






Abstract
Diffuse leptomeningeal glioneuronal tumor (DLGNT) occurs predominantly in children and is typically characterized by diffuse leptomeningeal lesions throughout the neuroaxis with focal segments of parenchymal involvement. Recent reports have identified cases without diffuse leptomeningeal involvement that retain classic glioneuronal features on histology. In this report, we present a case of a 4-year-old boy with a large cystic-solid intramedullary spinal cord lesion that on surgical biopsy revealed a biphasic astrocytic tumor with sparsely distributed eosinophilic granular bodies and Rosenthal fibers. Next-generation sequencing revealed a KIAA1549-BRAF fusion, 1p/19q codeletion, and lack of an IDH1 mutation. Methylation profiling demonstrated a calibrated class score of 0.98 for DLGNT and copy number loss of 1p. Despite the morphologic similarities to pilocytic astrocytoma and the lack of oligodendroglial/neuronal components or leptomeningeal dissemination, the molecular profile was definitive in classifying the tumor as DLGNT. This case highlights the importance of molecular and genetic testing in the characterization of pediatric central nervous system tumors.
Diffuse leptomeningeal glioneuronal tumor (DLGNT) was a provisional entity recognized under the neuronal and mixed neuronal-glial tumor class in the 2016 WHO classification of CNS tumors and is a distinct class in the 2021 WHO classification.1 DLGNTs typically present in children as diffuse leptomeningeal lesions throughout the neuroaxis with focal areas of parenchymal involvement.2 Cytopathologic findings of DLGNT resemble oligodendroglial tumors with glioneuronal differentiation. Molecular characterization typically reveals either a 1p/19q codeletion or isolated 1p deletion with mitogen-activated protein kinase (MAPK) activating alterations and a characteristic absence of isocitrate dehydrogenase (IDH) mutations.3 Despite classically described leptomeningeal involvement, recent cases of DLGNT have been reported without leptomeningeal dissemination, including several with isolated intramedullary spinal cord tumors.4–8 Here we present a case of a patient with an isolated intramedullary spinal cord tumor that presented histologically as a pilocytic astrocytoma but revealed to be a DLGNT based on advanced molecular and genetic testing.
Results
Case Presentation
The patient presented to our hospital at age 4. His past medical history was notable for mild left ventricular hypertrophy. He had 1 year of abdominal pain and constipation, then developed torticollis and difficulty ambulating with gait ataxia, patellar hyperreflexia, and decreased strength of bilateral lower extremities worse on the left. MRI of the spine revealed a solid-cystic intramedullary spinal cord lesion extending from C7 to T12 with multifocal areas of contrast enhancement and associated infiltration and edema down to the conus medullaris (Figure 1A and B). There were no intracranial lesions or leptomeningeal enhancement. He had associated syringohydromyelia from the medulla down to the C7/T1 level. He underwent hemilaminectomies for subtotal resection of the lesion in the upper thoracic region. Further tumor resection was halted due to changes in intraoperative neuromonitoring signals. Postoperatively, he had a symptomatic thoracic cord syrinx requiring syringopleural shunt placement. He also had symptomatic progression of an upper cervical syrinx that required placement of a syringoarachnoid shunt. He went on to develop communicating hydrocephalus without imaging evidence of leptomeningeal dissemination or intracranial involvement and underwent ventriculoperitoneal shunt placement. Based on pathology at the time as discussed below, the lesion appeared most consistent with a pilocytic astrocytoma, although the 1p/19q deletion raised concerns the tumor may be more oligodendroglial-like, and methylation was not yet completed. He was started on temozolomide 200 mg/m2 for 5 days given its activity against both low-grade glioma and oligodendroglioma; however, after 4 weeks, he developed worsening lower extremity weakness and increased torticollis. A repeat MRI of the spine revealed tumor progression, prompting treatment with radiotherapy. Radiotherapy (total dose 50.4 Gy over 28 fractions) was delivered to a volume encompassing the thecal sac from C6 to L1; the majority of treatment was given using pencil beam scanned proton therapy although the first 5 fractions were delivered using x-ray fields to avoid treatment delay in the setting of progressive neurologic symptoms. His symptoms improved during radiotherapy, and subsequent imaging showed a decrease in tumor size (Figure 1C and D). At 30 months after surgery and 25 months from the end of radiotherapy, the tumor remained stable on repeat imaging. He has regained strength in his lower extremities and is ambulatory without assistance.
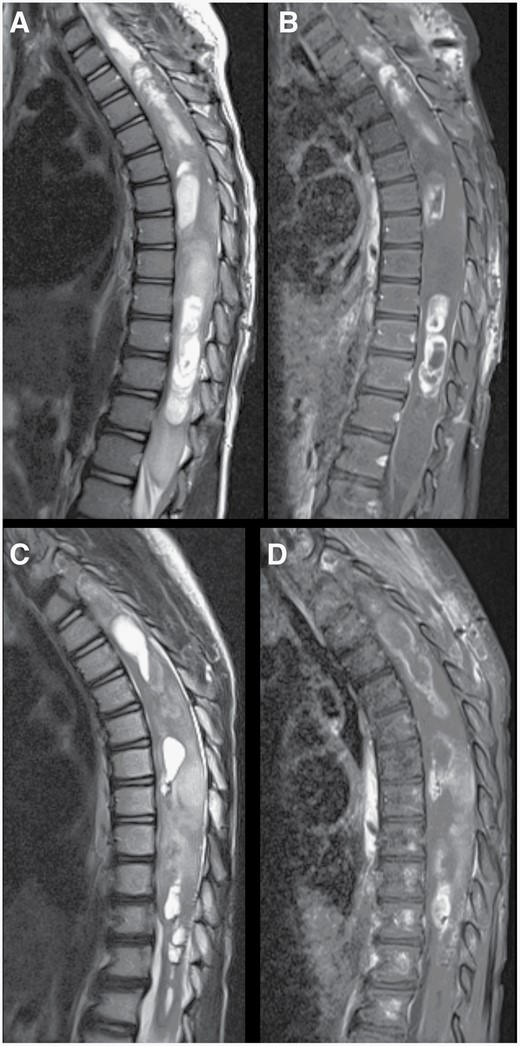
Changes following treatment of spinal cord tumor. Preoperative sagittal T2-weighted (A) and post-contrast T1-weighted (B) MR images of the thoracic spine demonstrating a large cystic-solid intramedullary spinal cord tumor with expansion of the spinal cord and multiple areas of contrast enhancement. Following treatment, repeat T2-weighted (C) and post-contrast T1-weighted (D) MR imaging showed a decrease in the enhancing portions as well as the cystic areas of the tumor. Expansion of the distal spinal cord and conus medullaris also decreased.
Histological Findings
On histological examination, hematoxylin and eosin-stained sections demonstrated a biphasic astrocytic tumor with occasional eosinophilic granular bodies and Rosenthal fibers (Figure 2A). Immunoperoxidase staining revealed GFAP-positive tumor cells with ATRX retention. Additional tumor markers for histone H3K27M alteration and mutant IDH-1 were negative, with P53 staining weakly in a subset of cells. There were no observed mitoses or areas of necrosis. Ki67 staining revealed a proliferative index of less than 5%.
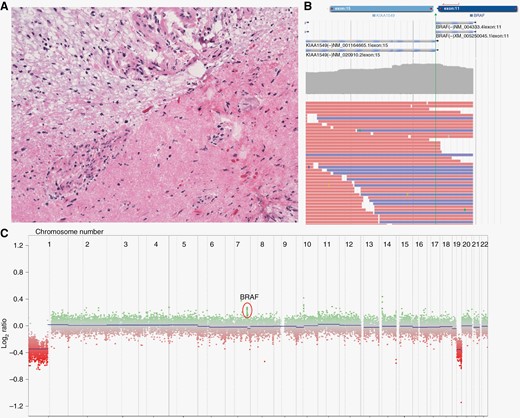
Histologic appearance and molecular profiling of the tumor. Hematoxylin and eosin stain of the excised neoplasm at 200× final magnification (A). The stain demonstrates a biphasic pattern, classic of pilocytic astrocytoma, with a compact component with numerous Rosenthal fibers in the right corner. The interface between the 2 components shows prominent vascularity. Next-generation sequencing identified a KIAA1549-BRAF fusion (B) as well as 1p/19q codeletion and gain of partial 7q (C).
Molecular Findings
A comprehensive targeted next-generation sequencing panel revealed a KIAA1549-BRAF fusion, a finding frequently associated with pilocytic astrocytomas (Figure 2B). Copy number changes revealed 1p/19q codeletion and gain of partial 7q involving exons 11–18 of the BRAF gene compatible with the BRAF fusion (Figure 2C). No IDH1 mutation was identified. The specimen was subsequently sent for methylation profiling, which revealed a high calibrated score for DLGNT of 0.98.
Discussion
DLGNT classically appears on MRI as nodular leptomeningeal enhancement, especially around the basal cisterns and cerebellar sulci.9,10 Recent series have described cases of DLGNT with isolated lesions in the spinal cord, similar to our case,6 but with the originally described histologic appearance of oligodendrocyte-like cells and variable numbers of neuronal elements.2,11 Although cases have been described with focal areas of pilocytic features,6 our case uniquely had isolated histologic features of a pilocytic astrocytoma without neuronal elements. This may be due to limited sampling, although a significant portion of the tumor was removed and evaluated.
Molecularly, DLGNT is defined by the presence of 1p deletion with or without 19q deletion in the presence of KIAA1549-BRAF fusions and the absence of IDH1 mutations.2,11KIAA1549-BRAF fusion is often associated with low-grade gliomas, and specifically with pilocytic astrocytoma, in the absence of a co-occurring 1p or 1p/19q deletion.12 1p/19q codeletions can be found in oligodendrogliomas,13 as well as adult diffuse gliomas where they have been reported to rarely occur concomitantly with KIAA-BRAF fusions and IDH1 mutations.14 While the present case presented histologically as a pilocytic astrocytoma, the co-occurrence of KIAA1549-BRAF fusion and 1p/19q codeletion prompted strong suspicion that the lesion in question was a DLGNT.
DNA methylation signatures are a reproducible way to classify pediatric CNS tumors that have overlapping histopathologic features or limited morphology due to small sample sizes. A study conducted by Deng et al. utilized methylation profiling on a large cohort of DLGNT tumors and identified 2 subdivisions, namely, DLGNT methylation class 1 (DLGNT-MC-1) and DLGNT methylation class 2 (DLGNT-MC-2).11 Loss of chromosome 1p was universal in the entire DLGNT cohort, but only DLGNT-MC-2 carried a gain of chromosomal arm 1q.11 Codeletion of 1p/19q was most frequently observed in DLGNT-MC-1 cases. Both subgroups had recurrent gene alterations affecting the MAPK pathway, with KIAA1549-BRAF fusion being the most common.11 Subsequent evaluation of our patient’s DNA on an expanded methylation model which subdivides DLGNT into MC-1 and MC-2 indicated our patient closely aligned with the DLGNT-MC-1 subgroup (calibrated score = 0.99). DLGNT-MC-1 has less dissemination at diagnosis, a significantly younger age at diagnosis (median of 5 vs 14 years, P < .01) and a less aggressive clinical course than the DLGNT-MC-2 group (5-year overall survival of 100% vs 43%, P < .05).11
As highlighted by our case, there can be considerable heterogeneity in the histologic appearance within tumors classified as DLGNT. This can increase the chance of misdiagnosis based on histology alone. In the work by Deng et al., cases molecularly defined as DLGNT were originally classified as primitive neuroectodermal tumors, pilocytic astrocytoma, anaplastic astrocytoma, anaplastic oligodendroglioma, extraventricular neurocytoma, and ganglioglioma.11
There are currently no established treatment protocols for children with DLGNT given the rarity of the entity and challenges related to accurate diagnosis. As demonstrated by our case, surgical resection is often not possible given the disseminated nature of the disease. Most reports describe the use of various chemotherapeutic regimens, including temozolomide and bevacizumab.15 As was the case with our patient, radiation can also play a role in controlling disease progression.15 Combined 1p deletion and MAPK/ERK pathway activation represent a diagnostic biomarker and potential therapeutic target for DLGNT, paving the way for the creation of future clinical trials now that clear molecular diagnostic criteria have been established.
Conclusion
Here we present the case of a child with an intramedullary spinal cord tumor that radiographically and histologically appeared consistent with pilocytic astrocytoma yet was subsequently confirmed to be DLGNT by molecular testing. Our study highlights the growing importance of methylation profiling in the current integrated histopathological classification of CNS tumors, particularly in cases with atypical morphologic or radiologic characteristics.
Acknowledgment
We thank our patient and his family for sharing this case presentation.
Conflict of interest statement
None declared.
Funding
No funding was received to aid the preparation of this manuscript.
Author Contributions
Conceptualization: PJM, JBF. Investigation: PJM, MLH, MS, LS, AV, BO, CHK, AMT, PBS, JBF. Formal analysis: PJM, MLH, MS, LS, AV, BO, CHK, AMT, PBS, JBF. Writing (original draft): PJM, MLH, JBF. Writing (review and editing): PJM, MLH, MS, LS, AV, CHK, BO, AMT, PBS, JBF. Visualization: MS, LS, AV, BO. Supervision: JBF.
References
Author notes
These authors jointly contributed to this work.

{kind=link}
{kind=link}