






Abstract
A genetic predisposition to tumor development can be identified in up to 10% of pediatric patients with central nervous system (CNS) tumors. For some entities, the rate of an underlying predisposition is even considerably higher. In recent years, population-based approaches have helped to further delineate the role of cancer predisposition in pediatric oncology. Investigations for cancer predisposition syndrome (CPS) can be guided by clinical signs and family history leading to directed testing of specific genes. The increasingly adopted molecular analysis of tumor and often parallel blood samples with multi-gene panel, whole-exome, or whole-genome sequencing identifies additional patients with or without clinical signs. Diagnosis of a genetic predisposition may put an additional burden on affected families. However, information on a given cancer predisposition may be critical for the patient as potentially influences treatment decisions and may offer the patient and healthy carriers the chance to take part in intensified surveillance programs aiming at early tumor detection. In this review, we discuss some of the practical and ethical challenges resulting from the widespread use of new diagnostic techniques and the most important CPS that may manifest with brain tumors in childhood.
Approximately 300,000 children worldwide are diagnosed with cancer every year.1 Central nervous system (CNS) tumors account for 20% of all pediatric cancers but are the major cause of morbidity and mortality in pediatric oncology.2 Over the recent years, there has been an increasing interest in the different factors contributing to the development of cancer. In pediatric, adolescent, and young adult (AYA) patients, for whom external carcinogens have a smaller role in tumorigenesis compared to adults, a genetic predisposition may in some cases explain susceptibility to cancer early in life. Studies examining large cohorts of pediatric oncology patients with different types of cancer have identified a genetic tumor predisposition in 7%-10% of patients. Patients suffering from CNS malignancies are overall more likely to have an underlying genetic predisposition than patients with other malignancies.3–6 For several reasons, 7%-10% may still underestimate the actual contribution of cancer predisposition syndrome (CPS) to pediatric/AYA CNS malignancies. Most of the present studies investigating cancer predisposition included only patients with availability of both tumor and healthy control tissue. However, some tumors that are typically associated with a genetic predisposition are not biopsied on a regular basis, eg, optic pathway gliomas (OPG) in neurofibromatosis 1 (NF1), thus respective alterations are most probably underrepresented. Further, diagnostic screening studies are often limited to a pre-defined list of established predisposition genes. However, there is growing evidence from analyses of international multicenter cohorts, including retrospective and prospective clinical studies and patient series, that additional genes predispose to pediatric brain cancer.6–8 Population-based studies using whole-exome sequencing (WES) or whole-genome sequencing (WGS) approaches like the INFORM study or the Zero Childhood Cancer Program may reveal further candidate genes.9,10
Dealing with a case of pediatric cancer may be one of the most severe stressors that parents endure. The diagnosis of a cancer predisposition puts an additional burden on the patients and their families. There are ethical, legal, and social issues to consider when investigations are initiated that may lead to the diagnosis of a genetic predisposition. The provision of adequate genetic counseling and psychological support for these families is paramount. In this non-comprehensive review, we want to
(1) sensitize the reader and raise the index of suspicion to identify children suffering from CNS malignancies on the ground of an underlying tumor predisposition,
(2) describe some of the ethical and practical issues to be considered when extensive molecular analysis is initiated that may result in identification of a tumor predisposition,
(3) describe the most common tumor predisposition syndromes encountered in pediatric neuro-oncology with a focus on the respective intracranial manifestations.
Clinical Recognition of Cancer Predisposition Syndrome
Sometimes a genetic disorder predisposing to tumor development is already known to a patient before a diagnosis of cancer is made, eg, NF1 can be known before an associated OPG becomes apparent, especially if other members of the family are affected. This is typically the case if the genetic disorder has additional characteristics besides tumor development, eg, dysmorphic features or malformations. Nevertheless, more often a cancer predisposition is not obvious to the clinician. Still, it is important to consider a predisposition at the time of cancer diagnosis as, in some cases, the underlying genetic disorder may influence the treatment regimen. Examples would be SHH-driven medulloblastoma in Gorlin or Li-Fraumeni syndrome (LFS) for which irradiation sparing approaches should be discussed. To help the identification of affected patients in pediatric oncology certain clinical criteria suggestive of a genetic predisposition have been defined, including11,12:
(1) a positive family history of cancer,
(2) certain types of tumors often associated with a genetic predisposition,
(3) multiple synchronous or metachronous tumors,
(4) extraordinary treatment toxicity,
(5) other clinical signs and symptoms indicative of a genetic disorder,
(6) somatic genetic findings indicating an underlying constitutional variant.
Many pediatric oncology centers now use standardized questionnaires to assess risk factors that may hint towards a genetic predisposition.11,12 Validation of these questionnaires is ongoing, but first results show that they succeed in identifying patients with a cancer predisposition.13 It is important to allow enough time for assessing the past medical history of the patient and the family and a thorough clinical examination. Some signs of genetic diseases like subtle skin changes or ear creases may be overlooked during routine clinical examinations. Thus, in some cases, involvement of a clinical geneticist experienced in the diagnosis of clinical syndromes may be helpful.
A growing number of techniques (eg, multi-gene panel sequencing, WES, WGS, and transcriptome or methylome analyses) are now available at reasonable costs for tumor analysis. Sometimes a genetic predisposition is not suspected clinically but the molecular analysis of the tumor may reveal features typical of a CPS. Somatic pathogenic variants in certain cancer-predisposing genes may warrant investigations to confirm or exclude if they are present constitutionally as the likelihood of the being constitutional may exceed 10%.14,15 Additionally, there are some specific findings in next-generation sequencing (NGS) that may indicate an underlying genetic predisposition that are independent of the genomic sequence. Prominent examples are chromothripsis in LFS-associated tumors or the presence of a hyper- or ultramutant phenotype in DNA repair disorders, especially constitutional mismatch repair deficiency (CMMRD).16,17 Somatic mutational signature analysis or assessment of tumor mutational burden can be helpful in this context.4,18 Methylation studies may lead to the diagnosis of molecular brain tumor subgroups often associated with constitutional variants like the sonic hedgehog (SHH) subgroup of medulloblastoma and guide further investigation.6
For several reasons, it is important to review the possibility of a CPS regularly during treatment. During the stressful time around diagnosis, the family may not be aware of or able to recall a family history. In addition, some information, like somatic genetic data or treatment toxicity will only be available after treatment has been initiated.
If there is clinical suspicion for a CPS genetic counseling should be offered. In some cases, a diagnosis can be clinically confirmed using established disease-specific criteria, eg, NF1.19 Directed genetic testing for the respective syndrome can be discussed with the patient and the family to confirm or rule out a clinically suspected diagnosis.
In our experience with population-based analyses within INFORM, a prospective, non-interventional, and multi-national registry collecting clinical and molecular data of relapsed, progressive, or high-risk pediatric cancer (www.dkfz.de/en/inform) and MNP2.0, which combines DNA methylation profiling and targeted gene panel sequencing at primary diagnosis of CNS malignancies (www.kitz-heidelberg.de/en/for-physicians/clinical-studies/molecular-diagnostics-studies/mnp-20), about half of the CPS identified by systematic sequencing approaches have not previously been clinically determined (unpublished data). Thus, it seems that clinical criteria may not be able to reliably identify all patients with a genetic predisposition, eg, patients with de novo variants.
Reporting of Constitutional Genomic Variants
Both the INFORM and MNP2.0 studies are designed to improve diagnostic classification and guide treatment by molecular analysis of tumor tissues. In these studies, as it is common practice, normal tissue control samples (most commonly blood) are analyzed in parallel with tumor samples to allow for more precise identification of true somatic variants, which are not detectable in the normal tissue control.
In such context, analysis of constitutional DNA is often not intended and the availability of the data creates ethical and practical challenges that need to be addressed. Thus, if healthy control tissue is analyzed for an associated cancer predisposition in the sense of an opportunistic screening it needs to be discussed beforehand if, to what extent, and how any constitutional variants will be communicated to the patient. Paramount guiding principles are that any decision has to be made in the best interest of the child and that any possible harm to the patient, eg, anxiety or unnecessary invasive investigations must be avoided. The child’s current needs and interests should be considered, but also the child’s future interests (eg, access to surveillance for adult-onset tumors, the child’s autonomy, and the right not-to-know, ie, “the child’s right to an open future”). This may also affect the interests of other family members involved, especially the interests of the child’s parents should also be considered. It may at least be conceivable that a potentially life-saving diagnosis of a cancer predisposition in a parent is indeed in the interest of the child.
To be able to inform the patient or the family about the diagnosis of a tumor predisposition, the patient and/or his guardians have to consent to investigation of constitutional DNA beforehand. Although not required by law in all jurisdictions, this should include the assent, ie, the agreement of older children and adolescents.20 Patient/parent counseling should include explanation and discussion of possible findings and respective consequences for the patient and other family members that may result from these. Most patients and parents choose to be informed about a possible cancer predisposition.21,22
Generally, variants that are reported to the patient should be analytically valid, they should have clinical significance and they should be actionable.
To ensure analytical validity, the validation of results generated in a research setting in an accredited laboratory may be useful. Variants of unknown significance (VUS) or variants in genes of unknown significance (GUS) are genetic variants that cannot be classified as either pathogenic/likely pathogenic or benign/likely benign in the context of the patient’s disease with the available evidence.23,24 In such cases, the functional consequence of the variant at protein level is not known or the role of the affected gene in the patient’s condition is unclear. With WES or WGS, and even with larger multi-gene panels, the chances of discovering VUS in genes related to cancer are high. Additional data generated from tumor analyses, such as mutational signatures, RNA sequencing, and expression analysis may help to increase diagnostic certainty and help to classify VUS.25,26 Practices for reporting VUS differ between laboratories.27 If the analysis of constitutional DNA is established as a part of a molecular diagnostic program it should be defined a priori, if, in what context, and how the presence of VUS in relevant genes will be reported. In our daily practice, we only report VUS in a diagnostic setting, ie, if directed testing of a specific set of genes guided by the patient’s medical and family history reveals a VUS. In a research context like in INFORM and MNP2.0, where analysis is often independent of the patient’s specific situation, we and others consider the identification of constitutional genetic variants to be secondary findings and do not report VUS to the patient.28–30 To that end, study protocols, patient information and informed consent often stipulate that the patient will only be informed about likely pathogenic or pathogenic variants related to the patient’s cancer diagnosis, but not about VUS. To make an informed decision whether a VUS is indeed rather likely pathogenic or pathogenic a thorough review of the clinical course and the family history may be helpful. However, especially in a research context, this information is often unknown to the person interpreting the constitutional variants. Requesting additional information from the patient once analyses are completed may already put an unnecessary burden of uncertainty on patients and their families.
Given these difficulties and challenges, the procedure for incidental or secondary findings should be discussed with the patient during the consent process. The 2016 guidelines of the American College of Medical Genetics (ACMG) recommend the analysis of 59 medically actionable genes causing severe, heritable diseases in every patient with an exome- or genome-wide analysis in a diagnostic setting. Twenty-four of these genes are cancer-related.31 It is of note that the actionability of the genes on the ACMG list has been assessed for adult patients and some genes may be considered not actionable in children.32
European guidelines also recommend reporting of secondary findings that are indicative of a serious health issue, but stop short of providing a list of genes for obligatory analysis or opportunistic genomic screening (“where the opportunity should be used to routinely and systematically look for secondary findings.” 33,34 Still, the patients’ right not-to-know should be taken into account and the current ACMG guidelines recommend an option to opt-out during the informed consent process.23 Notably, both American and European guidelines suggest that the patient’s right not-to-know does not automatically over-ride professional responsibilities when the patient’s own health or that of close relatives are at stake.30,33 In addition, national legislation always needs to be considered.35 Selective predictive testing of children for known familial genetic disorders is traditionally only permissible for childhood-onset diseases.36,37 However, the original 2013 ACMG guidelines on incidental findings stated “that the ethical concerns about providing children with genetic risk information about adult-onset diseases were outweighed by the potential benefit to the future health of the child and the child’s parent of discovering an incidental finding where intervention might be possible,” and recommended analyzing and reporting of the ACMG gene list in children.30 While the ACMG argue that relevant information may be lost to the families by not reporting predisposition for late-onset disorders, the ESHG recently claimed “further ethical scrutiny” in this context, eg, to decide whether “the possible future benefits for the child are sufficiently weighty to trump the remaining concerns about violating the minor’s right to informational privacy.” 34 This view is not shared by other expert groups arguing against predictive testing of adult-onset disorders in children.38
In pediatric oncology, situations may get complicated further, since it is sometimes not entirely clear whether a constitutional variant identified in a tumor predisposition gene with a typical adult-onset of cancer, eg, BRCA2, is indeed contributing to the development of the child’s tumor or not. These findings are in a gray zone between intended and secondary findings as well as between diagnostic and predictive testing. Further research, including the systematic analysis of somatic mutational processes as a putative result of a constitutional variant is required to delineate the role of adult-onset cancer predisposition genes in pediatric tumors. Patient registries may help to follow-up these patients and better understand the cancer risks for pediatric carriers and to investigate what other genetic or nongenetic factors that may modulate the tumor risk in individuals with a cancer predisposition.
Patients should also be made aware during consultation that molecular genetic testing does not necessarily deliver an explanation why the child has developed cancer. In many cases, even with suggestive family histories, no genetic predisposition can be detected. This does not fully exclude an underlying genetic change facilitating cancer development in the affected patient. Genetic alterations may be missed for technical reasons (sequencing methods or analytical pipelines), eg, intronic variants outside of the canonical splice sites or large deletions. Furthermore, our current understanding of cancer predisposition is limited and with few exceptions restricted to mostly autosomal-dominant inherited single-gene disorders caused by pathogenic variants in tumor suppressor genes or, in a few cases only oncogenes. Thus, many laboratories have employed a candidate gene approach, often only investigating 100-200 genes known to be associated with cancer.39 In consequence, variants in rare or unknown cancer-predisposing genes like the recently described GPR161-related predisposition syndrome associated with sonic hedgehog medulloblastoma in infants will not be identified.8 In some cases, postzygotic mosaicism may explain why a blood sample did not reveal an underlying tumor predisposition. Mosaicism is a well-described phenomenon in which individuals harbor 2 or more populations of genetically distinct cells as a result of an early postzygotic mutation. Depending on the timing in embryonic development, mosaicism may or may not be detectable across different tissues, including blood. Mosaicism is an important and most likely underreported phenomenon in cancer predisposition. It has been described for several cancer-predisposing genes, eg, for NF1 in patients with OPG and for TP53 in patients with choroid plexus carcinoma.40,41 If suspected, mosaicism can sometimes be detected in blood using very deep sequencing (coverage > ×1000) or using samples of otherwise affected tissue, like skin lesions or independent tumors.42 In addition, hundreds of single-nucleotide polymorphisms (SNP), ie, genetic variants that are common in the general population, have been associated with mildly increased risk for various tumor types in different populations.43 The role of these SNPs in pediatric/AYA cancers has not been intensively investigated yet. Furthermore, the role of sequence variants in noncoding regions and epigenetic changes are poorly understood and are not regularly analyzed.
Cancer Predisposition Syndromes With a Wide Tumor Spectrum Including CNS Malignancies
Li-Fraumeni Syndrome
LFS is a highly penetrant, early-onset tumor predisposition syndrome caused by heterozygous pathogenic variants in the TP53 gene.44 Clinical criteria to guiding directed genetic diagnosis are detailed in Table 1.45 Up to 40% of patients are diagnosed with cancer before their 18th birthday.46 Tumors classically associated with LFS are soft-tissue sarcoma, osteosarcoma, premenopausal breast cancer, brain tumors, and adrenocortical carcinoma. In pediatric neuro-oncology, some entities are highly associated with LFS. Constitutional TP53 variants can be identified in up to 50% of choroid plexus carcinomas,46 a disease-defining tumor of LFS. Further, LFS is also associated with medulloblastoma (SHH subgroup), glioma, especially K27 wildtype high-grade gliomas (HGG) and other rare CNS malignancies.6,47,48
Criteria for TP53 Testing in Cancer Patients
| Clinical Criteria for Li-Fraumeni Syndrome | |
|---|---|
| Classical criteria (all must apply) | A patient with a sarcoma diagnosed before age 45 yr |
| A first-degree relative with any cancer before age 45 yr | |
| A first- or second-degree relative with any cancer before age 45 yr or a sarcoma at any age | |
| Revised Chompret criteria (only one must apply) | Familial presentation: Proband with an LFS spectrum tumora prior to age 46 yr and at least 1 first- or second-degree relative with an LFS tumor (except breast cancer, if the proband has breast cancer) before the age of 56 yr or with multiple tumors |
| Multiple tumors: Proband with multiple malignancies (except 2 breast cancers), of which at least 2 belong to the LFS spectrum, before the age of 46 yr | |
| Rare tumors: Patients with ACC, choroid plexus carcinoma, or embryonal anaplastic subtype rhabdomyosarcoma independent of family history | |
| Early-onset breast cancer: Patients with breast cancer before the age of 31 yr |
| Clinical Criteria for Li-Fraumeni Syndrome | |
|---|---|
| Classical criteria (all must apply) | A patient with a sarcoma diagnosed before age 45 yr |
| A first-degree relative with any cancer before age 45 yr | |
| A first- or second-degree relative with any cancer before age 45 yr or a sarcoma at any age | |
| Revised Chompret criteria (only one must apply) | Familial presentation: Proband with an LFS spectrum tumora prior to age 46 yr and at least 1 first- or second-degree relative with an LFS tumor (except breast cancer, if the proband has breast cancer) before the age of 56 yr or with multiple tumors |
| Multiple tumors: Proband with multiple malignancies (except 2 breast cancers), of which at least 2 belong to the LFS spectrum, before the age of 46 yr | |
| Rare tumors: Patients with ACC, choroid plexus carcinoma, or embryonal anaplastic subtype rhabdomyosarcoma independent of family history | |
| Early-onset breast cancer: Patients with breast cancer before the age of 31 yr |
Abbreviations: ACC, adrenocortical carcinoma; LFS, Li-Fraumeni syndrome.
aLFS spectrum tumors: premenopausal breast cancer, soft-tissue sarcoma, brain tumor, and adrenocortical carcinoma.
Criteria for TP53 Testing in Cancer Patients
| Clinical Criteria for Li-Fraumeni Syndrome | |
|---|---|
| Classical criteria (all must apply) | A patient with a sarcoma diagnosed before age 45 yr |
| A first-degree relative with any cancer before age 45 yr | |
| A first- or second-degree relative with any cancer before age 45 yr or a sarcoma at any age | |
| Revised Chompret criteria (only one must apply) | Familial presentation: Proband with an LFS spectrum tumora prior to age 46 yr and at least 1 first- or second-degree relative with an LFS tumor (except breast cancer, if the proband has breast cancer) before the age of 56 yr or with multiple tumors |
| Multiple tumors: Proband with multiple malignancies (except 2 breast cancers), of which at least 2 belong to the LFS spectrum, before the age of 46 yr | |
| Rare tumors: Patients with ACC, choroid plexus carcinoma, or embryonal anaplastic subtype rhabdomyosarcoma independent of family history | |
| Early-onset breast cancer: Patients with breast cancer before the age of 31 yr |
| Clinical Criteria for Li-Fraumeni Syndrome | |
|---|---|
| Classical criteria (all must apply) | A patient with a sarcoma diagnosed before age 45 yr |
| A first-degree relative with any cancer before age 45 yr | |
| A first- or second-degree relative with any cancer before age 45 yr or a sarcoma at any age | |
| Revised Chompret criteria (only one must apply) | Familial presentation: Proband with an LFS spectrum tumora prior to age 46 yr and at least 1 first- or second-degree relative with an LFS tumor (except breast cancer, if the proband has breast cancer) before the age of 56 yr or with multiple tumors |
| Multiple tumors: Proband with multiple malignancies (except 2 breast cancers), of which at least 2 belong to the LFS spectrum, before the age of 46 yr | |
| Rare tumors: Patients with ACC, choroid plexus carcinoma, or embryonal anaplastic subtype rhabdomyosarcoma independent of family history | |
| Early-onset breast cancer: Patients with breast cancer before the age of 31 yr |
Abbreviations: ACC, adrenocortical carcinoma; LFS, Li-Fraumeni syndrome.
aLFS spectrum tumors: premenopausal breast cancer, soft-tissue sarcoma, brain tumor, and adrenocortical carcinoma.
Up to 60% of SHH medulloblastoma associated with LFS may show chromothripsis, ie, massive chromosomal rearrangements.16 Thus, a finding of chromothripsis in SHH medulloblastoma should prompt investigation for constitutional variants.
Although there are concerns about the risk of second malignancies after radiochemotherapy there are currently no specific treatment recommendations for patients with LFS. The European medulloblastoma SIOP PNET5 (NCT02066220) protocol is the first addressing the question of whether avoiding alkylating agents and reduction of radiation allows for better survival and less treatment-related toxicity in patients with a constitutional pathogenic TP53 variant.
Surveillance recommendations have been established for both children and adults with clinical examinations and regular imaging including whole-body and dedicated cranial MRI as well as ultrasound (US).49,50 They have been shown to allow for early detection of tumors that is associated with prolonged survival and to be cost-efficient.51,52 To reduce possible false-positive and false-negative results, early-detection imaging should ideally be carried out in a center with experience in the assessment of whole-body MRIs of asymptomatic patients.53
It is generally accepted that there is a genotype-phenotype correlation in LFS, ie, that missense variants in the crucial DNA binding domain can have a dominant-negative effect and lead to a higher burden of disease than nonsense variants and variant-specific surveillance has been proposed.54,55 In our opinion, there are currently not enough data yet to customize screening protocols taking a genotype-phenotype correlation into account, but with more data collected in designated registries, this will be possible in the future.
Constitutional Mismatch Repair Deficiency (CMMRD)
In 1959, Turcot first reported 2 patients presenting with polyposis, colorectal carcinoma (CRC), and CNS malignancies.56 Since then, the combination of colorectal cancer and CNS tumors had been referred to with the meanwhile outdated term Turcot syndrome. This led to an overlap in terminology between CMMRD, Lynch syndrome (LS), and familial adenomatous polyposis (FAP); all of which can present with colorectal cancer and CNS tumors in a single patient or family.
CMMRD is a rare but highly penetrant, autosomal-recessive CPS defined by early-onset or synchronous malignancies and loss of DNA mismatch repair (MMR). It is caused by bi-allelic pathogenic variants in one of the MMR genes MSH2, MSH6, MLH1, and PMS2 or rarely EPCAM or MSH3.57–60 Mono-allelic variants in these genes cause the autosomal-dominant LS (see below).
Tumors in CMMRD include CRC, brain tumors, especially glioblastoma and hematological and lymphoid malignancies. In 2014, clinical criteria for the diagnosis of CMMRD using a scoring system have been proposed (Table 2).61 Importantly, the clinical phenotype may overlap with neurofibromatosis, and patients with CMMRD may fulfill the clinical criteria of NF1.62
Criteria for CMMRD Testing in Cancer Patients
| Indication Criteria for CMMRD Testing in Cancer Patients: Test If ≥3 Points | |
|---|---|
| Malignancies/premalignancies: One is mandatory; if more than one is present in the patient, add the points | |
| Carcinoma from the Lynch syndrome (LS) spectruma at age <25 yr | 3 points |
| Multiple bowel adenomas at age <25 yr and the absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 yr | 3 points |
| WHO grade III or IV glioma at age <25 yr | 2 points |
| NHL of T-cell lineage or sPNET at age <18 yr | 2 points |
| Any malignancy at age <18 yr | 1 point |
| Additional features: Optional; if more than one of the following is present, add the points | |
| Clinical sign of NF1 and/or ≥2 hyperpigmented and/or hypopigmented skin alterations Ø > 1 cm in the patient | 2 points |
| Diagnosis of LS in a first- or second-degree relative | 2 points |
| Carcinoma from LS spectruma before the age of 60 in first-, second-, and third-degree relative | 1 point |
| A sibling with carcinoma from the LS spectruma, high-grade glioma, sPNET, or NHL | 2 points |
| A sibling with any type of childhood malignancy | 1 point |
| Multiple pilomatricomas in the patient | 2 points |
| One pilomatricoma in the patient | 1 point |
| Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient | 1 point |
| Consanguineous parents | 1 point |
| Deficiency/reduced levels of IgG2/4 and/or IgA | 1 point |
| Indication Criteria for CMMRD Testing in Cancer Patients: Test If ≥3 Points | |
|---|---|
| Malignancies/premalignancies: One is mandatory; if more than one is present in the patient, add the points | |
| Carcinoma from the Lynch syndrome (LS) spectruma at age <25 yr | 3 points |
| Multiple bowel adenomas at age <25 yr and the absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 yr | 3 points |
| WHO grade III or IV glioma at age <25 yr | 2 points |
| NHL of T-cell lineage or sPNET at age <18 yr | 2 points |
| Any malignancy at age <18 yr | 1 point |
| Additional features: Optional; if more than one of the following is present, add the points | |
| Clinical sign of NF1 and/or ≥2 hyperpigmented and/or hypopigmented skin alterations Ø > 1 cm in the patient | 2 points |
| Diagnosis of LS in a first- or second-degree relative | 2 points |
| Carcinoma from LS spectruma before the age of 60 in first-, second-, and third-degree relative | 1 point |
| A sibling with carcinoma from the LS spectruma, high-grade glioma, sPNET, or NHL | 2 points |
| A sibling with any type of childhood malignancy | 1 point |
| Multiple pilomatricomas in the patient | 2 points |
| One pilomatricoma in the patient | 1 point |
| Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient | 1 point |
| Consanguineous parents | 1 point |
| Deficiency/reduced levels of IgG2/4 and/or IgA | 1 point |
Abbreviations: CMMRD, constitutional mismatch repair deficiency; NHL, non-Hodgkin lymphoma; sPNET, supratentorial primitive neuroectodermal tumors.
aLS spectrum tumors: colorectal, endometrial, small bowel, ureter, renal pelvis, biliary tract, stomach, and bladder carcinoma.
Adapted from Adam et al60.
Criteria for CMMRD Testing in Cancer Patients
| Indication Criteria for CMMRD Testing in Cancer Patients: Test If ≥3 Points | |
|---|---|
| Malignancies/premalignancies: One is mandatory; if more than one is present in the patient, add the points | |
| Carcinoma from the Lynch syndrome (LS) spectruma at age <25 yr | 3 points |
| Multiple bowel adenomas at age <25 yr and the absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 yr | 3 points |
| WHO grade III or IV glioma at age <25 yr | 2 points |
| NHL of T-cell lineage or sPNET at age <18 yr | 2 points |
| Any malignancy at age <18 yr | 1 point |
| Additional features: Optional; if more than one of the following is present, add the points | |
| Clinical sign of NF1 and/or ≥2 hyperpigmented and/or hypopigmented skin alterations Ø > 1 cm in the patient | 2 points |
| Diagnosis of LS in a first- or second-degree relative | 2 points |
| Carcinoma from LS spectruma before the age of 60 in first-, second-, and third-degree relative | 1 point |
| A sibling with carcinoma from the LS spectruma, high-grade glioma, sPNET, or NHL | 2 points |
| A sibling with any type of childhood malignancy | 1 point |
| Multiple pilomatricomas in the patient | 2 points |
| One pilomatricoma in the patient | 1 point |
| Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient | 1 point |
| Consanguineous parents | 1 point |
| Deficiency/reduced levels of IgG2/4 and/or IgA | 1 point |
| Indication Criteria for CMMRD Testing in Cancer Patients: Test If ≥3 Points | |
|---|---|
| Malignancies/premalignancies: One is mandatory; if more than one is present in the patient, add the points | |
| Carcinoma from the Lynch syndrome (LS) spectruma at age <25 yr | 3 points |
| Multiple bowel adenomas at age <25 yr and the absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 yr | 3 points |
| WHO grade III or IV glioma at age <25 yr | 2 points |
| NHL of T-cell lineage or sPNET at age <18 yr | 2 points |
| Any malignancy at age <18 yr | 1 point |
| Additional features: Optional; if more than one of the following is present, add the points | |
| Clinical sign of NF1 and/or ≥2 hyperpigmented and/or hypopigmented skin alterations Ø > 1 cm in the patient | 2 points |
| Diagnosis of LS in a first- or second-degree relative | 2 points |
| Carcinoma from LS spectruma before the age of 60 in first-, second-, and third-degree relative | 1 point |
| A sibling with carcinoma from the LS spectruma, high-grade glioma, sPNET, or NHL | 2 points |
| A sibling with any type of childhood malignancy | 1 point |
| Multiple pilomatricomas in the patient | 2 points |
| One pilomatricoma in the patient | 1 point |
| Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient | 1 point |
| Consanguineous parents | 1 point |
| Deficiency/reduced levels of IgG2/4 and/or IgA | 1 point |
Abbreviations: CMMRD, constitutional mismatch repair deficiency; NHL, non-Hodgkin lymphoma; sPNET, supratentorial primitive neuroectodermal tumors.
aLS spectrum tumors: colorectal, endometrial, small bowel, ureter, renal pelvis, biliary tract, stomach, and bladder carcinoma.
Adapted from Adam et al60.
Pathogenic variants in PMS2 are a rare cause of LS (<5%-15%) but account for a majority (60%) of CMMRD cases.63 The less aggressive course of PMS2-associated CMMRD may lead to a higher rate of detection, as patients with secondary and tertiary malignancies are more likely to be referred for genetic diagnostics.62 Special attention needs to be paid when sequencing PMS2 as the presence of pseudogenes with large overlapping sequences may interfere with the interpretation of sequencing results.64 For that reason, PMS2 is often excluded from diagnostic multi-gene panels for CNS tumors and may have to be requested separately.
Glioblastoma in CMMRD can acquire a somatic mutation in POLD1 or POLE that leads to an ultra-mutator phenotype with a typical molecular signature characterized by single-nucleotide changes present in exponentially high numbers (>100/mb).17 Even if this ultra-mutator phenotype is not present, CMMRD-related tumors often display a high mutational load and microsatellite instability (MSI) that can now be assessed reliably using NGS data.65–67 The presence of MMR mutational signatures or MSI markers in the sequencing data should trigger a thorough review of the MMR genes. On the other hand, it is important to be aware that especially the non-GI tumors in CMMRD may not display MSI.68,69 New techniques also allow for the detection of MSI in blood to suggest a diagnosis of CMMRD.63,70,71 Despite new technologies becoming increasingly available tried and tested methods like immunohistochemistry and detection of MSI by PCR still play an important role in diagnostics.
A surveillance protocol for patients with CMMRD has recently been published recommending regular imaging with MRI and US, blood tests, and gastrointestinal endoscopy from age 6.72
Interestingly, despite the loss of MMR function, there are thus far no reports on extensive toxicity following radio- or chemotherapy.72 Several common chemotherapeutic agents, eg, temozolomide that is heavily used in the treatment of glioblastoma, require adequate MMR to exert their tumor damage. While the response to temozolomide is limited in CMMRD patients, the drug could still further increase the mutational load and drugs should be avoided in CMMRD patients. Instead, hyper-mutant tumors may respond to treatment with immune-checkpoint inhibitors.73 CMMRD may be amendable to chemoprevention, possible agents include aspirin to reduce adenoma-associated inflammation or immune-checkpoint inhibitors.72,74
Lynch Syndrome
In most cases, parents of a child with CMMRD will both have LS, as they will be heterozygous for the MMR gene variant. Interestingly, especially in PMS2-related CMMRD, the family history may be unremarkable due to the low penetrance.69 Families of affected individuals should be offered genetic counseling and if necessary, surveillance for LS-associated tumors.
The cancer risk for pediatric heterozygous carriers is not very well delineated. CNS malignancies, especially glioblastoma are known tumor entities within LS but these usually occur in adult patients. Nonetheless, there are several case series and reports of pediatric carriers developing brain cancers like glioblastoma or oligodendroglioma and this is in keeping with our own experience.75–77 Loss of heterozygosity (LOH) in the tumor, loss of protein expression in immunohistochemistry and MSI may be helpful to determine if the constitutional MMR variant is indeed contributing to tumorigenesis.
Familial Adenomatous Polyposis
FAP is an autosomal-dominant CPS caused by pathogenic variants in the APC gene. It is characterized by the development of numerous (often >1000) gastrointestinal polyps at young age and a high risk for the development of CRC. The wider tumor spectrum includes medulloblastoma, typically of the WNT subgroup. While only a small fraction of all medulloblastoma are caused by APC alterations (~1%), they account for a majority of CTNNB1-wildtype WNT-driven medulloblastoma.6,78 Thus, patients with a medulloblastoma of the WNT group and the absence of a somatic CTNNB1 exon 3 mutation should be routinely tested for pathogenic APC variants following genetic counseling.6
Neurofibromatosis 1
NF1 is an autosomal-dominantly inherited neuro-cutaneous disorder characterized by typical skin findings and the development of multiple neurofibromas. Clinical criteria for the diagnosis of NF1 rely on the typical skin manifestations (café-au-lait macules, freckling), ocular manifestations (Lisch nodules), the presence of tumors (neurofibromas, plexiform neurofibromas, OPG), osseous lesions (sphenoid dysplasia or tibial pseudarthrosis), and family history.19
NF1 is associated with various mostly benign tumors. In the CNS, the predominant tumor entity is OPG, but low-grade gliomas (LGG) in other locations including brainstem are more prevalent than in the general population, especially in patients presenting with OPG.79 Spinal tumors are reported in up to 60% of NF1 patients but similar to OPG many affected patients remain asymptomatic.62
Malignant neoplasia includes malignant peripheral nerve sheath tumors (MPNSTs) in 8%-13% of NF1patients. These usually arise from preexisting plexiform neurofibromas80 but also juvenile myelomonocytic leukemia (JMML) and embryonal rhabdomyosarcoma.81 HGGs are rare (<1%) and if a diagnosis of NF1 based on clinical characteristics outlined above cannot be confirmed by molecular genetics in a patient with HGG, a possible diagnosis of CMMRD should be considered.
Notably, while 15% of NF1 patients have asymptomatic (mostly unilateral) OPGs on routine screening MRIs only 0.7%-6% of NF1 patients present with symptomatic OPG requiring treatment.79,82,83 In most cases, symptomatic OPG present in children younger than 6 years, although adult-onset symptomatic OPGs can occur. As many OPG remain asymptomatic and associated CNS lesions may remain stable or even devolve, routine MRI surveillance is not generally recommended. Instead, surveillance for optic pathway disturbances by ophthalmologic assessment is warranted every 6-12 months until the age of 8 years.84
Cancer Predisposition Syndromes With Characteristic CNS Malignancies
There is a range of CPS associated with very specific tumor entities. In fact, the association is sometimes so strong that if the entity is encountered in a pediatric patient, further investigations for the respective CPS should be considered. Figure 1 displays common cancer-predisposing genes in various CNS malignancies.
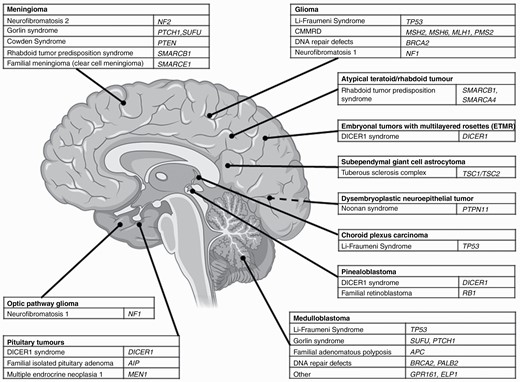
CNS malignancies and associated cancer predisposition syndrome. Abbreviation: CMMRD, constitutional mismatch repair deficiency; created with BioRender®.
Rhabdoid Tumor Predisposition Syndrome (RTPS)
Rhabdoid tumors include malignant extracranial rhabdoid tumors, that arise most commonly in the kidney and atypical teratoid/rhabdoid tumor (AT/RT) that occur in the CNS. Both entities are characterized by aggressive behavior and early-onset. The rhabdoid family of tumors is characterized by somatic loss of SMARCB1 or SMARCA4. Up to 35% of patients carry a constitutional loss-of-function variant or deletion in SMARCB1 or occasionally SMARCA4, that cause RTPS1 and RTPS2, respectively.85 RTPS2 is associated with the development of small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) in female patients and it is less likely to lead to the development of AT/RT than RTPS1. Constitutional variants are de novo in the majority of cases. If a sibling of a patient with an apparent de novo variant is affected germ cell mosaicism is a relevant mechanism to consider.85,86 Whole gene deletions have been described frequently in RTPS. As larger deletions can be missed with NGS, it may be helpful to employ a second method like multiplex ligation-dependent probe amplification (MLPA) to screen for large deletions, especially if there is a clinical suspicion for a genetic predisposition.
Both RTPS1 and 2 are usually caused by loss-of-function variants or deletions. Missense variants, often activating in character, are more likely to lead to syndromes with developmental delay like Coffin-Siris syndrome and Nicolaides-Baraitser syndrome or schwannomatosis.87–89 Unfortunately, these correlations are not complete, eg, there are reports of RTPS caused by missense variants and of families with 1 member suffering from RTPS and other members developing schwannomas only.
The early-onset and aggressive progression of malignant rhabdoid tumors warrant close surveillance, especially during the first years of life. An ambitious surveillance protocol, if deemed feasible locally, would start with 4-weekly MRI (whole body or CNS and spine) supplemented by US and clinical examination until an age of 6 months, as up to this age it may be possible without general anesthesia.90 A less intense recommendation for surveillance includes 3-monthly cranial MRI and abdominal ultrasound scan (whole-body MRI may be considered) up to the age of 5 years.91 Due to the rarity of patients with RTPS2 no specific screening recommendations are available, but surveillance along the recommendations for RTPS1 is possible, supplemented by pelvic US to detect SCCOHT.90
DICER1 Tumor Predisposition Syndrome
DICER1 tumor predispositions syndrome is an autosomal-dominantly inherited CPS caused by pathogenic variants in DICER1. The encoded protein is an RNase III endonuclease that has a central role in processing micro-RNAs. Neoplasms associated with DICER1 predisposition are pleuropulmonary blastoma (PPB), ovarian sex cord-stromal tumors (esp. Sertoli-Leydig cell tumor) and more rarely lung cysts, cystic nephroma, that may progress to anaplastic sarcoma, Wilms tumor, nodular hyperplasia of the thyroid, nasal chondromesenchymal hamartoma, ciliary body medulloepithelioma, and genitourinary embryonal rhabdomyosarcoma. The most common intracranial DICER1-associated tumors are pineoblastoma, and pituitary blastoma. Recent studies identified constitutional pathogenic variants in DICER1 in 3/18 patients with pineoblastomas and in 9/10 patients with pituitary blastomas.92,93 More recently, CNS sarcomas and embryonal tumors with multilayered rosettes (ETMR) have been described as a entity within the DICER1 predisposition syndrome.94,95 Recommendations for testing for constitutional DICER1 variants and surveillance have recently been published.96,97
Gorlin Syndrome
Gorlin syndrome, also known as nevoid basal cell carcinoma syndrome (NBCCS) is a rare, autosomal-dominant inherited CPS caused by pathogenic variants in the PTCH1 or SUFU genes (SHH pathway). There is an increased risk for multiple tumors, especially basal cell carcinoma. Diagnostic criteria to allow for clinical diagnosis have been published.98 About 5% of patients with Gorlin syndrome develop medulloblastoma of the SHH subgroup that mostly occurs at a young age. Constitutional pathogenic variants of PTCH1 or SUFU have been identified in 11% of SHH medulloblastoma and in 21% of SHH medulloblastoma younger than 3 years.6 Radiation-sparing treatment techniques are recommended given the high risk to induce skin cancer.
At least 14%-21% of SHH medulloblastoma carry a constitutional variant in a known CPS gene including PTCH1 and SUFU, BRCA2, PALB2, and TP53, and recent publications showed that an additional 2% and 14% of SHH medulloblastoma are caused by constitutional variants in GPR161 and ELP1, respectively.7,8 Directed genetic testing should be considered for all SHH medulloblastoma, especially if there is evidence of genomic instability or the patient is of young age (<3 years).6
Tuberous Sclerosis Complex
Subependymal giant cell astrocytoma (SEGA) are a rare subtype of LGG (1%-2% of all pediatric brain tumors). They occur almost exclusively in patients diagnosed with the autosomal-dominant inherited tuberous sclerosis complex (TSC). TSC is an autosomal-dominant inherited disorder caused by pathogenic variants in TSC1 (hamartin) or TSC2 (tuberin). About 10%-20% of TSC patients are affected by SEGA. LOH events in TSC1 or TSC2 have been detected in a large proportion of SEGAs.99 Guidelines for management, including the use of mTOR inhibitors and surveillance of, have been published.100,101
Familial Retinoblastoma
Retinoblastoma is a popular example of hereditary cancer. In 1971, Knudsen’s seminal paper on familial retinoblastoma and the 2-hit hypothesis was published, leading the way for the discovery of RB1, the gene causing familial retinoblastoma, and other tumor suppressor genes.102 Retinoblastoma is the most common intraocular tumor in childhood, in most cases, presenting before the age of 2 years. About 40% of retinoblastoma patients carry a pathogenic constitutional variant in RB1 mostly resulting in bilateral disease. In 80% of cases, the underlying variant occurs de novo. Thus, family history is often not hinting at a CPS.103 Around 5% of retinoblastoma patients with a pathogenic RB1 variant develop “trilateral retinoblastoma.” This term describes the co-occurrence of uni- or more commonly bilateral retinoblastomas and pineoblastoma or other midline primitive neuroectodermal tumors (formerly PNET).104 Interestingly, methylation analysis shows that pinealoblastoma associated with RB1 alterations form a distinct subgroup (PIN-RB) and cluster together with retinoblastoma likely indicating commonalities in the cellular origin of these tumors.105 Thus, screening recommendations for known carriers of RB1 variants include, besides regular ophthalmologic examination, an MRI of the CNS at time of retinoblastoma diagnosis.106
Conclusions/Outlook
Our understanding and awareness of cancer predisposition in pediatric neuro-oncology have increased steadily over the recent years. Recommendations and guidelines for diagnosis and management have now been published for most of the more common CPS.
Our currently applied workflow to identify patients affected by a CPS is illustrated in Figure 2. Depending on the local setting, we suggest to routinely use published checklists with signs and symptoms indicative of a possible genetic cancer predisposition. Any suspicious findings should be discussed with the family and followed up by genetic counseling and potentially directed genetic investigations. If available, molecular analysis of tumor samples with parallel analysis of healthy control tissue, as exemplarily described for the brain tumor-focused diagnostic research projectMNP2.0, can identify additional patients, who may not have fulfilled clinical criteria for genetic counseling or directed testing. Comprehensive information about possible genetic findings and consequences for the patient and their family is essential prior to any genetic investigations, especially in a research context, where unexpected findings may be revealed.
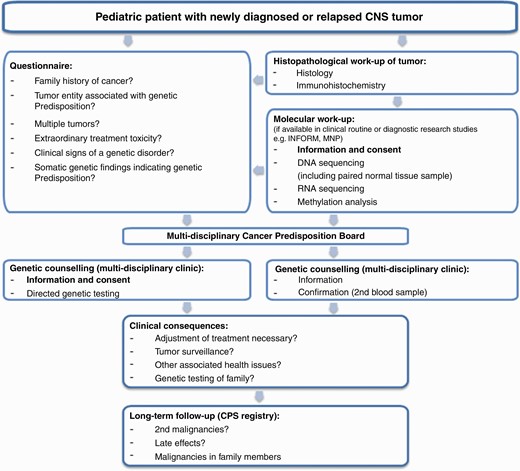
Our current diagnostic approach for the identification of a genetic cancer predisposition. This workflow should be adjusted according to the local setting and diagnostic possibilities. Abbreviations: CNS, central nervous system; CPS, cancer predisposition syndrome.
Patients with a CPS and their families need to be supported on various levels, especially around the time of diagnosis of the respective CPS or an associated malignancy that is based on it. A close cooperation between clinical geneticists, pediatric oncologists, and psychosocial professionals is of paramount importance to provide the family with required information on cancer risks, surveillance, potential treatment options and possible toxicities, and the risks and possibilities associated with genetic testing of other family members as well as support structures. In our experience, it is helpful to see patients and families affected by a CPS in a joint consultation with pediatric oncology and human genetics. In this forum, most of the questions the patients and families may have at the time of diagnosis can be directly addressed by the interdisciplinary team. Yet there are still many questions to be answered. We have recently implemented a registry for cancer patients and carriers with a CPS to prospectively collect clinical data and to establish an interface to other entity-focused studies or registries (www.krebs-praedisposition.de/en/). Patient registries and population-based approaches will help to understand the incidence of cancer in the various CPS and inform future updates to surveillance recommendations. Data from patient registries may also help to guide treatment in patients developing cancer in the context of a CPS. In the future, surveillance strategies may be adapted to accommodate genotype-phenotype relationship. Intensive surveillance may be necessary for some variants in a cancer predisposition gene, while other variants in the same gene may permit a less strict or even no surveillance. Also, the modalities of surveillance may change in the future. Currently, different imaging techniques (MRI, US, and endoscopy) are the mainstays of surveillance. The use of liquid biopsies, eg, cell-free–circulating tumor DNA in peripheral blood or cerebrospinal fluid, for early detection of malignancies is currently investigated by us and several other groups.107 New techniques and adapted use of surveillance may allow to reduce false-positive findings, reducing the physical and psychological harm these entail for the patient, while improving early diagnosis and treatment of developing malignancies.
Funding
This work was supported by Deutsche Kinderkrebssstiftung (INFORM and MNP), BILD hilft e.V. „Ein Herz für Kinder“ and the German Cancer Consortium (DKTK) (INFORM). We acknowledge additional funding from „Ein Kiwi gegen Krebs.“
Conflict of interest statement. The authors declare that they have no conflict of interest.
References

{kind=link}
{kind=link}