]. Copyright 2022 Nature Publishing Group. (f) Schematic diagram of the proton transfer from diffuse layer to Helmholtz layer across the H-bond gap in the alkaline EDL. Reproduced with permission from Ref. [48]. Copyright 2022 Nature Publishing Group. (g) HER/HOR polarization curves of Pt(111) and Pt(111) + caffeine in 0.1 M KOH. Reproduced with permission from Ref. [49]. Copyright 2020 American Chemical Society. (h) Atomic configuration of an equilibrated H2O–Me-N1C2–Pt(100) interface. The grey, brown, blue, red and white spheres represent Pt, C, N, O and H atoms, respectively. (i) Schematic illustration of an interfacial water dimer on Pt(100) interface with Me-N1C2. (h, i) Reproduced with permission from Ref. [22]. Copyright 2023 Nature Publishing Group.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nsr/11/12/10.1093_nsr_nwae315/17/m_nwae315fig3.jpeg?Expires=1750214452&Signature=qzpPqA2b0Kr~rYBqRaMyaLgXOG6545E6IJl4vx~rLLD4m7qFnyaTvrKJglpWe-3KN7JJFJeM2nwJnyqL8jV9ODo48LzQ454HcNC3b1gs82IGz3dT~0m1kbpVHMHKZXbWUe08cbGgSUNeoDUP-4PT22YYqki9Dp4vn~-J88Znsvi82zo0vNtZ7TKVrDbevhh-vIpfAPK-iR5fVKTaAD46UmsEwrcbTuqYgKbk2ZFvcmJsuMNKLHjYAVqUymDqmO5b3KDocxJRIJ4vBuQOXVhPif1CCKvFmDvtGHVnGAv37w57Aok~8GPDoTJIlik4z92bODX4za9z1JDWeCGgD2eEew__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Microenvironmental regulation for hydrogen electrocatalysis. (a) The exchange current density of HOR/HER on Pt/C, Ir/C, Pd/C and Rh/C as a function of Epeak. Reproduced with permission from Ref. [12]. Copyright 2016 American Association for the Advancement of Science. (b) The O–H stretching wavenumber of interfacial water as a function of potential. Reproduced with permission from Ref. [43]. Copyright 2020 American Chemical Society. (c) KIE of HER/HOR on Pt(111) and Pt(110) in HClO4/DClO4 and KOH/KOD. Reproduced with permission from Ref. [44]. Copyright 2020 American Chemical Society. (d) Schematic of operando ETS measurements and (e) the normalized ETS conductance of the PtNW device in 0.1 M MOH (M = Li, Na and K) electrolyte solutions as a function of potential. Reproduced with permission from Ref. [47]. Copyright 2022 Nature Publishing Group. (f) Schematic diagram of the proton transfer from diffuse layer to Helmholtz layer across the H-bond gap in the alkaline EDL. Reproduced with permission from Ref. [48]. Copyright 2022 Nature Publishing Group. (g) HER/HOR polarization curves of Pt(111) and Pt(111) + caffeine in 0.1 M KOH. Reproduced with permission from Ref. [49]. Copyright 2020 American Chemical Society. (h) Atomic configuration of an equilibrated H2O–Me-N1C2–Pt(100) interface. The grey, brown, blue, red and white spheres represent Pt, C, N, O and H atoms, respectively. (i) Schematic illustration of an interfacial water dimer on Pt(100) interface with Me-N1C2. (h, i) Reproduced with permission from Ref. [22]. Copyright 2023 Nature Publishing Group.
However, it does not seem sufficiently rigorous to directly associate the measured Hupd peak with HBE. The similar pH dependence of Hupd peaks on different metals suggests that this is not related to the innate properties of electrodes, but rather dominated by the interfacial microenvironment. Therefore, the apparent HBE (HBEapp), which takes the Gibbs free energy of interfacial water adsorption (WBE) into consideration, was proposed as a more proper descriptor for hydrogen electrocatalysis. Koper et al. suggested that the non-Nernstian pH shift of Hupd peak is due to the fact that the co-adsorbed alkali metal cations along the step weaken the OH adsorption at the step sites on Pt(553) [41]. Xu and Yan et al. combined cyclic voltammetry and surface-enhanced infrared absorption spectroscopy (SEIRAS), and proposed that the pH-dependent shift of the Hupd peak on Pt may be attributed to the altered structure of interfacial water by the electrode potential, rather than by cations [42]. Shao et al. tracked the changes in HBE and WBE on Pt thin film under different pH conditions, and claimed that the experimentally determined HBE is actually the combined result of electric field, Had coverage, Pt–water and Had–water interactions [43]. In electrolytes with higher pH values, the lowered WBE leads to an increased distance between the Pt surface and the interfacial water layer, thus hindering charge transfer and resulting in slower reaction kinetics (Fig. 3b). The results on the kinetic isotope effect (KIE) of HER/HOR on single-crystalline Pt(111) and Pt(110) also support the significant influence of pH on water structure at the interface [44]. No significant KIE for hydrogen reactions was observed in acidic media, but a KIE as high as 3.4 was observed for HOR on Pt(111) in alkaline solution, indicating that interfacial water plays a crucial role in promoting proton adsorption and overall hydrogen reaction kinetics in alkaline media (Fig. 3c). The interfacial electric field is also considered an intrinsic origin of the pH effect on hydrogen electrocatalysis. Surendranath et al. quantified the interfacial electric field strength on Pt as a function of pH by using a non-Faradaic reaction probe and tracking the pH sensitivity of H2/H+ catalysis [45]. The results show that with each unit increment in pH, the electrostatic potential at the Pt surface increases significantly, indicating a distinct difference in electric field strengths between acidic and alkaline conditions. The pronounced difference in interfacial electric field strength at RHE would influence the rate of solvent molecule reorganization, thereby contributing to the pH dependence of HER/HOR kinetics.
Different alkali metal cations (AM+) have been found to significantly affect the HER in alkaline solutions, but the cation effect is rather insignificant in acidic media. This may be due to the presence of a large amount of OH species in alkaline solutions, leading to a more complex interfacial microenvironment, with the cations believed to influence the interfacial solvent layer through these OHad species. Jia et al. investigated the cation effect on hydrogen electrocatalysis in Pt, Ni and PtNi systems, and found that Li+ does not affect the HER on Pt, but significantly improves the HER on Ni and PtNi [46]. This implies that Li+ could not only facilitate the OHad removal by forming a OHad–(H2O)x–Li+ structure, but also hinders HOR by weakening the adsorption strength of OHad. It was also observed that increasing the Li+ concentration only promotes HER, whereas changing the identity of AM+ affects both HER/HOR on Pt. These experimental results collectively indicate that the cation effect could alter the rate of hydrogen electrocatalytic reactions by influencing the OHad on Pt. Duan et al. investigated the cation effects of HER on Pt via electrochemical impedance spectroscopy and electrical transport spectroscopy (Fig. 3d, e) [47]. In situ experimental evidence on surface adsorbates and charge-transfer resistance (Rct) at the electrode/electrolyte interface indicates that the high polarity of OHad allows it to act as electrically favored proton acceptors and geometrically favored proton donors, thereby accelerating the kinetics of the Volmer step. Cations with smaller atomic numbers (such as Li+) could minimize the interference with OHad on Pt, resulting in a higher OHad coverage and thus a better HER activity than Na+ and K+. These works imply that the cation effect is often closely related to the OHad. Although the bifunctional mechanism of hydrogen electrocatalysis in alkaline is still a topic of much debate, it is almost unanimously agreed that OHad has a significant impact on the kinetics of HER/HOR. Chen et al. conducted in situ surface-enhanced infrared absorption spectroscopy and AIMD simulations, and argued that the main origin for the suppressed activity of HER/HOR on Pt in alkaline electrolytes is the presence of water gaps in the EDL and the weakened connectivity of hydrogen bond networks (Fig. 3f) [48]. Furthermore, OHad could significantly improve the connectivity of the hydrogen bond network, which explains the fact that the hydrogen reaction activity of PtRu is higher than that of Pt in alkaline media.
In addition to electrolytes, some recent studies have shown that organic additives adsorbed on electrode surfaces can fine-tune the interfacial microenvironment and improve the kinetics of alkaline hydrogen reactions. Snyder et al. used caffeine adsorbed on Pt surface as a ‘double-layer dopant’ and enhanced the rates of HER/HOR on Pt by more than 5-fold (Fig. 3g) [49]. Fourier Transform Infrared Spectrometer (FTIR) spectroscopy provides evidence that the caffeine molecules are adsorbed on the Pt surface and can be stabilized and regenerated through electrochemical deposition during load/potential cycling. The hydrophobic and oxophobic nature of caffeine can inhibit electrode corrosion and thus enhance catalytic stability. In addition, its slight polarity weakens the OHad, leading the team to speculate that it may affect the hydrogen bonding network and the mobility of surrounding water molecules, thus leading to an elevated rate of hydrogen reactions. Inspired by the intriguing results regarding caffeine, Xu et al. used theophylline derivatives with partial structural similarities to caffeine and investigated the effect of different molecular fragments of caffeine on the HOR/HER activity on Pt in alkaline media [50]. In situ Raman spectroscopy provided the intensity of weakly hydrogen-bonded water on Pt decorated with 7-alkyl substituted theophylline derivatives of varying alkyl chain lengths, consistent with the trend of corresponding HOR/HER exchange current densities. This indicates that theophylline derivatives can enhance the HOR/HER activity by promoting interface water reorganization through increased weakly hydrogen-bonded water. In addition to caffeine-like reagents, Jia et al. found that N-methylimidazoles at the Pt–water interface can also accelerate the hydrogen reaction [22]. The more negatively charged pyridinic nitrogen (N3) in N-methylimidazole forms a strong N3···H2O bond with interfacial H2O, keeping the second layer of water molecules close to the Pt surface to facilitate OH− diffusion at the interface, thereby accelerating the Volmer step on Pt (Fig. 3h, i). This organic interface strategy has been shown to be quite feasible for practical devices, with a 40% performance enhancement achieved in an anion exchange membrane electrolyzer by adding 1,2-dimethylimidazole to the Pt cathode.
As manifested in the non-Nernstian pH effect, the interface microenvironment could have profound influences on the HER/HOR kinetics. Current research suggests that the underlying mechanisms may involve alteration in the EDL structure (such as the interfacial water layer) and the electric field. Considering the scientific and practical importance of hydrogen electrocatalytic reactions, delving into the regulation mechanisms of interface effects can not only aid in optimizing the design of hydrogen-related energy devices, but also help in understanding the intrinsic connections between interfacial structure and catalytic performance.
MICROENVIRONMENTAL REGULATION FOR OXYGEN ELECTROCATALYSIS
Hydrogen and oxygen electrocatalysis form the foundation of water electrolyzers and hydrogen−oxygen fuel cells. Compared with hydrogen-related reactions, OER/ORR have much slower kinetics, and the high overpotential for the oxygen reaction side is the primary origin of device polarization. Therefore, optimizing the efficiency of oxygen electrocatalytic reactions is crucial for further improving device performance.
Currently, it has been found that properly designed electrolyte compositions can accelerate OER, known as the electrolyte effect. Zhang et al. showcased that the addition of selenites (SeO32−) or sulfate (SO42−) in the electrolyte could significantly improve the OER performance of transition metal hydroxides such as Ni(OH)2, Cu(OH)2 and Co(OH)2 (Fig. 4a) [14]. In situ Raman spectroscopy and theoretical calculations indicate that these surface-adsorbed chalcogenates can significantly lower the Gibbs free energy of the intermediate of OER (*OOH), thus promoting OER activity. Similarly, surface-adsorbed carboxylate ligands (CLs) on bi/trimetallic layered double hydroxides (LDHs)/MOFs materials also exhibit a universal boosting effect for OER [15]. In situ spectroscopic evidence shows that the negatively charged CLs adsorbed on NiFeMn MOFs can act as mediators for proton transfer, accelerating and facilitating the adsorption, activation, and dissociation of OH− to promote the OER. Besides these individual anions, the combinations of different anions have been found to exhibit interesting synergistic effects. Gong et al. discovered that the combination of borate and fluoride anions can have an intriguing synergistic effect for OER under neutral conditions, with the water oxidation current almost an order of magnitude higher than that for individual anions (Fig. 4b) [16]. This synergy was considered to be due to the hierarchical arrangement of these two anions at the interface, where borate anions in the inner Helmholtz layer promote proton-coupled electron transfer (PCET) steps, lowering the onset potential, while fluoride anions located further outside effectively disrupt the polymer hydrogen-bonding network in water, sustaining a larger current.
![Microenvironmental regulation for oxygen electrocatalysis. (a) OER polarization curves of Ni(OH)2 in 1 M KOH with different concentrations of SeO32−. Reproduced with permission from Ref. [14]. Copyright 2020 John Wiley & Sons, Inc. (b) OER polarization curves of Co(OH)2 in neutral electrolytes with different concentrations of borate and fluoride anions. Reproduced with permission from Ref. [16]. Copyright 2022 Elsevier Inc. (c) OER polarization curves of the NiFe-based catalysts recorded in alkaline electrolytes with different alkali metal cations. Reproduced with permission from Ref. [52]. Copyright 2022 John Wiley & Sons, Inc. (d) The schematic illustration of alkali cation intercalation and elongated Co–O bonds in CoOOH during the OER. Reproduced with permission from Ref. [53]. Copyright 2023 John Wiley & Sons, Inc. (e) The schematic illustration of tuning local proton activity for the ORR via protic cations of ionic liquids with different pKa. Reproduced with permission from Ref. [23]. Copyright 2021 Nature Publishing Group. (f) Cyclic voltammograms of Pt(111) in HClO4 solutions of different concentrations. (g) Summary of specific activities at 0.9 V (vs. RHE) of Pt(111) in HClO4 and methanesulfonic acid (MSA) of different concentrations. (f, g) Reproduced with permission from Ref. [55]. Copyright 2022 Nature Publishing Group. (h) ORR polarization curves of Pt/C in phosphate-buffered water (0.5 M KPi) solutions with different concentrations of zeolitic nanocrystals. Reproduced with permission from Ref. [57]. Copyright 2023 Nature Publishing Group.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nsr/11/12/10.1093_nsr_nwae315/17/m_nwae315fig4.jpeg?Expires=1750214452&Signature=i6RgeJ0RMDunYVR~Ha599MkpvtJO39pPrfkDp0SDbU~ghRUecndZmRX73lOgDQf9Z1LEKiiX0~raLBm9-k7k4G99eRv53c6wIgrdP1d3x9gQ9oU0eVmBBLKkS-kiWxmP5ZRDKEXRK3K9IQ-5Gt-nqiAEllvgHNCpXZhXmFSYpgeX-02U1aU6G1i58PmVYyr69d2PGOlTMkn3W40ioGMnR6vt7ENawW57iccvElg~OMxXA-Kr6lSmBFG7tdAmPVWBfAFOBKqgv~DQTwwsS8R0NHzHEwLq2zSefZmba~LSmyiuJEF9mk1A5VoTxdFm~FhTCnofZcfOH43emi9dTP0sOw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Microenvironmental regulation for oxygen electrocatalysis. (a) OER polarization curves of Ni(OH)2 in 1 M KOH with different concentrations of SeO32−. Reproduced with permission from Ref. [14]. Copyright 2020 John Wiley & Sons, Inc. (b) OER polarization curves of Co(OH)2 in neutral electrolytes with different concentrations of borate and fluoride anions. Reproduced with permission from Ref. [16]. Copyright 2022 Elsevier Inc. (c) OER polarization curves of the NiFe-based catalysts recorded in alkaline electrolytes with different alkali metal cations. Reproduced with permission from Ref. [52]. Copyright 2022 John Wiley & Sons, Inc. (d) The schematic illustration of alkali cation intercalation and elongated Co–O bonds in CoOOH during the OER. Reproduced with permission from Ref. [53]. Copyright 2023 John Wiley & Sons, Inc. (e) The schematic illustration of tuning local proton activity for the ORR via protic cations of ionic liquids with different pKa. Reproduced with permission from Ref. [23]. Copyright 2021 Nature Publishing Group. (f) Cyclic voltammograms of Pt(111) in HClO4 solutions of different concentrations. (g) Summary of specific activities at 0.9 V (vs. RHE) of Pt(111) in HClO4 and methanesulfonic acid (MSA) of different concentrations. (f, g) Reproduced with permission from Ref. [55]. Copyright 2022 Nature Publishing Group. (h) ORR polarization curves of Pt/C in phosphate-buffered water (0.5 M KPi) solutions with different concentrations of zeolitic nanocrystals. Reproduced with permission from Ref. [57]. Copyright 2023 Nature Publishing Group.
Similar to anions, alkali metal cations have also been found to significantly affect the OER performance of electrode materials, although the intrinsic role of the cations is still rather vague thus far. Koper et al. found that regardless of the presence of Fe impurities in alkaline electrolytes, the OER activities of NiOOH always show an intrinsic cation dependency [51]. In situ surface-enhanced Raman scattering (SERS) results suggest that the cation effect is related to the interaction with the superoxo OER intermediate (NiOO−). Cations with larger atomic numbers (such as Cs+) are supposed to be capable of better stabilizing the NiOO−−M+ species, thereby promoting O2 evolution. Advanced in situ characterization technologies provide new opportunities to probe the cations effect. Fischer et al. further demonstrated the correlation between the cations effect on the NiFe(OOH)-based electrocatalysts and the potential of maximum entropy (PME) using the laser-induced current transient (LICT) technique (Fig. 4c) [52]. The results suggest that Cs+ cations may accelerate the kinetics of the OER by altering the structure of the interfacial water layer. Combining the results from operando powder X-ray diffraction (PXRD), high-resolution transmission electron microscopy (HRTEM) and X-ray absorption spectroscopy (XAS), Luo et al. demonstrated that cations enter the layers of the Co-OOH catalyst through intercalation, leading to an increase in interlayer spacing (Fig. 4d) [53]. Cations with larger atomic numbers could facilitate the elongation of Co–O bonds, promoting the formation of high-valence OER-active *O–Co(IV) species. Co-OOH–Cs exhibits the lowest energy barrier for the rate-determining step (RDS), which explains the enhanced OER activity.
Similarly, the cations effects for ORR are also quite significant. Marković et al. proposed that hydrated alkali metal cations M+(H2O)x form OHad–M+(H2O)x clusters at the interface via non-covalent interactions with OHad [18]. Cations with larger atomic numbers have lower hydration energies, leading to the higher ORR activities on Pt(111). Moreover, organic cations have been found capable of effectively promoting the ORR on Pt. Nakamura et al. studied the effect of tetraalkylammonium (TAA) cations with different alkyl chain lengths on the ORR on Pt(111) [54]. The high hydrophobicity of TAA cations with longer alkyl chains significantly alters the hydration structure of TAA. Among them, THA with the longest alkyl chain (n = 6) boosted the performance of Pt(111) by 8-fold. The high hydrophobicity of THA and its hydration shell weakens the binding of surrounding OHad and H2Oad through weaker interactions, lowering the coverage of OHad and disrupting the stable hydrogen-bonding network, thus facilitating the entry of oxygen molecules onto the Pt(111) surface. Shao-Horn et al. recently reported the acceleration of the ORR kinetics of Au and Pt using protic cations of ionic liquids (Fig. 4e), where the ORR on Au mainly proceeds via the 2e− pathway, whereas on Pt it follows the 4e− pathway [23]. The ORR specific kinetic currents exhibit a volcano-shaped relationship vs. the pKa of protic cations in the ionic liquids, with the maximum enhancement obtained when the pKa values of the protic cations and the reaction intermediate in the RDS are similar. The optimal pKa for Pt is around 15, close to the pKa value of H2O, whereas the optimal pKa for Au is around 11, close to the pKa value of H2O2. In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and kinetic simulation results indicate a strong hydrogen bond interaction between the protic cations and the ORR intermediate, leading to faster proton tunneling kinetics and accelerated oxygen catalysis.
Earlier research suggested that the non-specifically adsorbed anions act as spectator species and have little impact on ORR. Yet recently, Koper et al. unearthed an unexpected effect of non-specifically adsorbed anions (ClO4−) on the ORR kinetics on Pt(111) (Fig. 4f, g) [55]. The role of the non-specifically adsorbed anions was explained by introducing the *O ↔ *OH transition as a new kinetic descriptor for ORR on Pt(111). This descriptor also successfully elucidated the impact of perfluorosulfonic acid (PFSA) ionomer and cations in alkaline media on the ORR activity on Pt(111). In addition, the mechanism for the significantly lower activity of M-N-C materials in acid (than in alkaline media) has also attracted attention. Chen et al. used FeCo-N6-C DAC as a model system and combined AIMD simulation with in situ ATR-SEIRAS to study the pH effect on its ORR activity [56]. The results indicate that the difference in activity stems from the distinct EDL structures in acidic and alkaline environments, where the orientation of interfacial water on electrode surfaces varies, impacting the formation of hydrogen bonds between the oxygenated intermediates and the interfacial water molecules, eventually leading to differences in ORR kinetics. In addition to the above ion effects, introducing microporous nanocrystals with hydrophobic internal surfaces and hydrophilic external surfaces into the electrolyte generates ‘microporous water’, which exhibits an O2-carrying capacity two orders of magnitude higher than pure water [57]. This enhancement in O2 supply at the electrode interface mitigates mass transport limitations (Fig. 4h), and thus facilitates the direct measurement of intrinsic ORR activity on Pt. This optimized mass transport strategy may offer a straightforward and effective intermediate testing method to bridge the gap between RDE and MEA measurement.
MICROENVIRONMENTAL REGULATION FOR CO2 REDUCTION REACTION
Electrochemical CO2RR powered by renewable energies is a promising route to transforming CO2 into value-added products and achieving sustainable carbon recycling. The microenvironment of electrocatalysis has a significant impact on CO2RR activity and selectivity. In comparison to HER and ORR, most CO2RR cases occur in a more negative potential range, where the cations tend to migrate to the cathode under the driving force of the electric field. These cations, serving as crucial components at the reaction interface, play a significant role in the CO2RR process. It has been noted in the majority of studies that the influence of alkali metal cations on CO2RR activity follows the order of Cs+ > K+ > Na+ > Li+ [58,59]. Although this order is consistent across various research works, the intrinsic mechanism of the cation effect remains controversial. Hori et al. proposed that cations could alter the OHP potential through specific adsorption, which in turn influences the concentration of H+ on the electrode surface, leading to different product selectivities [60]. On the other hand, Chan et al. refuted the possibility of cations undergoing specific adsorption (Fig. 5a) [61]; instead they suggested that cations would accumulate on the electrode surface through non-covalent interactions under CO2RR conditions, thus generating a high interfacial electric field. Weakly hydrated cations, with their smaller hydrated ionic radii, exhibit the least repulsion near the electrode. The resulting high concentration of cations leads to a higher surface charge density and a stronger interfacial electric field. Such modifications on the potential across the electron transfer plane could affect the apparent activation energy of the entire reaction, thereby influencing the adsorption and activation processes of CO2.
![Microenvironmental regulation for CO2 reduction reaction. (a) Illustration of the origin of cation effects in field-driven electrocatalysis. Reproduced with permission from Ref. [61]. Copyright 2019 the Royal Society of Chemistry. (b) The schematic illustration of the distribution of pH and CO2 concentration in the boundary layer, and FE for CO increases and for H2 decreases with increasing cation size due to a decrease in polarization. Reproduced with permission from Ref. [58]. Copyright 2016 American Chemical Society. (c) The recorded anodic scans performed to oxidize any CO produced in argon and CO2 atmosphere. (d) Average CO2 adsorption Gibbs free energy at U = 0 V vs. standard hydrogen electrode (SHE) in the absence (grey) or presence (light to dark brown) of an alkali cation, with CO2 and the environment (Au–H2O or Au–H2O–M+) as the energy references. (e) αCO2 and CO2 Bader charge, qCO2, are shown in red and black, respectively. (c–e) Reproduced with permission from Ref. [64]. Copyright 2021 Nature Publishing Group. (f) Plotting the selectivity toward formic acid against the contact angle yields a marked relationship, with modifiers 2 and 4 as outliers at 65° and 97°, respectively. Reproduced with permission from Ref. [71]. Copyright 2019 American Chemical Society.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nsr/11/12/10.1093_nsr_nwae315/17/m_nwae315fig5.jpeg?Expires=1750214452&Signature=jeBhlgC3NRCkIBqqSMs4JWi3ecKq31bYF7F7RTgN-dfjf--CWhv-8EsvIx5bOMy3o2BuE7D0Xpi~gLjHdOQ0ZsAMMJHcjDYcaUJku1q4YZFBVPxIxFKVVxjqBT3bhY8Ly-EwACsRW-80ChuBJsqLW9fDx0NZfLKGjd59-XaO~4sbLPm6gqyeVjnPMNWFYpwSWjPIWZ9PFY6xiHHp0IqFeXrWrdkjySDDqvRFX3VRh9QBCaOTMJAAPlSInLmWTg-n~Pb2NqH-jZsNiyw7MWVNObkMTDUPNGboJAwG3hBP-7Js9Ij1Hr3qajS-dIJaK6d3eYh6CUCYDSPJsnbGPPnBKQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Microenvironmental regulation for CO2 reduction reaction. (a) Illustration of the origin of cation effects in field-driven electrocatalysis. Reproduced with permission from Ref. [61]. Copyright 2019 the Royal Society of Chemistry. (b) The schematic illustration of the distribution of pH and CO2 concentration in the boundary layer, and FE for CO increases and for H2 decreases with increasing cation size due to a decrease in polarization. Reproduced with permission from Ref. [58]. Copyright 2016 American Chemical Society. (c) The recorded anodic scans performed to oxidize any CO produced in argon and CO2 atmosphere. (d) Average CO2 adsorption Gibbs free energy at U = 0 V vs. standard hydrogen electrode (SHE) in the absence (grey) or presence (light to dark brown) of an alkali cation, with CO2 and the environment (Au–H2O or Au–H2O–M+) as the energy references. (e) αCO2 and CO2 Bader charge, qCO2, are shown in red and black, respectively. (c–e) Reproduced with permission from Ref. [64]. Copyright 2021 Nature Publishing Group. (f) Plotting the selectivity toward formic acid against the contact angle yields a marked relationship, with modifiers 2 and 4 as outliers at 65° and 97°, respectively. Reproduced with permission from Ref. [71]. Copyright 2019 American Chemical Society.
Distinct from the local electric field effect, Bell et al. proposed another theory, suggesting that cations could modulate the process by buffering the local pH (Fig. 5b) [58]. Due to electrolyte polarization during the CO2RR process, the local pH at the electrode surface tends to increase, and a large amount of OH− would react with CO2 to form (bi)carbonate, thus lowering the accessibility of CO2. At the negatively charged cathode surface, the hydration shell of cations with larger atomic numbers experiences a stronger electrostatic field, thereby lowering its pKa of hydrolysis. This lowered pKa enables the hydrated cations to act as a buffering agent, suppressing the rise of local pH near the cathode. This leads to an increase in the local concentration of dissolved CO2, thereby facilitating the CO2RR process. Zhang et al. detected the changes in local pH during the CO2RR process on Au [62]. They found that cations with larger atomic numbers have a stronger buffering effect, with the interfacial pH values following the order of Li+ > Na+ > K+ > Cs+, which is in good agreement with Bell's results. Nørskov et al. suggested that the high local electric field produced by solvated cations can stabilize adsorbed intermediates with large dipole moments, thereby influencing the CO2RR process [63]. According to the GCS model, the strength of the electric field within the EDL under a given PZC is influenced by the size of the hydrated cations in the electrolyte. Cations with larger atomic numbers would experience stronger driving forces and accumulate near the electrode surface, generating a stronger interfacial electric field, which enhances the adsorption stability of some important CO2RR intermediates. Therefore, a higher CO2RR activity on copper was observed in the presence of cations with larger atomic numbers, which is attributed to their higher concentration in the OHP [59]. Koper et al. further refined this model and discovered that CO2RR does not occur in the absence of metal cations on gold, copper and silver electrodes (Fig. 5c) [64]. Only models considering the electrostatic interaction between metal cations and key intermediates can explain this observation. Through AIMD, the team further confirmed the stabilizing effect of partially desolvated metal cations on CO2 adsorption, activation, and the formation of CO2− intermediates (Fig. 5d, e).
The anion effects differ from those for cations, with most studies focusing on adjusting the local pH (through distinct buffering capacities) and inducing catalyst restructuring. For instance, Hori et al. investigated the impact of anions with different buffering capacities (pKa: H2PO4− < HCO3− < ClO4−) on CO2RR [65]. They found that H2PO4−, having a strong buffering capacity, could easily neutralize OH− near the copper electrode surface, maintaining a lower local pH, which correlates with high selectivity for H2 and CH4, while suppressing the formation of C2H4. In contrast, ClO4−, which lacks the buffering ability, led to a higher local pH at the copper electrode surface and enhanced the selectivity for C2H4. Cuenya et al. explored the influence of different halide anions on the restructuring process of metallic Cu surfaces [66]. Their findings indicated that different halide ions induce varying degrees of surface roughness. Compared with the original copper catalyst, the halide-ion–induced restructured catalysts exhibited improved selectivities for C2+ products.
The pH effect has been indirectly demonstrated through the effects of alkali metal cations and anions, significantly influencing reaction rate and product selectivity [58]. In CO2RR, the local pH near the electrode surface is typically higher than the pH of the bulk electrolyte, owing to the consumption of protons and the subsequent production of OH− during surface reactions. Thus, the impact of local pH on CO2RR is more pronounced than that of bulk pH. For instance, Koper et al. discovered that as the CO2RR proceeds, the local pH increases, which promotes the HER on gold electrodes [67]. Controlling the local pH through mass transport conditions can regulate the rate of competing HER, thereby facilitating the CO2RR process on gold electrodes. In addition, CO2 as a reactant itself also acts as a buffering agent by reacting with OH− at the electrode surface to form (bi)carbonates, thus significantly suppressing the rise in local pH during the CO2RR process. Therefore, the pH effect of CO2RR can be better observed in CORR with similar reaction pathways and RDS. For instance, the selectivity for acetate products in CORR can be significantly improved under conditions of high alkalinity. This may be due to the higher local pH facilitating the reaction of OH− with certain intermediates to produce acetate [24]. To sum up, understanding the impact of local pH is of increasing importance for distinguishing between product selectivities.
Strongly alkaline microenvironments are generally believed to inhibit the HER and promote the formation rate of C2+ products on Cu electrodes, but this inevitably leads to a crossover between pH and cation effects. Sargent et al. suggested that OH− on or near the copper surface can effectively lower the activation energy barriers for CO2RR and CO–CO coupling [68]. Experimental results showed that the onset of ethylene evolution at −0.165 V vs. RHE in 10 M KOH occurs almost simultaneously with the production of CO. This study proposed a plausible strategy to enhance the formation of C2+ products by increasing the pH of the electrolyte. However, this hypothesis contradicts the current understanding of the mechanisms involved in the formation of C2+ products. Recent research indicated that the RDS for C2+ product formation involves the dimerization of two adsorbed CO molecules through the Langmuir–Hinshelwood process, which does not involve proton transfer [69,70]. Thus, the formation rate of C2+ products theoretically should not depend so strongly on the pH of the electrolyte. Lu et al. discovered that at the same SHE potential, the concentration of Na+ has a greater promoting effect on the formation rate of C2+ products in CORR than does the concentration of OH− [69]. This finding suggests that in strongly alkaline electrolytes, the primary factor enhancing the C2+ product formation rate could be the high concentration of cations, rather than OH−.
In addition to altering the composition of electrolytes to regulate cations, anions and pH, recent studies have shown that the modulation of reactant composition, catalyst surface functionalization (including modifications with organic molecules and polymers), and alterations in electrolysis methods can also regulate the interfacial microenvironment, thereby improving the activity and selectivity of the CO2RR [10,28,71]. Lu et al. demonstrated that the introduction of a moderate amount of O2 into the CO2RR system increased the production rate of oxygenates and hydrocarbons by 216 times [72]. Mechanistic studies revealed that the introduction of O2 created a hydroxyl-rich microenvironment on the surface of the Cu catalyst, thereby lowering the energy barriers for the formation of oxygenates and hydrocarbons. Bao et al. employed a co-feeding method of CO and CO2, whereby increasing the pressure of CO in the feeder resulted in a shift of the primary electrolysis product from ethylene to acetate [73]. Further studies revealed that this switching in selectivity is closely related to the increased coverage of CO intermediates and the rise in local pH. Specifically, ethylene is preferentially formed at low *CO coverage, whereas a high *CO coverage and elevated local pH would favor the formation of acetate. In terms of catalyst surface functionalization, Wang et al. innovatively developed a universal amino acid modification strategy, whereby copper electrodes functionalized with amino acids exhibited enhanced capability for hydrocarbon production regardless of electrode morphology [74]. Mechanistic studies indicated that the –NH3+ group of the adsorbed zwitterionic glycine could effectively facilitate the stable adsorption of the key intermediate CHO*. Bell et al. used bilayer cation- and anion-conducting ionomer coatings to control the catalytic microenvironment on the surface of copper electrodes [28]. Anion-exchange ionomers (AEIs), on account of their high CO2 solubility, increase the local ratio of CO2 to H2O. Cation-exchange ionomers (CEIs), on the other hand, raise the local pH by capturing the OH− produced during the CO2RR and blocking the transport of buffering carbonate species into the catalyst microenvironment (via Donnan exclusion). In combination with pulsed electrolysis, the local ratio of CO2/H2O and pH were further increased on the ionomer-layer–modified Cu electrode, thereby improving the selectivity for C2+ products. Toma et al. employed a range of polymeric and molecular modifiers with different hydrophilicity and hydrophobicity to adjust the selectivity of CO2RR on Cu surfaces [71]. Their research revealed that protic species could enhance the selectivity for H2, hydrophilic additives could facilitate the formation of formic acid, and cationic hydrophobes would improve the selectivity for CO (Fig. 5f). The molecular dynamics simulations indicated that hydrophilic and hydrophobic modifiers would influence the formation of surface hydrides. Specifically, surface hydrides are more stable in the presence of hydrophobic species compared with hydrophilic ones. Consequently, the team suggested that the promotion of formic acid formation on hydrophilic surfaces is due to the weakening of the M–H bond. In contrast, hydrophobic species would lead to stronger M–H bonds, thus inhibiting the formation of formic acid and allowing the formation of CO to prevail. Pulsed electrolysis, by applying periodically alternating potentials, offers new possibilities for regulating the reaction microenvironment and the electrocatalytic process. For example, Cuenya et al. studied the effects of different anodic oxidation potentials (Ea) during pulsing on CO2RR. They found that at Ea = 0.9 V vs. RHE, a significant number of grain boundaries and defect sites formed on the surface of copper particles, leading to a substantial increase in the Faradaic efficiency for C2+ products [75]. At Ea = 1.2 V vs. RHE, the regeneration of Cu2O consumed an excess of OH−, causing a decrease in the local pH at the catalyst surface, which favored the reaction pathway towards CH4 formation.
CO2RR could give a variety of products, which significantly adds to the complexity of its reaction pathways. The microenvironment not only affects the CO2RR kinetics, but also alters the selectivity of the final products. This provides a broader stage for microenvironment regulation strategies, but also increases the difficulty of understanding their intrinsic mechanisms.
MICROENVIRONMENT REGULATION FOR PRACTICAL ELECTROCHEMICAL DEVICES
In practical electrochemical devices (often featuring high current densities), gases are often used directly (rather than in the dissolved form) as the reactants to ensure an efficient mass transport, which, on the other hand, also leads to a more complex reaction microenvironment. Consequently, investigations into microenvironments relevant to practical electrochemical devices are of great necessity. It is encouraging that some progress has been made in this field recently.
Water electrolyzers and hydrogen–oxygen fuel cells
Producing green hydrogen via water electrolysis is generally considered an ideal solution for achieving a future of renewable energy and carbon neutrality. Anion exchange membrane water electrolyzers (AEMWEs), benefiting from the use of inexpensive metal catalysts, offer notable advantages in cost. Typically, electrolyzing alkaline solutions may improve the performance and stability of AEMWEs in contrast with directly electrolyzing pure water, yet the exact mechanism underlying this performance enhancement remains uncertain. Weber et al. employed experimental and mathematical modeling approaches to expound the impact of hydroxide concentration on the performance of water electrolyzers [76]. They demonstrated the functional relationship between polarization curves and high-frequency resistance with hydroxide concentration. The findings indicated that increased hydroxide concentration is vital not only for the membrane and catalyst layer's ohmic resistance, but also for the reaction kinetics. Direct electrolysis of seawater is another cost-effective strategy, but it faces major challenges such as chloride corrosion and electrode passivation. Ling et al. introduced a Lewis acid layer (Cr2O3) on the surface of the catalyst (CoOx) to electrolyze water molecules and capture the in situ generated OH− [77]. This in situ enrichment of OH− effectively inhibits chloride corrosion on the catalyst surface and mitigates the issue of precipitate formation, achieving long-term stability of seawater electrolyzer exceeding 100 h at 500 mA cm−2. Compared with the same pH environments at both the cathode and anode in conventional electrolyzers, bipolar membrane (BPM) devices enable the independent selection of ideal pH conditions for each half-reaction, along with compatible catalysts. Boettcher et al. designed a BPM water electrolyzer that can provide an acidic environment for the HER side to accelerate its kinetics (Fig. 6a), while providing an alkaline environment on the OER side to allow for the utilization of low-cost base metal catalysts [78].
![Microenvironmental regulation for practical electrochemical devices. (a) BPM water-dissociation measurements. WD is studied in an electrolyzer fed by only pure water. Reproduced with permission from Ref. [78]. Copyright 2020 American Association for the Advancement of Science. (b) Illustration of the conventional direct-contact Pt/ionomer interface and the proposed non-contact Pt/ionomer space for the cathode catalyst layer. Reproduced with permission from Ref. [25]. Copyright 2023 Nature Publishing Group. (c) Schematic illustration of FTDT cell. (d) Electrode stability tests for 1 cm2 and 100 cm2 FTDT cells at different cell voltages. (c, d) Reproduced with permission from Ref. [85]. Copyright 2022 Nature Publishing Group. (e) Molecular structures of the polyelectrolytes and schematic drawing of the membrane structure. Reproduced with permission from Ref. [86]. Copyright 2021 Nature Publishing Group. (f) Schematic illustration of ionic environment and transport near the catalyst surface functionalized by the PFSA ionomer. Reproduced with permission from Ref. [17]. Copyright 2021 American Association for the Advancement of Science.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nsr/11/12/10.1093_nsr_nwae315/17/m_nwae315fig6.jpeg?Expires=1750214452&Signature=LSXQm7Lb-ms1-ym74R-pjD2En9Abn385uwD8YobMkoLfY1cy0D4jY~4H36LlGipGOI1DTIx2fQtqZcf8KP6YXL4Vg9~Wcsthc6OXeIVFrqx6eH49xFTGA~9RYRfNkKAPcKVOhAvgFSuwW-OvT7OxgzAoGYk8L69IYNexu9VrFuJcLWPAt1yu-ezB02PkXSWuvgHbHQ2IJx2l36OjehldUjtEx~SNkjIpGl~IdKcXDZDA9XiNLyQIOcb2-wTOPVwGijcEPzKGzgwsvm89p11V5j7ypFRtC~~StGtTt9x3UqKxqE0Wf~~~0uSVKoxgUzicH5~gS1zaD4crxfqAd2XPBQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Microenvironmental regulation for practical electrochemical devices. (a) BPM water-dissociation measurements. WD is studied in an electrolyzer fed by only pure water. Reproduced with permission from Ref. [78]. Copyright 2020 American Association for the Advancement of Science. (b) Illustration of the conventional direct-contact Pt/ionomer interface and the proposed non-contact Pt/ionomer space for the cathode catalyst layer. Reproduced with permission from Ref. [25]. Copyright 2023 Nature Publishing Group. (c) Schematic illustration of FTDT cell. (d) Electrode stability tests for 1 cm2 and 100 cm2 FTDT cells at different cell voltages. (c, d) Reproduced with permission from Ref. [85]. Copyright 2022 Nature Publishing Group. (e) Molecular structures of the polyelectrolytes and schematic drawing of the membrane structure. Reproduced with permission from Ref. [86]. Copyright 2021 Nature Publishing Group. (f) Schematic illustration of ionic environment and transport near the catalyst surface functionalized by the PFSA ionomer. Reproduced with permission from Ref. [17]. Copyright 2021 American Association for the Advancement of Science.
The performance and durability of fuel cells are influenced not only by the inherent properties of the catalysts but also by the microenvironment at the catalyst/ionomer interface. In proton exchange membrane fuel cells (PEMFCs), the adsorption of sulfonate groups in the ionomer on the surface of Pt-based catalysts would hamper their catalytic activities. To address this issue, Wei et al. employed cyclohexanol with the chair or boat conformations to coordinate with the ionomer, thus effectively blocking the way in which the ionomer is adsorbed onto the Pt surface (Fig. 6b) [25]. This approach led to the liberation of Pt active sites and significantly enhanced mass transport. On the other hand, the uniform distribution of ionomer can effectively improve oxygen mass transport at the Pt/ionomer interface, allowing for a lower Pt loading without incurring high voltage losses. Strasser et al. achieved this by introducing balanced sp2 pyridinic/pyrrolic/graphitic N functional groups interacting with the ionomer chains, ensuring homogeneous coverage of the ionomer on carbon supports [26]. Furthermore, considering the poor dispersion characteristics of conventional ionomers, Sung et al. employed supercritical isopropyl alcohol (IPA) in conjunction with water as cooperative polar co-solvents to boost the polarity of the solvent and intensify solvation by creating hydrogen bonds. This, in turn, led to the formation of an ionomer dispersion featuring colloidal particles significantly smaller on average than those present in commercially accessible dispersions [79]. For PEMFCs, water is solely formed at the cathode; in contrast, for AEMFCs, water is generated at the anode, while the water at the cathode is consumed. This poses more challenges for water management in the operation of AEMFCs. Mustain et al. incorporated hydrophobic components (PTFEs) into the gas diffusion layer and catalyst layer, resulting in gas diffusion electrodes (GDEs), and the water management was optimized by regulating the hydrophilicity/hydrophobicity of the catalyst/ionomer interface [80]. This design led to a stable operation, demonstrating continuous functioning for more than 1000 h at 600 mA cm−2.
CO2RR electrolyzers
GDEs enhance the mass transport of CO2 to the active electrocatalysts, leading to greater current densities and carbon conversion efficiencies, thus constituting a key technology that is expected to achieve large-scale application of CO2RR. The GDE-based CO2RR electrolyzer could fail typically owing to carbonate deposition. Specifically, this is due to the high local concentration of carbonates formed from the reaction of CO2 with generated OH−, combined with the crossover of cations such as K+ and Na+ from the anolyte [81,82]. The carbonate deposition in GDEs would block the transport pathways of CO2, thus leading to device failure. Therefore, precise control of the microenvironment in GDEs, especially the regulation of carbonate chemistry, is crucial for CO2RR devices.
Janáky et al. developed an operando activation and regeneration process in which a solution containing alkaline metal cations is periodically injected into the cathode of a zero-gap electrolyzer to alleviate carbonate deposition [83]. This enables water-fed electrolyzers to operate with a CO2RR efficiency comparable to that using alkaline electrolytes (CO partial current density of 420 ± 50 mA cm−2, sustained for over 200 h). Additionally, the team demonstrated the scalability of this method across multiple electrolyzer stacks, each with an effective area of 100 cm2. Recently, Zhuang et al. employed a bifunctional ionomer as the polymer electrolyte to assemble an MEA electrolyzer capable of operating in pure water without adding any alkali metal cations, thereby avoiding the issue of carbonate deposition from the start [84]. When operated at a cell voltage of 3.54 V, ethylene is produced with an industrial-scale partial current density of 420 mA cm−2 without any electrolyte consumption. Chen et al. addressed the carbonate deposition issue by modifying the method of CO2 feeding [85]. They developed an electrolyzer that utilizes the forced convection of an aqueous CO2-saturated catholyte throughout a porous electrode (Fig. 6c). The decrease in local pressure led to increased exsolution of gaseous CO2 from dissolved CO2 and bicarbonate, which in turn significantly decreased the thickness of the diffusion layer (by a factor of ten). They assembled a scaled-up 4 × 100 cm2 electrolyzer stack that produced CO at a remarkable rate of 90.6 L h−1 and maintained good stability (Fig. 6d).
BPMs composed of a cation exchange layer (CEL) and an anion exchange layer (AEL) have also been explored for managing ion transport within the CO2RR electrolyzer [82]. A reverse-bias BPM can effectively control the ion transport across the bulk of the membrane. Here, cations from the anode are repelled by the AEL, while anions from the cathode are repelled by the CEL. This configuration effectively regulates the microenvironment at the GDL surface, suppressing the deposition of carbonates and the crossover of products. However, the acidic environment at CEL usually results in a low-pH microenvironment at the CEL/cathode interface, resulting in low CO2RR efficiency. In light of this, Mallouk et al. utilized ratiometric indicators and layer-by-layer polyelectrolyte assembly to deposit weakly acidic polyelectrolyte layers on the strongly acidic CEL (Fig. 6e) [86]. This arrangement allows for the measurement and manipulation of local pH at the GDL surface with a spatial resolution of tens of nanometers. The results indicate that the presence of the weak acid layer suppresses HER without adversely affecting CO2RR.
Compared with electrolyzers working under alkaline or neutral conditions, those working under acidic conditions typically exhibit higher carbon conversion efficiencies by inhibiting the formation of carbonates. However, the kinetically favored HER tends to dominate under acidic electrolytes. Sargent et al. addressed this challenge by modifying the catalyst surface with a cation-augmenting layer, maintaining a high concentration of K+ around the GDL surface (Fig. 6f) [17]. This approach enabled an efficient CO2RR process on Cu catalysts at pH <1 with a single-pass CO2 utilization of 77%. The research by Hu et al. further demonstrated that in acidic electrolytes, the hydrated alkali cations physisorbed on the cathode could alter the distribution of the electric field within the EDL [87]. This alteration inhibits the migration of hydronium ions towards the cathode, thereby impeding hydrogen evolution and facilitating the reduction of CO2. Moreover, Sinton et al. reported the development of a heterogeneous catalyst adlayer composed of covalent organic framework (COF) nanoparticles and cation exchange ionomers [27]. The imine and carbonyl-functionalized COFs formed uniformly distributed cation-carrying and hydrophilic–hydrophobic nanochannels. This structure led to a high local alkalinity and cation-enriched catalytic microenvironment near the GDL surface, which suppresses HER and enhances the conversion of CO2 to multi-carbon products on copper in strongly acidic conditions.
In summary, the research on the microenvironment effects at the EEI provides guidance for the design of practical electrochemical devices. However, it remains to be verified whether these anions, cations, and organic compounds would affect the long-term stability of ionomers or membranes. Therefore, we encourage the validation of strategies based on planar electrode systems in practical devices, with particular emphasis on long-term stability tests to drive the practicality of these devices.
IN SITU/OPERANDO CHARACTERIZATION TECHNIQUES AND THEORETICAL SIMULATIONS
Conventional characterization techniques are often inadequate for observing the dynamic changes in mass/charge transport occurring at the EEI during electrocatalysis. To address this issue, a range of in situ/operando characterization techniques (including in situ visualization and spectroscopic methods) have been developed. Meanwhile, significant progress has been made in theoretical computational techniques based on EDL (AIMD) and electrochemical devices (continuum modeling) recently.
In situ visualization methods
In situ visualization can help monitor the bubble behavior at the interface, which is crucial for guiding the design of more efficient devices, especially for water/CO2RR electrolyzers. Sun et al. developed a simple method called bubble pump consumption chronoamperometry (BPCC) involving the use of a gas supply unit and a data acquisition system (comprising high-speed camera, light source, and electrochemical workstation) [88]. By injecting individual oxygen bubbles into GDEs and monitoring the diffusion behavior along real-time current evolution, the exact correlation between the actual current over time and the dynamics of oxygen diffusion consumption was established. This enabled the determination of key parameters such as the gas diffusion coefficient and density of effective catalytic sites of GDEs. Zhang et al. observed the process of H2 evolution in proton exchange membrane electrolyzer cells (PEMECs) through a specialized high-speed microscopic visualization setup [89]. They demonstrated that gas evolution and bubble nucleation predominantly take place on catalyst layers at the edges of pores in the thin/tunable liquid/gas diffusion layers (TT-LGDLs). In addition to bubble behavior, in situ visualization can also help observe the wetting characteristics at the interface. Tileti et al. employed in situ liquid-phase TEM with an electrochemical approach to uncover the variation of wetting characteristics of cobalt-based oxide catalysts during potential cycling [90]. This method establishes the real-time correlations between the wetting behavior of materials and OER, providing visual evidence for the study of solid–liquid interfacial interactions in electrochemistry.
The establishment of real-time characterization of component morphology and fluid dynamics is crucial for optimizing the performance and stability of fuel cells. Notably, Bazylak et al. combined advanced dual-modality tomography (simultaneous neutron and X-ray tomography) with sophisticated techniques for processing images (iterative reconstruction and metal artifact reduction) for characterization of fuel cell components (Fig. 7a) [91]. This method allows for the simultaneous, high-contrast, yet independent spatial and temporal analysis of both the morphology and moisture content of MEA components. In addition, Mustain et al. employed neutron imaging with operando micro–X-ray computed tomography (CT) to observe the distribution of liquid water over time and space within the functioning device [80]. This could lay the basis for optimizing water management.
![In situ/operando characterization techniques and theoretical simulations. (a) Fuel cell imaging schematic and sample neutron and X-ray images. Reproduced with permission from Ref. [91]. Copyright 2020 American Association for the Advancement of Science. (b) Model of shell-isolated nanoparticles (Au@SiO2 NPs, SHINs) at a Pt(111) surface and the mechanism of the ORR process revealed by the EC-SHINERS method. Reproduced with permission from Ref. [96]. Copyright 2019 Nature Publishing Group. (c) Representative structures of hydrated ions near the electrodes. Reproduced with permission from Ref. [102]. Copyright 2022 Nature Publishing Group. (d) Surface sites for the 3 × 4-(111) (top left) and 3 × 1-(211) (top right) slab models used for the site-partitioning of the AIMD trajectories. Symmetrically equivalent sites are shown in the same color. (Bottom) Schematic of Gaussian weights on surface sites with the Gaussian weighting function (fg) against the distance d. The weighting function overlap is depicted for two sites separated by the minimum distance dmin. Reproduced with permission from Ref. [103]. Copyright 2020 AIP Publishing LLC. (e) Standard electrochemical compression cell and corresponding physical model schematic representation of a typical experimental setup, and (f) the corresponding modeled domain for the BL model. Reproduced with permission from Ref. [107]. Copyright 2023 Elsevier Inc.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nsr/11/12/10.1093_nsr_nwae315/17/m_nwae315fig7.jpeg?Expires=1750214452&Signature=0-HlRMm85zni5mDk72u2cBs47uDDWhSBc~uPCA8piCqZY73w7L548f6AJbK8w3Frxn7ANJGOIgglaWyseyZyxUeRWyoxZCmX~8fiya3xpPrQXdvxpqT406jNjq7KechFYnNgjuX3~OyA6C6SgbiBi3xOazwapphA4JGigYL5XO~9AjllohHX7YCtGFqnQJcgodkt-x0XZHJhrVcqDOHdqimiHKFcOab7ku9qXXt0xLFc3N8urIZ2LIBIoeoWLQ97SgA6g-GsEomqFyy89xvK8SpNoqbMGxXnYvaB4tTR~tv5zamJANjOGefWnef7cNE5aUtdeCttL-sp2hv~gWVkNg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
In situ/operando characterization techniques and theoretical simulations. (a) Fuel cell imaging schematic and sample neutron and X-ray images. Reproduced with permission from Ref. [91]. Copyright 2020 American Association for the Advancement of Science. (b) Model of shell-isolated nanoparticles (Au@SiO2 NPs, SHINs) at a Pt(111) surface and the mechanism of the ORR process revealed by the EC-SHINERS method. Reproduced with permission from Ref. [96]. Copyright 2019 Nature Publishing Group. (c) Representative structures of hydrated ions near the electrodes. Reproduced with permission from Ref. [102]. Copyright 2022 Nature Publishing Group. (d) Surface sites for the 3 × 4-(111) (top left) and 3 × 1-(211) (top right) slab models used for the site-partitioning of the AIMD trajectories. Symmetrically equivalent sites are shown in the same color. (Bottom) Schematic of Gaussian weights on surface sites with the Gaussian weighting function (fg) against the distance d. The weighting function overlap is depicted for two sites separated by the minimum distance dmin. Reproduced with permission from Ref. [103]. Copyright 2020 AIP Publishing LLC. (e) Standard electrochemical compression cell and corresponding physical model schematic representation of a typical experimental setup, and (f) the corresponding modeled domain for the BL model. Reproduced with permission from Ref. [107]. Copyright 2023 Elsevier Inc.
In situ spectroscopies
As a good complement to in situ visualization methods, in situ spectroscopies (including in situ X-ray scattering, surface-enhanced infrared/Raman spectroscopies) provide molecular-level experimental evidence for the chemical composition and adsorption–desorption information at the EEI.
In situ X-ray scattering can probe deep into condensed matter (such as solid phase and liquid phase) without hindering mass transport and electrochemical reactions, making it relatively easy to operate. The MEA components of CO2RR electrolyzers have long been struggling with poor stability attributed to cathode flooding and salt precipitation. However, the understanding of the mechanisms behind these local environmental changes remains rather limited. Seger et al. employed operando X-ray analysis combined with online gas chromatography and mass spectrometry to monitor electrolyte precipitation and salt deposition in the GDE of CO2RR electrolyzers [92]. Concurrently, direct evidence of bicarbonate salt precipitation at the cathode was identified, which exacerbates the issue of electrolyte flooding and shifts the reaction selectivity towards HER. Furthermore, the team employed operando wide-angle X-ray scattering (WAXS) to reveal how salt formation in the MEA cathode varies with changes in alkaline cations [93]. The findings indicate that salts with lower solubilities (e.g. Li/Na-based) would crystalize faster, which limits the CO2 from accessing the catalyst and enhances the competing HER. The key to avoiding salt precipitation is to use highly soluble alkali metal salts (e.g. CsHCO3) in the anolytes along with an optimal salt concentration ranging from 0.01 to 0.1 M.
Infrared/Raman spectroscopies serve as potent tools for molecular fingerprinting based on vibration modes. In previous sections discussing hydrogen/oxygen reactions and CO2RR, many works have demonstrated the importance of these two spectroscopies in analyzing microenvironmental effects [14,15,23,42,43,48,50]. Here, we will further elaborate on their respective characteristics, drawing from recent representative studies. Generally, IR spectroscopy detects dipole-moment-induced light absorption, whereas Raman spectroscopy assesses polarizability-induced inelastic scattering. Raman active centrosymmetric molecules are typically IR-inactive in IR owing to the mutual exclusion rule, and vice versa. Consequently, in the majority of instances, Raman and IR spectroscopies provide supplementary information to each other [94]. In order to address the enduring constraints of SERS related to structure and material applicability, Tian et al. introduced the shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) technique [95]. In SHINERS, the application of an ultra-thin and consistent silica shell on gold nanoparticles proves effective in amplifying the Raman signals from molecules near the nanoparticle surface without disruption. This enables Raman signal acquisition from various substrates. Li et al. investigated the ORR process at Pt(hkl) single-crystal surfaces using the SHINERS technique (Fig. 7b) [96]. They acquired direct spectral proof of *OH, *HO2 and O2−, elucidating the ORR mechanism at both the molecular and atomic levels under acidic and alkaline conditions. SHINERS’ distinct benefit lies in its specific adaptability to investigate the adsorption arrangement and catalytic mechanisms of probe molecules on surfaces of single crystals. The team further employed SHINERS to study the structure and dissociation procedure of interfacial water on Pd single-crystal surfaces, offering insights into the mechanism of structural arrangement of interfacial water and the influence of local cation disruption on the water structure [97].
ATR-SEIRAS is another commonly used in situ spectroscopic technique supplementary to SERS. Smith et al. investigated the pH gradients at the polycrystalline Cu and phosphate buffer interface during CO2RR using in situ SEIRAS [98]. The results indicate that at the potential of −1 V vs. RHE, where hydrocarbons are formed, there is a significant difference of 5 pH units between the local pH near the electrode surface and the bulk solution pH. This suggests that activity analysis under mass transport limitations must consider the impact of the large concentration gradients. Xu et al. employed ATR-SEIRAS to investigate the impact of different alkali metal cations on the Au electrode surface CO2 concentration [99]. At −0.8 V vs. RHE, the interfacial CO2 concentrations were found to be lowest in the presence of Cs+, providing a plausible explanation for the cation effect. Recently, Yang et al. combined in situ infrared nanospectroscopy (nano-FTIR) with in situ SERS and successfully revealed the formation pathway of NP/ordered-ligand interlayer induced by bias-induced dissociation of tetradecylphosphonic acid (TDPA) ligands on Ag NPs [100]. They also found that the accumulation of non-covalent interactions in the interfacial microenvironment during ligand dissociation promotes the development of surface-constrained electric fields, eventually enhancing the selectivity and activity for CO2RR. The combined spectroscopic approach provides an excellent example for investigating the dynamic response of microenvironments with nanoscale resolution, offering guidance for studying the interfacial microenvironments of nanomaterials.
Ab initio simulation and continuum modeling
The progress of in situ characterization techniques has helped deepen the understanding of microenvironmental effects. However, these techniques still require further refinement to enhance the applicability in different EEIs and the reliability of data analysis. The theoretical simulation method can supplement, to some extent, the deficiency of experimental methods.
AIMD can efficiently establish the atomic-scale structure of the EDL, adjust the basic characteristics of the electrochemical interface, and deliberately introduce specific ions and solvents [101]. Kim et al. reproduced the experimentally observed characteristic peaks of electrochemical impedance spectroscopy (EIS) in dilute aqueous electrolyte through AIMD, and revealed that the origins for the two peaks observed at anodic and cathodic potentials are the electrosorption of ions and structural phase transition, respectively (Fig. 7c) [102]. This work demonstrates the advantages of the AIMD approach in building complex EDL structures, enabling direct validation of simulation results through comparison with experimental findings. In addition, AIMD can also predict solvent effects at solid/liquid interfaces with good accuracy. Chan et al. explored the solvation at charge-neutral metal/water interfaces using extensive AIMD simulations with commonly used continuum solvent models (Fig. 7d) [103]. They considered multiple metals (including Cu, Au, Pt) and various adsorbates relevant to electrochemical reactions (including CO2 reduction and oxygen redox), and reasonably quantified the solvation energies. These results provide a reference for establishing effective/parameterizable solvation models. Additionally, AIMD can effectively distinguish the configuration of the electrolyte at the interface and in the bulk liquid phase. Grob et al. analyzed the atomic configurations of water solvation layers on Pt(111) electrodes using AIMD [104]. The results revealed the difference of water molecule configurations between the surrounding solvation layer (through the oxygen atom bounding on Pt) and the bulk electrolyte (via H-up and H-down configurations), and less dense water solvation layers on hydrogen-covered Pt.
Benefiting from its advantages in simulating the EDL structure, AIMD is well suited as a powerful tool for analyzing microenvironment effects based on EEI. However, it is not as applicable for practical electrochemical devices working at high current densities because the corresponding microenvironment of porous electrodes often involves intricate multiphase fluid, mass transport, temperature, pressure, and other physical variables. Physics-based continuum modeling can play a crucial role in analyzing the microenvironment effects of electrochemical devices (include water electrolyzers, hydrogen–oxygen fuel cells, and CO2 electrolyzers), as it can link the device performance with electrode characteristics and operational conditions, and obtain theoretical product distributions and other device parameters [105]. This is crucial for the rational utilization of microenvironment effects to drive the design and preparation of high-performance devices. For instance, Weber et al. simulated the performance of Cu-based MEA for CO2RR through continuum modeling [106]. In this model, the team systematically investigated the effects of gas and liquid water transport, temperature, the properties of ionomer and membrane, heat transfer, and different feed conditions (liquid-fed and vapor-fed) on product distribution. The results indicate that higher temperatures (350 K) and sufficient water supply are advantageous for the formation of C2+ products. In addition, Bell et al. integrated continuum modeling and the covariance matrix adaptation evolutionary strategy (CMA-ES) to fit Tafel parameters for electrochemical CO2RR over Ag and Sn catalysts (Fig. 7e, f) [107]. This method eliminates subjective errors in fitting Tafel equations, establishing a more rigorous benchmark test. These results underscore the importance of accounting for mass transport and buffer kinetics in the analysis of the product of CO2 electrolyzers.
In summary, advanced in situ/operando techniques provide evidence of adsorption/desorption of reactants, products and, in particular, the intermediates, as well as real-time monitoring on the microenvironment status of device components. Further integration with the new theoretical calculation techniques can reasonably simulate the process of catalytic reactions and the operating status of devices. The development of these technologies and methods could well assist the study of microenvironment effects.
CONCLUSION AND PERSPECTIVE
The past few decades have witnessed the booming development of sustainable electrocatalysis, covering both fundamental research in laboratories and commercializable applications in industry. The regulation of the microenvironment (by means of pH, cation/anion effects and constructing organic compound interface, for instance, caffeine) has proven successful in boosting the reaction kinetics and stability and improving the selectivity for specific target products in the hydrogen/oxygen reaction and carbon dioxide reduction reaction. The rapid advance of in situ/operando characterization techniques and theoretical simulation methods offers powerful tools to probe the catalytic pathways in further depth, which adds to understanding of EDL structure and microenvironment effect. Thanks to these advanced methodologies, scientists have begun to try unveiling the underlying mechanisms of microenvironment regulation, and found that the interfacial electric field, interfacial structures (such as water layer and hydrogen bond network) and the local concentration/coverage of relevant species (such as protons, hydroxide ion, reactants and intermediates) may play notable roles. On the basis of a more in-depth understanding on the electrode/electrolyte interface, the strategies for microenvironment regulation could readily translate into the design and fabrication of high-performance electrochemical devices for practical applications, including water/CO2 electrolyzers and fuel cells, promising a grand picture of large-scale implementation for green energy and sustainable development. Nevertheless, some related topics are still under debate, and a few subareas may need to be explored in further depth. At the end of this review, we share our views on several aspects that call for more thorough scrutiny.
Emphasis on a more accurate model for the electrical double layer model. A clear and complete model for the EDL structure is the key to deciphering the microenvironment effect. It is of great necessity to conduct standardized measurements on the EDL properties (for instance, the electric fields for different EEIs, PZC) and to establish a more comprehensive database. The complexity in electrode preparation in metallic single-crystal electrode and the high requirements on the precision of electrocatalytic measurements call for advanced, high-efficiency characterization methodologies. In addition, the emerging in situ/operando characterization and theoretical simulation techniques are of great significance in providing more specific information on the EDL (such as the solvation of electrode surface, the location and interaction of solvated anions/cations and impact of the electric field).
Emphasis on the research on microenvironment regulation for low-cost catalysts based on non-platinum–group metals. Historically, research on the microenvironment regulation of noble metal electrodes such as Pt electrodes helped to establish the correlation between EEI and hydrogen/oxygen reaction kinetics, yet the generality of this correlation for different electrode surfaces needs to be further validated. Currently, a vast number of low-cost, high-performance catalysts based on non-precious metals have been developed, and the research on microenvironment regulation for these electrode materials is of high necessity.
Emphasis on the research on microenvironment regulation of advanced electrochemical devices operating at industrially-relevant current densities. For those advanced electrochemical devices such as MEA, the experimental tests are more complicated than those for planar electrodes, and the corresponding results are more subject to stochastic errors. Therefore, it is necessary to standardize the measurements for these advanced devices and to conduct multiple parallel tests so as to minimize errors. In addition, we noted that a few readily implementable ‘intermediate test techniques’ (between the conventional three-electrode configuration and the MEA) have emerged recently, such as promoting the mass transport for RDE [57], and using the floating electrode setup [108]. From a technoeconomic perspective, achieving high activity, selectivity, and stability at low overpotentials and at industrial-level current densities in advanced electrochemical devices presents greater challenges and significance. Another topic that needs particular attention is the effect of microenvironment on the long-term stability of devices. A typical example is the CO2 electrolyzer designed on the basis of cation effects, where the high-concentration cations could elevate catalytic efficiency and improve product selectivity, yet this could also exacerbate the problem of carbonate precipitation, thereby shortening the life cycle of the device. Therefore, in practice, a subtle balance needs to be maintained between the two conflicting aspects, so as to ensure the long-term operation of the devices featuring optimized reaction microenvironments.
FUNDING
This work was supported by the National Key R&D Program of China (2021YFF0500500), the National Natural Science Foundation of China (21925202 and U22B2071), Yunnan Provincial Science and Technology Project at Southwest United Graduate School (202302AO370017), and the International Joint Mission On Climate Change and Carbon Neutrality.
Conflict of interest statement. None declared.
REFERENCES
Author notes
Equally contributed to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}