






Embryonal tumors with multilayered rosettes (ETMR) are rare but aggressive cancers, commonly occurring in children under 3 years of age. They are an under-recognized entity and the current World Health Organization (WHO) diagnostic evaluation can be challenging to implement in a timely manner to allow prompt treatment, particularly in resource-limited healthcare settings. ETMRs also represent a therapeutic challenge as there are no uniform treatment protocols. Chemotherapy-only strategies may be employed aiming to avoid or delay the deleterious effects of radiation to the developing brain. Here, we describe 2 cases of ETMR from different healthcare settings, which were presented at the Society for Neuro-Oncology Pediatric Molecular Tumor Board Quarterly Series (March 2022) to highlight and discuss the challenges for timely diagnosis and management. These cases highlight the heterogeneous responses to treatment, which remain unpredictable based on current knowledge. One patient died despite use of multimodality therapy including surgery, intensive chemotherapy, and radiotherapy. The second patient is a long-term survivor, treated with chemotherapy only after surgery. We discuss the minimal set of key pathological and molecular findings required in order to establish the timely diagnosis of ETMR, the role of different therapies, and future perspectives on the management of this rare and aggressive condition, with the aim of improving clinical outcomes.
Case 1
A 21-month-old male presented with raised intracranial pressure (ICP), including unsteadiness and irritability. Initial MRI showed a large, localized, poorly enhancing 52×46 mm posterior fossa mass, with associated hydrocephalus (Figure 1A), containing calcification on non-contrast CT (Figure 1B). Despite steroids, he became drowsier and experienced brief generalized seizures. An extraventricular drain was inserted with clinical stabilization prior to tumor surgery, where gross total resection (GTR) was achieved. There were no metastases on imaging and cerebrospinal fluid (CSF) cytology obtained by lumbar puncture was clear.
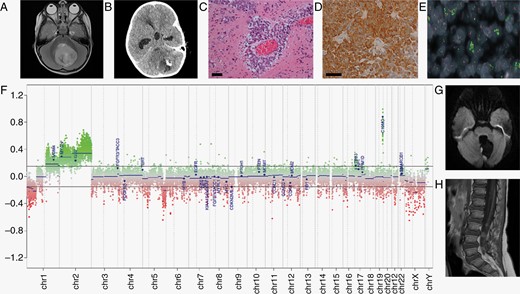
Radiological, pathological, and molecular features of ETMR for case 1. (A) At presentation, axial T2 sequences demonstrated a well-defined midline posterior fossa mass mainly involving the cerebellar vermis with extension into the fourth ventricle. The mass showed increased T2 signal and contained small cystic areas. (B) Non-contrast CT revealed that the mass contained a small area of calcification. (C) Histology showed an embryonal tumor comprising cells in sheets and multilayered arrangements around vessels on a neuropil background. Scale bar=100 µm. (D) Immunohistochemistry revealed LIN28A immunoreactivity. Scale bar = 100μm. (E) FISH image of formalin-fixed paraffin-embedded tissue section demonstrated interspersed cells which displayed amplification of the C19MC locus (19q13.42: green fluorochrome) compared with the control locus (19p13.3: red fluorochrome). Amplified signals appear as large clusters as the amplification that has occurred are focal (intra-chromosomal). (F) The copy number plot generated from the methylation array, shows amplification in the region of the C19MC and gain of chromosomes 1q and 2. (G) MRI head scan performed 5 months after diagnostic scan, after GTR and while on induction chemotherapy, showing first (localized) recurrence. Diffusion-weighted imaging (DWI) sequences showed new nodules at the margins of the surgical resection cavity consistent with recurrence. (H) MRI post-contrast sagittal T1 sequence 4 months after first recurrence, and after further GTR and focal proton radiotherapy, showing multiple new enhancing nodules overlying the spinal cord and cauda equina nerve roots consistent with spinal drop metastases. Key: GTR, gross total resection.
Histology showed an embryonal tumor with cellular areas composed of pleomorphic cells with hyperchromatic nuclei and scant cytoplasm, arranged in nodules, sheets, and multilayered perivascular rosettes on an abundant neuropil-like stroma (Figure 1C). Immunohistochemistry (IHC) revealed LIN28A positivity (Figure 1D) and patchy synaptophysin and GFAP positivity. BAF47 (INI1) was retained. Amplification of the C19MC locus was detected by interphase fluorescence in situ hybridization (FISH) (Figure 1E). Illumina EPIC methylation profiling, analyzed using the DKFZ Heidelberg classifier (MNP v11.6), confirmed methylation class ETMR with a calibrated score of 1. The methylation array copy number plot demonstrated C19MC amplification and gain of chromosomes 1q and 2 (Figure 1F). NHS-England commissioned standard-of-care whole-genome sequencing (WGS) confirmed C19MC amplification and a TTYH1-C19MC fusion, and also revealed copy number gains of chromosomes 1q, 2, and 4, but no targetable change. The combined histological and molecular data were thus consistent with ETMR. Germline DICER1 mutation, particularly associated with non-C19MC-amplified ETMR,1 was excluded.
After discussion in the multidisciplinary team (MDT) and with family, a chemotherapy-only approach was favored, sparing radiotherapy for any recurrence. Following stem cell harvesting, HEADSTART-II chemotherapy was commenced2,3 (Figure 2A). This involved 5 cycles of induction chemotherapy with vincristine, cisplatin, cyclophosphamide, etoposide, and methotrexate, with imaging after 2, 4, and 5 courses, prior to planned high-dose chemotherapy and stem cell rescue. These courses were delivered in a timely fashion but resulted in multiple episodes of sepsis. Unfortunately, MRI scan after the fifth course showed nodular areas of restricted diffusion at the margins of the surgical cavity in keeping with localized tumor recurrence (Figure 1G). There were no spinal drop metastases and CSF cytology was again clear. A surgical GTR was again achieved, and 54 Gy focal proton radiotherapy (30 daily 1.8 Gy fractions) was initiated within 5 weeks of surgery. WGS on the recurrence sample confirmed that the C19MC amplification was retained with the known TTYH1-C19MC fusion, but no newly acquired driver variants were observed. The relapse sample had an elevated accumulation of somatic mutations compared with the primary tumor, particularly for substitutions (primary 837, relapse 4511) and structural variants (primary 31, relapse 247). The pattern of substitution (mutational signature) indicated an excess attributable to platinum-based chemotherapy, as previously described.1
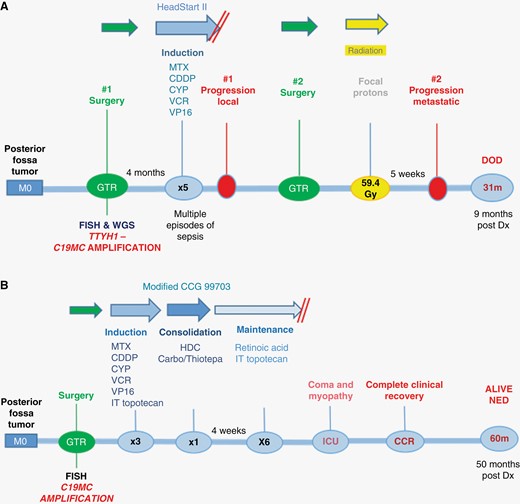
Patient timelines of treatment journeys and outcomes for case 1 (A) and case 2 (B). Key: C19MC, Chromosome 19 microRNA cluster (C19MC) amplification; CDDP, Cisplatin; CYP, Cyclophosphamide; DOD, died of disease; Dx, diagnosis; FISH, fluorescence in situ hybridization; GTR, gross total resection; HDC, high-dose chemotherapy; ICU, intensive care unit; IT, intrathecal; MTX, Methotrexate; NED, no evidence of disease; VCR, Vincristine; VP16, Etoposide; TTYH1, tweety family member 1 gene; WGS, whole-genome sequencing.
After irradiation, MDT discussion deemed it reasonable to commence a 12-month maintenance chemotherapy schedule with intrathecal topotecan every 28 days (d), oral retinoic acid differentiation therapy d1-15 and continuous oral valproate d1-28. This received local drugs and therapeutics board approval as it was felt likely to be well tolerated with minimal side effects, despite no published evidence of efficacy. Five weeks following proton radiotherapy however, baseline MRI revealed asymptomatic metastatic deposits, with spinal lesions present (Figure 1H), in addition to lesions within the ventricular system, eg, adjacent to the septum pellucidum, and bilaterally within the auditory canals (leptomeningeal tumor deposits). Consequently, maintenance chemotherapy was not initiated, and the patient died of disease 3 weeks later at the age of 31 months, 9 months following primary diagnosis.
Case 2
A 10-month-old male infant presented with a short history of vomiting, lethargy, and head tilt. CT scan revealed a posterior fossa tumor, subsequently confirmed on MRI (Supplementary Figure 1). GTR was achieved and a diagnosis of ETMR was made based on the presence of embryonal tumor cells with rosette features on morphology, LIN28A positive IHC, and FISH confirming C19MC amplification (Supplementary Figure 1). A modified CCG 99703 approach was used, with three courses of HEADSTART II induction chemotherapy with methotrexate intensification, and additional intrathecal topotecan, followed by high-dose chemotherapy with carboplatin and thiotepa.4 After MDT discussion, it was decided to deliver maintenance chemotherapy with intrathecal topotecan and retinoic acid (Figure 2B). Following the sixth dose of intrathecal chemotherapy, sudden neurological deterioration occurred with hypotonia and coma, requiring 2-week intensive care unit admission, with severe neurological sequelae at discharge. CSF culture was negative, ICP was within normal range, and CT head was unremarkable. Focal radiotherapy planning at age 18 months was abandoned due to this major complication. Complete neurological recovery was made within 6 months. The patient is disease-free 50 months following initial diagnosis and remains neurologically intact.
Discussion
Based on the latest WHO Classification of Tumors of the Central Nervous System (fifth edition; 2021), ETMR are recognized as a distinct tumor type and are classified as a WHO grade 4 brain tumor. They are aggressive in nature and typically occur in children under 3 years of age,5,6 as seen in both cases here. ETMRs are likely underdiagnosed7 and epidemiological data are lacking, with limited numbers of reported cases having complete treatment and outcome data. The presenting symptoms of ETMR vary and depend on tumor size and location, but the most common clinical sign is raised ICP,7 again seen in both cases here. Tumor location is heterogeneous, although the majority are supratentorial, with rare spinal cases described.7,8
Key Diagnostic Criteria
ETMRs typically present as large, well-defined masses with diffusion restriction on MRI.5 Imaging for both cases here showed areas of calcification, a frequent radiological finding for ETMRs.9 Histologically, characteristic ETMR features include multilayered and pseudo-stratified rosette structures and large areas of neuropil, containing a mixture of dendrites, glial cells, and unmyelinated axons.10,11 In some cases, ETMRs may display diverse histological features, rarely including divergent differentiation patterns.5 Therefore, a set of key molecular findings is essential for the diagnosis. In most cases, however, morphological appearances are strongly suggestive of ETMR, and molecular testing is used to prove the diagnosis. Molecular studies have shown recurrent amplification of C19MC, an oncogenic miRNA cluster containing>40 miRNAs at chromosomal locus 19q13.42, as a genetic hallmark and likely main driver of ETMR, occurring in ~90% of cases regardless of histology.1,10 Prediction of the downstream targets of C19MC miRNAs is challenging due to the large number of miRNAs within the cluster, each with multiple predicted binding sites. Furthermore, both oncogenic and tumor suppressive functions have been identified for C19MC miRNAs, implying context-dependent downstream effects.5,12 In the majority of C19MC-positive ETMR cases, C19MC expression is driven by translocation and fusion with Tweety Family Member 1 (TTYH1) gene.5,13 This TTYH1-C19MC fusion was confirmed in case 1 with WGS. Interestingly, amplification of another miRNA cluster, miR-17~92 (MIR17HG), has been identified in 3 ETMR patients,1 associated with increased proliferation and invasion in other malignancies.14 Furthermore, C19MC and MIR17HG cluster miRNAs share sequence homology and thus may have overlapping targets and functions.5
While C19MC amplification is the most common genetic abnormality in ETMR, biallelic mutations of the miRNA processing gene, DICER1, are the second most common, occurring in ~5% of cases and exclusively in cases lacking C19MC or MIR17HG amplifications.1 Of note, 2 reported cases of histologically diagnosed pineoblastoma had DICER1 mutations and were reclassified as ETMRs after methylation profiling.15 It should be noted that the 2 cases presented here were from different healthcare settings. Consequently, methylation profiling was completed for case 1 as standard-of-care, but not performed for case 2. At the time of investigation for case 1, methylation profiling was analyzed using the DKFZ Heidelberg classifier version 11.6, which included only a single ETMR grouping. It is important to highlight that the most recent version of the classifier (v12.5) now includes 2 distinct methylation subclasses of ETMR, namely embryonal tumor with multilayered rosettes, C19MC altered and embryonal tumor with multilayered rosettes, atypical. Contemporaneous re-analysis of methylation profiling for case 1 on the v12.5 classifier confirmed the C19MC altered subclass with a calibrated score of 0.9998, as expected.
ETMRs also display high expression of the RNA-binding protein LIN28A, which regulates the let-7 miRNA family.16,17 While high expression levels of LIN28A/LIN28B occur in ETMRs, and are straightforward to demonstrate through IHC, this is also observed in other high-grade tumors, such as atypical teratoid/rhabdoid tumors (AT/RT) and high-grade gliomas.10 Therefore, LIN28A immunopositivity is supportive of an ETMR diagnosis, but neither sufficient nor specific,10 hence the need for additional molecular testing. Thus, a combination of neuroradiological (restricted diffusion on MRI), histological (typical multilayered and pseudo-stratified rosette structures and large areas of neuropil), immunohistochemical (LIN28A immunopositivity), and FISH (C19MC amplification) studies are considered the minimal set of key WHO investigations to reach a timely diagnosis in the majority of ETMR cases (Figure 3), including case 2 presented here. However, the exact diagnostic pathway will depend on the healthcare setting. In England, for example, the NHS-England target for turnaround time (TAT) for methylation array is 21 days, and in practice is often less than this. Indeed, a very recent German study has also shown a median TAT of 21 days for methylation and WGS data.18 As a result, in some UK centers, FISH is no longer routinely performed as the relevant molecular information can be obtained in a timely fashion from methylation and a rapid next-generation sequencing panel, which confirms the C19MC amplification or DICER1 variants if present. However, while methylation profiling and WGS can also confirm the diagnosis, as for case 1, these techniques are not necessarily widely available. The key, regardless of healthcare setting, is ensuring a rapid TAT on molecular results to avoid unnecessary delay in initiating treatment for this aggressive condition.19
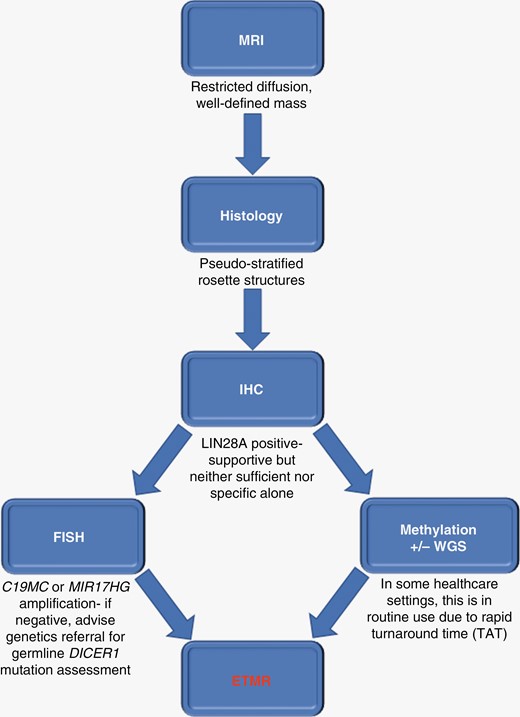
Minimal set of key radiological, pathological, and molecular findings required to establish the efficient and timely diagnosis of ETMR, as per the current WHO classification. Currently, typical radiology, histology, positive LIN28A IHC, and FISH demonstrating C19MC or MIR17HG amplification are sufficient for ETMR diagnosis. Note that LIN28A IHC positivity is neither sufficient nor specific for ETMR, and is not a diagnostic criterion in the WHO classification. However, positive LIN28A IHC testing, which is simple and rapid to perform, along with typical morphology, highlights ETMR as a diagnostic likelihood, allowing clinicians to instigate pragmatic and timely management for patients whilst awaiting confirmatory FISH and/or other investigations. It should be noted that methylation profiling and/or whole-genome sequencing (WGS) are already routinely used in some healthcare settings for ETMR diagnosis, due to rapid turnaround time (TAT). Key: FISH, fluorescence in situ hybridization; ETMR, Embryonal tumor with multilayered rosettes; IHC, immunohistochemistry.
To date, clinical and molecular data on ETMR has provided insufficient insight to explain the heterogeneous responses observed with current therapies, as exemplified by the cases described here. It is possible that further routine methylation profiling and wider adoption of WGS could help identify ETMR subtypes20 and, in turn, potential reasons for this heterogeneity.
Role of Different Therapies
To date, there remains no standard treatment for ETMR and a paucity of prospective clinical data on which to base clinical decision-making. Univariate and multivariate analyses of 38 patients showed better overall survival (OS) with GTR, high-dose chemotherapy, and radiotherapy.7 Primary GTR is frequently attempted as the tumors are often localized and well-demarcated. However, surgery at a young age has a high risk for perioperative complications and the aggressive nature of ETMR can lead to poor pre- and post-operative status with impaired neurological status.5,21 Second resections are sometimes attempted for recurrence, such as in case 1 presented here, and there has been a report of long-term survival following secondary resection.22
Current chemotherapy strategies are based on protocols for young children with CNS primitive neuroectodermal tumor (CNS-PNET),23 with different intensive combinations used.5 There is also limited published evidence on the efficacy of different chemotherapy regimens for ETMR. Thus, for case 1, despite the lack of efficacy data, a 12-month maintenance chemotherapy schedule was deemed reasonable to offer. Of note, the recently published prospective P-HIT study of 30 patients with ETMR showed a 5-year OS of 47% after treatment with carboplatin/etoposide induction and high-dose chemotherapy, compared with 8% with other treatments.23
Due to the deleterious effects of radiation to the developing brain, radiotherapy is not always delivered to ETMR patients.5 This was the rationale in case 1, where, after MDT and family discussion, a chemotherapy-only approach was favored, sparing radiotherapy for treatment of any recurrence. However, survival benefits from radiotherapy have been reported.5 Mayr et al reviewed the ETMR literature and found 228 published cases, where, including their cohort of 9 patients, 26 (11%) of patients survived>36 months and all but 2 of these long-term survivors received radiotherapy.24 Given that ETMR commonly arises during early childhood, the potential benefit of radiotherapy needs to be balanced against the high risk for impairment of neurocognitive function.7 Large prospective ETMR trials are needed to fully evaluate this but will be challenging to perform. Unfortunately, despite intensive surgical, chemotherapy, and radiotherapy treatments, 5-year OS remains poor, highlighting the need for novel therapeutic strategies.25
Future Perspectives
Previous studies have utilized ETMR cell lines to screen for novel therapeutic agents and several potentially effective drugs have been identified, including IGF1R inhibitors, mammalian target of rapamycin (mTOR) inhibitors, topoisomerase inhibitors, PI3K inhibitors, actinomycin D, polo-like kinase inhibitors, aurora kinase inhibitors, anthracyclines, decitabine, and panobinostat.5 Developing a better understanding of ETMR pathogenesis has led to some preclinical studies testing therapies that target potential downstream pathways. Such examples include arsenic trioxide (an inhibitor of the SHH pathway) inhibiting ETMR growth in vitro and in vivo,26 and topoisomerase and PARP inhibitors targeting R-loops and chromosomal instability in vitro.1 Furthermore, the bromodomain inhibitor JQ1 has been shown to be effective in ETMR cell lines through likely targeting of a MYCN-driven super-enhancer network.27
Conclusion
Over the last decade, advances in understanding the fundamental genetic abnormalities in ETMR have led to its recognition as a distinct disease entity and resulted in improved diagnostic tools. However, due to limited clinical data with complete treatment and outcome results, and lack of both prospective clinical trials and appropriate preclinical models, there is no established consensus on the best management approach for these patients. Many unanswered questions remain, such as consensus on the optimal chemotherapy regimen, and when, or if, to deliver radiotherapy. This Society for Neuro-Oncology Pediatric Molecular Tumor Board Quarterly Series Report describes 2 cases of ETMR, with different treatment journeys and outcomes in different healthcare settings. The 2 cases highlight the heterogeneous responses to treatment which remain unpredictable based on our current knowledge; improved clinical/molecular data may help further our understanding. International collaboration will be essential to capture and utilize such growing datasets, as well as preclinical results, to establish prospective ETMR clinical trials to improve outcomes for these young children.
Acknowledgments
This research was made possible through access to the data and findings generated by the 100 000 Genomes Project. The 100 000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100 000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The 100 000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. TSJ is grateful for funding from Great Ormond Street Children’s Charity, The Brain Tumour Charity, Children with Cancer UK, Cancer Research UK, NIHR and the Olivia Hodson Cancer Fund. All research at GOSH NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR GOSH Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Conflict of interest statement
The cases described in this Report were presented at the Society for Neuro-Oncology Pediatric Molecular Neuro-Oncology Tumor Board Quarterly Series, March 22, 2022. The authors have nil else to declare.

{kind=link}
{kind=link}
{kind=link}