






Abstract
Recent large-scale genomic studies have revealed a spectrum of genetic variants associated with specific subtypes of central nervous system (CNS) tumors. The aim of this study was to determine the clinical utility of comprehensive genomic profiling of pediatric, adolescent and young adult (AYA) CNS tumors in a prospective setting, including detection of DNA sequence variants, gene fusions, copy number alterations (CNAs), and loss of heterozygosity.
OncoKids, a comprehensive DNA- and RNA-based next-generation sequencing (NGS) panel, in conjunction with chromosomal microarray analysis (CMA) was employed to detect diagnostic, prognostic, and therapeutic markers. NGS was performed on 222 specimens from 212 patients. Clinical CMA data were analyzed in parallel for 66% (146/222) of cases.
NGS demonstrated clinically significant alterations in 66% (147/222) of cases. Diagnostic markers were identified in 62% (138/222) of cases. Prognostic information and targetable genomic alterations were identified in 22% (49/222) and 18% (41/222) of cases, respectively. Diagnostic or prognostic CNAs were revealed by CMA in 69% (101/146) of cases. Importantly, clinically significant CNAs were detected in 57% (34/60) of cases with noncontributory NGS results. Germline cancer predisposition testing was indicated for 27% (57/212) of patients. Follow-up germline testing was performed for 20 patients which confirmed a germline pathogenic/likely pathogenic variant in 9 cases: TP53 (2), NF1 (2), SMARCB1 (1), NF2 (1), MSH6 (1), PMS2 (1), and a patient with 47,XXY Klinefelter syndrome.
Our results demonstrate the significant clinical utility of integrating genomic profiling into routine clinical testing for pediatric and AYA patients with CNS tumors.
Demonstrate the utility of complementary NGS profiling and CMA for pediatric and AYA CNS tumors.
Highlight the importance of identification of a subset of pediatric and AYA patients with germline cancer predisposition.
This article details our institution’s prospective clinical experience employing molecular profiling of CNS tumors from pediatric and AYA neuro-oncology patients. The identification of DNA sequence variants, RNA fusions, CNAs, and loss of heterozygosity events enhanced the pathologic diagnosis, provided prognostic information and molecular targets for therapy, and revealed germline alterations in cancer predisposition loci important for genetic counseling in high-risk families. Routine genomic profiling will aid in the identification of patients for clinical trials and will ultimately result in improved outcomes for children and AYAs with CNS tumors.
Central nervous system (CNS) tumors are the most common solid tumors in children1 and represent the leading cause of cancer-related mortality in children younger than 14 years of age. Brain tumors are the third most common cancer in adolescent and young adults (AYAs).2 In recent years, molecular profiling of pediatric CNS tumors has been shown to define diagnoses and predict outcomes more accurately than standard histopathology alone.3–5 Additionally, molecular testing has revealed several therapeutic targets and agents that have demonstrated promising results in clinical trials.3,5,6
Approximately 8–10% of pediatric cancer patients harbor mutations in germline cancer predisposition genes,7–9 which has significant implications for therapy, surveillance, and family counseling. However, these studies underestimate the true incidence, as copy number alterations (CNAs), epigenetic modification and variations in noncoding regions of the genome that modify the expression of tumor-related genes have yet to be fully characterized. Germline mutations are well-described in CNS tumors: 50% of patients with choroid plexus carcinomas (CPCs) have underlying TP53 mutations, up to 35% of those with atypical teratoid/rhabdoid tumors have germline mutations or CNAs in SMARCB1,10,11 and up to 20% of patients with SHH-activated medulloblastoma (MB) have germline mutations in PTCH1, SUFU, TP53, BRCA2, or PALB2.8
We developed a comprehensive, clinically validated DNA- and RNA-based next-generation sequencing (NGS) panel, OncoKids, to detect diagnostic, prognostic, and therapeutic markers across the spectrum of pediatric and AYA tumors.12 In the present study, we describe our institutional experience employing OncoKids chromosomal microarray analysis (CMA) and routine histology and immunohistochemistry (IHC) to profile 222 CNS tumors from 212 pediatric patients. We demonstrate the complementary nature of these 2 molecular platforms to provide actionable information for patient management in a clinical setting.
Methods
Case Selection
Two hundred and twenty-two CNS tumors from 212 patients (122 males, 90 females) were prospectively analyzed using NGS over a 2-year period. CMA was performed when there was sufficient DNA for the assay, and if CNAs, loss of heterozygosity (LOH) events, ploidy changes, and/or gene amplification for a particular histologic subtype were anticipated to have clinical significance. CMA was ultimately performed for 66% (146/222) of cases. The median age was for all patients was 9 years (range 14 weeks to 38 years) and 83% were younger than 15 years. Adolescents (15–19 years of age) and young adults (20–39 years of age) accounted for 17% of our cohort. Patient demographics and tumor types are given in Table 1 and Supplementary Figure 1. This study was reviewed and approved by the Children’s Hospital Los Angeles Institutional Review Board.
Patient/Tumor Characteristics of 222 CNS Samples From 212 Patients
| Characteristics | n (%) |
|---|---|
| Sex | |
| Male | 122 (58) |
| Female | 90 (42) |
| Age (years) | |
| ≤3 | 46 (22) |
| >3 and ≤10 | 74 (35) |
| >10 and <15 | 55 (26) |
| ≥15 and <39 | 37 (17) |
| Tumor category | |
| Low-grade glioma | 56 (25) |
| High-grade glioma | 52 (23) |
| Medulloblastoma | 31 (14) |
| Neuronal and mixed glioneuronal | 19 (9) |
| Ependymoma | 17 (8) |
| Embryonal non-medulloblastoma | 7 (3) |
| Choroid plexus tumor | 7 (3) |
| High-grade neuroepithelial tumor | 7 (3) |
| Glial, not otherwise specified | 5 (2) |
| Meningioma | 5 (2) |
| Adamantinomatous craniopharyngioma | 4 (2) |
| Schwannoma | 4 (2) |
| Other | 8 (4) |
| Sample type | |
| FFPE | 101 (45) |
| Frozen tissue | 121 (55) |
| Characteristics | n (%) |
|---|---|
| Sex | |
| Male | 122 (58) |
| Female | 90 (42) |
| Age (years) | |
| ≤3 | 46 (22) |
| >3 and ≤10 | 74 (35) |
| >10 and <15 | 55 (26) |
| ≥15 and <39 | 37 (17) |
| Tumor category | |
| Low-grade glioma | 56 (25) |
| High-grade glioma | 52 (23) |
| Medulloblastoma | 31 (14) |
| Neuronal and mixed glioneuronal | 19 (9) |
| Ependymoma | 17 (8) |
| Embryonal non-medulloblastoma | 7 (3) |
| Choroid plexus tumor | 7 (3) |
| High-grade neuroepithelial tumor | 7 (3) |
| Glial, not otherwise specified | 5 (2) |
| Meningioma | 5 (2) |
| Adamantinomatous craniopharyngioma | 4 (2) |
| Schwannoma | 4 (2) |
| Other | 8 (4) |
| Sample type | |
| FFPE | 101 (45) |
| Frozen tissue | 121 (55) |
Patient/Tumor Characteristics of 222 CNS Samples From 212 Patients
| Characteristics | n (%) |
|---|---|
| Sex | |
| Male | 122 (58) |
| Female | 90 (42) |
| Age (years) | |
| ≤3 | 46 (22) |
| >3 and ≤10 | 74 (35) |
| >10 and <15 | 55 (26) |
| ≥15 and <39 | 37 (17) |
| Tumor category | |
| Low-grade glioma | 56 (25) |
| High-grade glioma | 52 (23) |
| Medulloblastoma | 31 (14) |
| Neuronal and mixed glioneuronal | 19 (9) |
| Ependymoma | 17 (8) |
| Embryonal non-medulloblastoma | 7 (3) |
| Choroid plexus tumor | 7 (3) |
| High-grade neuroepithelial tumor | 7 (3) |
| Glial, not otherwise specified | 5 (2) |
| Meningioma | 5 (2) |
| Adamantinomatous craniopharyngioma | 4 (2) |
| Schwannoma | 4 (2) |
| Other | 8 (4) |
| Sample type | |
| FFPE | 101 (45) |
| Frozen tissue | 121 (55) |
| Characteristics | n (%) |
|---|---|
| Sex | |
| Male | 122 (58) |
| Female | 90 (42) |
| Age (years) | |
| ≤3 | 46 (22) |
| >3 and ≤10 | 74 (35) |
| >10 and <15 | 55 (26) |
| ≥15 and <39 | 37 (17) |
| Tumor category | |
| Low-grade glioma | 56 (25) |
| High-grade glioma | 52 (23) |
| Medulloblastoma | 31 (14) |
| Neuronal and mixed glioneuronal | 19 (9) |
| Ependymoma | 17 (8) |
| Embryonal non-medulloblastoma | 7 (3) |
| Choroid plexus tumor | 7 (3) |
| High-grade neuroepithelial tumor | 7 (3) |
| Glial, not otherwise specified | 5 (2) |
| Meningioma | 5 (2) |
| Adamantinomatous craniopharyngioma | 4 (2) |
| Schwannoma | 4 (2) |
| Other | 8 (4) |
| Sample type | |
| FFPE | 101 (45) |
| Frozen tissue | 121 (55) |
OncoKids NGS Panel
The OncoKids (Oncomine Childhood Cancer Research Assay; Thermo Fisher Scientific) panel uses low input amounts of DNA (20 ng) and RNA (20 ng) and is compatible with formalin-fixed paraffin-embedded (FFPE) and frozen tissue. The percentage of tumor cells in a given specimen was documented by a neuropathologist prior to extraction. DNA was extracted from samples with greater than 30% tumor from fresh frozen tissue (Qiagen Gentra Puregene Tissue Kit; Qiagen) or FFPE (Qiagen QIAamp DNA FFPE Tissue Kit; Qiagen). RNA extraction was performed in parallel using Qiagen RNeasy Mini RNA Extraction Kit for frozen tissue and Agencourt FormaPure Kit for FFPE. DNA and RNA libraries were generated using custom-designed Ampliseq primers targeting 3069 amplicons and 1701 targeted fusions and were sequenced using the Ion S5XL sequencing system (Thermo Fisher Scientific). All cases in this series passed established quality control metrics12 and generated sufficient data for analysis. Cases that had insufficient material and were not sent for testing (3 cases) and those that had insufficient DNA/RNA to proceed with testing (2 cases) during the 2-year period were excluded from this study. Following the mapping of the read data to the human genome (reference build GRCh37/hg19), single-nucleotide variants with an allele fraction greater than 6%, insertions and deletions with a variant allele frequency (VAF) greater than 10%, and high-level amplifications (>8 copies) were annotated and reported according to CAP/AMP/ASCO guidelines.13 Reported variants include variants of strong clinical significance (Tier I), variants of potential clinical significance (Tier II), and variants of unknown clinical significance (Tier III). Benign and likely benign variants (Tier IV) were not included. For novel fusions or fusions with low total RNA sequencing reads, RT-PCR and Sanger sequencing were performed for confirmation.
Chromosomal Microarray Analysis
CMA was performed using OncoScan for FFPE or CytoScan HD for fresh frozen tissue (Thermo Fisher Scientific) according to the manufacturer’s protocols. CNAs encompassing at least 25 consecutive probes and LOH exceeding 5000 kb were reviewed using Chromosome Analysis Suite (Thermo Fisher Scientific).
Targeted Analysis for Follow-up Germline Testing
Clinical, pathology, and molecular results were discussed at a weekly multidisciplinary tumor board. Recommendations for germline testing were made based on family history and one of the following criteria: (1) presence of Tier I or Tier II variant in a tumor-suppressor gene at a VAF of approximately 50% with no CNAs or LOH event in the variant locus; (2) a Tier I or Tier II variant in a tumor-suppressor gene with LOH encompassing the locus in the tumor; (3) the presence of 2 Tier I or Tier II variants in a cancer predisposition gene in the tumor; (4) a Tier I or Tier II variant in the tumor sample that had previously been reported in the germline setting; (5) a Tier I or Tier II variant in a cancer predisposition gene for patients with a clinical or family history suggestive of a cancer predisposition syndrome. Targeted Sanger sequencing or multiplex ligation-dependent probe amplification (MLPA; MRC-Holland) was performed according to the manufacturer’s protocols using the patient’s peripheral blood to confirm the germline variants.
Results
NGS revealed clinically significant Tier I and II sequence variants, gene amplifications, or RNA fusions in 66% (147/222) of cases. Diagnostic markers were identified in 62% (138/222) of cases that resulted in new, refined, or confirmed diagnoses. Prognostic information and potentially targetable alterations were identified in 22% (49/222) and 18% (41/222) of cases, respectively (Supplementary Table 1). Overall, the most common alterations were mutations in TP53, followed by KIAA1549–BRAF fusions, NF1 mutations, BRAF V600E mutations, and mutations in PIK3CA, ATRX, and H3F3A (K27M) (Supplementary Figure 2).
CMA was performed on 146 specimens from 138 patients. CNAs that provided supportive evidence for the pathologic diagnosis or prognosis were detected in 69% (101/146) of cases. In 57% (34/60) of cases, clinically significant CNAs were observed in tumors with noninformative NGS results. Clinically significant alterations detected in low-grade glioma (LGG) and high-grade glioma (HGG) and MBs are shown in Figures 1–3.
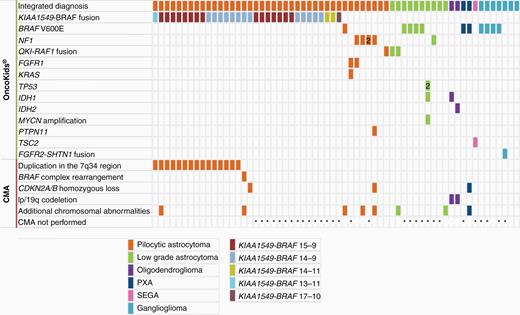
Genomic alterations of pediatric and AYA low-grade glioma and ganglioglioma. Genetic alterations, including Tier I and Tier II sequence variants, RNA fusions, as well as copy number alterations, identified by NGS (n = 62) and chromosomal microarray analysis (n = 32).

Genomic alterations of pediatric and AYA high-grade glioma identified by NGS (n = 46) and chromosomal microarray (n = 26). Each color in the rows corresponds to a specific genetic alteration as noted above.
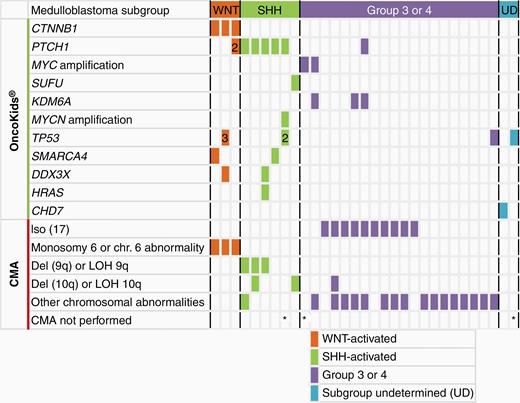
Genomic alterations of pediatric medulloblastomas in this cohort (n = 31). CMA was performed for 28 cases of medulloblastoma.
Genomic Profiling of LGGs
The most common CNS tumors in our cohort were LGGs (WHO grade I and II) including pilocytic astrocytoma (PA), diffuse astrocytoma, pleomorphic xanthoastrocytoma (PXA), oligodendroglioma, or subependymal giant cell astrocytoma (SEGA) and accounted for 26% (55/212) of patients. NGS demonstrated clinically significant genomic alterations in 95% (52/55) of LGGs (Figure 1). The most commonly observed gene fusion was KIAA1549–BRAF (n = 32, age 10 months to 15 years). Fusion breakpoints in KIAA1549 (ENST00000242365) and BRAF (ENST00000288602) were between exons 15-9 (15/32), 14-9 (13/32), and 14-11 (2/32). Single cases harboring breakpoints at 13-11 and 17-10 accounted for the remaining 2 fusion-positive cases. Fusion-positive cases were most commonly identified in the cerebellum (56%, 18/32), consistent with what has been previously described.14 Additional extra-cerebellar locations included suprasellar/chiasmatic region (5), medulla (4), temporal lobe (2), spinal cord (2), and basal ganglia (1). Eighteen of 27 cerebellar LGGs (67%) demonstrated a KIAA1549–BRAF fusion. RNA-sequencing reads were low (<250 total reads) for KIAA1549–BRAF in 3 cases, 1 was confirmed by RT-PCR, and the other 2 were concordant with the 1.9 Mb-duplication in 7q34 detected by CMA. In 2 cases a KIAA1549–BRAF fusion was detected by NGS but not by CMA, likely due to its presence in a small percentage of the cells (<20%). In 3 of 17 cases in which the KIAA1549–BRAF fusion was detected by both methods, CMA revealed additional chromosomal abnormalities, including a homozygous CDKN2A/B deletion, suggestive of inferior survival (Case 127)15; gain of chromosomes 5, 6, and 12 in a tumor from a 12-year-old patient (Case 144); and the third case with an unusual 1p deletion, 19p gain, and an interstitial deletion in 2q13 that encompassed the BUB1 gene (Case 126). The third case had a deletion that included the 5′ region of BRAF (Supplementary Figure 3, Case 126). The KIAA1549–BRAF fusion was confirmed by NGS and was likely due to a complex intrachromosomal rearrangement.16 DNA methylation profiling of this case supported an integrated diagnosis of PA (methylation class LGG, subclass PA and ganglioglioma).
QKI–RAF1 fusions were observed in LGGs in 3 patients (Patient #97, #98, #139, Supplementary Figure 4). The tumors all arose in young children (<6 years) and involved the brain stem and cerebellar peduncle(s). Rosenthal fibers were not evident in the initial biopsies of these patients but were observed in 2 recurrences, approximately 3 years after the original biopsy. Follow-up showed stable disease at 2 years for all patients and slow interval progression by 4–5 years. All 3 patients were alive at the last clinical follow-up.
Other genomic alterations in LGGs included mutations in BRAF (V600E), NF1, FGFR1, and PTPN11, as well as IDH1/IDH2 hotspot mutations in combination with 1p/19q codeletion in 2 oligodendrogliomas and a TSC2 mutation in a SEGA. A brainstem infiltrating astrocytoma from a 17-year-old male demonstrated an IDH1 hotspot mutation, MYCN amplification, and 2 TP53 mutations that did not favor a low-grade tumor; however, there was no evidence of high-grade histology features (Patient #95).
Genomic Profiling of HGGs
HGGs comprised 23% (52/222) of specimens tested in our series and included anaplastic astrocytoma (WHO grade III), glioblastoma (GBM; WHO grade IV), and anaplastic PXA. Combined NGS and CMA demonstrated clinically significant genomic alterations in 91% (42/46) of patients (Figure 2), frequently with multiple genomic alterations per sample. Recurrent oncogenic driver mutations included H3F3A K27M (12), HISTIH3B K27M (4), H3F3A G34R (5), mismatch repair deficiency with a hypermutation profile (3), NF1 loss-of-function mutations (9), NTRK fusion (1), PTPRZ1–MET fusions (2), other MET alterations (2), IDH1 hotspot mutations (2), BRAF V600E (4), as well as 2 or more co-occurring mutations in TP53 (21), ATRX (10), PIK3CA (7), ACVR1 (3), PDGFRA (2), PTEN (2), RB1 (2), PIK3R1 (2), PPM1D (2), MAP2K1 (1), KRAS (1), APC (1), or PTPN11 (1). TP53 was the most frequently altered gene in our cohort of HGGs with mutations observed in 46% (21/46) of cases. BRAF V600E, frequently seen in pediatric LGGs, was detected in 2 HGGs with either an H3F3A K27M or HISTIH3B K27M mutation. Gene amplification events involving CCND1, MET, EGFR, MYCN, or PDGFRA/KIT/KDR were observed in 5 of 46 patients with HGG. Two patients, with a pathologic diagnosis of GBM and K27M-mutant diffuse midline glioma, respectively, exhibited amplification of multiple regions. In addition, complex copy number profiles (19/46), chromosome 7 gain (6/46), chromosome 10 loss (2/46), biallelic CDKN2A/B loss (9/46), 17p loss or LOH including TP53 (9/46), 13q loss including RB1 (3/46), gain of 1q (2/46), and hypodiploidy (5/46) were recurrent findings. Chromosomal instability or chromothripsis was seen in a GBM, a gliosarcoma, and an anaplastic PXA.
In our series, we identified 5 patients with an H3F3A G34R mutation. The patients ranged in age from 11 to 18 years and the tumors showed variable histologic appearances: 1 was histologically an anaplastic astrocytoma, 1 was a GBM, and 2 cases were originally diagnosed as high-grade neuroepithelial tumor (HGNET), reflecting the difficulty of categorizing this tumor on the basis of histopathologic examination alone. The fifth case was an HGG containing a neoplastic ganglion cell component. Among the H3F3A G34R cases, frequent co-occurring mutations were identified in ATRX (5/5) and TP53 (4/5). Three cases had an additional mutation in PTEN, PDGFRA, or APC. CMA for 2 of these G34R-mutated tumors demonstrated complex copy number profiles that included homozygous loss of CDKN2A, LOH for 17p13 that unmasked a TP53 mutation on the remaining allele, deletion of RB1, and/or amplification of PDGFRA and KIT.
While the NGS panel has not yet been clinically validated to measure tumor mutational burden (TMB), 6 HGGs from 4 patients showed an average of 48 variants per specimen (~300 kb in total length), suggestive of high TMB. A homozygous MSH6 mutation consistent with autosomal recessive constitutional mismatch repair deficiency was found in 1 patient with a family history remarkable for an older sister who had died from a brain tumor at 15 years. A second patient’s tumor with histological features of GBM and a history of cancer in the family demonstrated loss of nuclear PMS2 by IHC. The patient was subsequently confirmed to harbor a PMS2 germline heterozygous (NM_000535.7:c.2521del, p.Trp841Glyfs*10) pathogenic variant. Notably, a recurrent HGG from a patient whose original tumor had an H3F3A G34R mutation, and mutations in TP53, ATRX, and PTEN, demonstrated a hypermutated profile in addition to the mutations detected in the original tumor. Subsequent whole-exome sequencing analysis of the recurrent tumor specimen confirmed the hypermutation profile (~103/Mb) and revealed a somatic MSH6 NM_000179.2:c.3656C>T (p.Thr1219Ile) variant that was not present in the patient’s blood. Furthermore, the specific G:C>A:T transition pattern accounted for more than 95% of variants detected. Given prior temozolomide exposure, these findings were suggestive of a temozolomide-induced O6-methylguanine lesion resulting in a previously described signature 11 type high tumor mutation (COSMIC signature v2).17–19
Genomic Profiling of MBs
Twenty-nine of the 31 MBs (94%) harbored at least one genetic alteration characteristic of the WNT, SHH, or Group 3 or 4 subgroups (Figure 3). CTNNB1 exon 3 activating mutations and monosomy 6 were detected in 3 WNT-activated MBs with CTNNB1 nuclear expression by IHC. Each of these 3 cases also harbored additional mutations, including a SMARCA4 mutation (Case 156), 3 different TP53 mutations with different allele fractions, possibly in different clones, and a DDX3X mutation (Case 157), and the third case with a PTCH1 mutation (Case 180). Five MBs had a PTCH1 mutation and 1 case showed a SUFU mutation consistent with the SHH subgroup. The SHH-activated MBs also demonstrated additional genomic events including MYCN amplification (1), TP53 (1), SMARCA4 (1), and DDX3X and HRAS mutations (1). Deletion and/or LOH of 9q or 10q co-occurred with these sequencing variants in 4 of 5 SHH-activated MBs with available CMA data. Finally, loss of 17p and gain of 17q indicative of an isodicentric chromosome 17q were detected in 50% (10/20) of Group 3 and 4 MBs, often with additional alterations (MYC amplification [2 cases], KDM6A mutations [3 cases], and a TP53 mutation [1 case]).
NTRK and Other Rare Rearrangements Detected by OncoKids
An ETV6–NTRK3 fusion and a TPM3–NTRK1 fusion were identified in a 3-year-old female with an unusual biphasic spindle and epithelioid glial neoplasm and in a 4-month-old male with desmoplastic infantile astrocytoma with focal anaplastic features, respectively. Both cases were otherwise difficult to diagnose and assign to a standardized treatment approach. For the first time in the pediatric setting, we identified a poorly differentiated cerebellar tumor with a YAP1–MAML2 fusion in a 4-year-old patient. This fusion has previously been observed in nasopharyngeal carcinoma, ovarian cancer, poroma, and metaplastic thymoma.20–22 Other uncommon alterations included an FGFR2–SHTN1 fusion in a right anteromedial occipital lobe ganglioglioma, an MN1–PATZ1 fusion in 1 case of HGNET-MN1 located in the right parietal lobe, and ASXL1 amplification in an intraventricular HGNET NOS. Additional studies of a larger number of patients are required to further clarify the clinical significance of these findings.
Diagnostic or Prognostic Markers Identified by CMA Alone
CMA demonstrated diagnostic or prognostic CNAs or LOH in 69% (101/146) of samples (Supplementary Table 1), and specifically, in 34 of 60 patients for which OncoKids was noninformative. For example, trisomy 2 and gain of the chromosome 19 miRNA cluster (C19MC) in 19q13.42 were diagnostic for ETMR (Case #16 and #17). A chromothripsis-like pattern and/or chromosomal instability was observed in a clival chondroid chordoma as well as a temporal lobe anaplastic PXA at the initial diagnosis (Figure 4) and recurrence. A SMARCB1 biallelic deletion was found in a poorly differentiated chordoma, consistent with previously reported cases.23
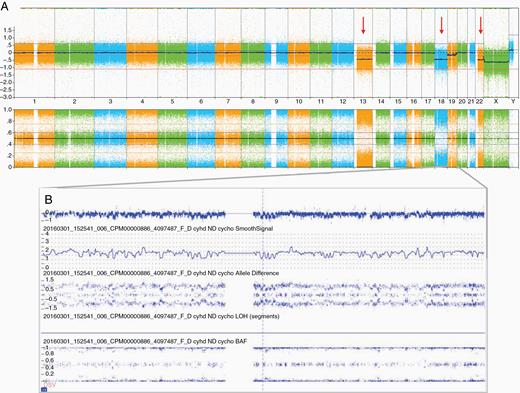
Copy number alterations in an anaplastic pleomorphic xanthoastrocytoma, WHO grade III, for a 5-year-old male patient (Patient 83, Case 91): (A) chromosomal microarray whole-genome view (from left to right: chromosomes 1–22 followed by X and Y) showed loss of chromosomes 13, 18, and 22 indicated by red arrows and (B) chromosome 19 chromothripsis.
Prognostic markers were also identified with CMA. In our cohort, a gain of chromosome 1q, which has been reported as a poor prognostic marker for posterior fossa ependymoma, was seen in 2 cases of ependymoma. Other recurrent CNAs in ependymomas included loss of 6q and homozygous deletion of CDKN2A/B. Complex chromosomal abnormalities, including gain of chromosome 7, loss of chromosome 10, and deletions or LOH that involve TP53 in 17p and RB1 in 13q, were frequent events in HGGs. Importantly, +7/−10 chromosomal CNAs are considered to be a high-grade molecular feature for IDH-wildtype GBM.24 Although the 7q34 duplication and KIAA1549–BRAF fusion were the most common CNAs in LGG, 5 cases demonstrated additional pathogenic CNAs, including homozygous loss of CDKN2A/B in 2 PAs, 1p/19q codeletion in combination with IDH1/2 hotspot mutations in 2 cases of oligodendroglioma, and a deletion in the 7q34 region involving the 5′ region of the BRAF gene in a PA that was confirmed by NGS to have a KIAA1549–BRAF fusion.
Germline Cancer Predisposition
Fifty-seven of 212 patients (27%) had a history or laboratory results suggestive of an underlying mutation in a cancer predisposition gene. Follow-up germline testing was performed for 20 of these patients and confirmed the germline pathogenic/likely pathogenic variants in 9 cases: TP53 (2), NF1 (2), SMARCB1 (1), NF2 (1), PMS2 (1), or MSH6 (1). One 6-year-old patient had a constitutional 47,XXY karyotype consistent with Klinefelter syndrome detected by CMA of the PXA (Case 151, Supplementary Table 1). The remaining 11 cases were negative for the variants identified with tumor profiling. Of the 37 patients who did not undergo germline testing, 6 had a clinical or family history highly suggestive of cancer predisposition (Supplementary Figure 5). These included a patient with a hypermutated HGG suggestive of a mismatch repair disorder, a patient with meningioma and LOH encompassing SMARCE1, 2 hemangioblastoma patients, 1 who had a clinical diagnosis of Von Hippel–Lindau syndrome and the second with a loss of 3p that included VHL, and a child with CPC and likely germline TP53 variant.
Discussion
Brain tumors in pediatric and AYA patients are distinct from adult CNS tumors with respect to the incidence of specific tumor types, cellular origins, and spectrum of oncogenic driver events. We therefore designed and implemented a targeted NGS panel, OncoKids, that would capture the majority of mutations or gene fusions that characterize these tumors. This approach, in combination with a whole-genome copy number array, reveals a broad spectrum of genomic abnormalities, including DNA sequence variants, RNA fusions, gene amplifications, as well as CNAs and LOH events with minimal sample input from frozen or FFPE tissue, to aid in diagnosis and prognosis as well as identify underlying genetic risk factors and targets for therapy. Our experience demonstrates that genomic profiling of pediatric and AYA CNS tumors using this integrated approach is feasible with turn-around times that allow the results to be used in a clinical setting at the time of diagnosis or relapse.
Since the publication of the 2016 WHO classification of CNS tumors, there have been multiple updates highlighting the impact of molecular genetics on CNS tumor classification and grading.25,26 In our series, diagnoses were refined in more than 50% of cases where the molecular data allowed for more specific subcategorization, even if the interpretation of overall tumor classification was not altered. Most importantly, the molecular results led to the reclassification of several patients’ tumors, including 2 with a diagnosis of CNS HGNET-MN1 and 1 with an IDH1-mutant 1p/19q-codeleted oligodendroglioma originally classified as glioma. Rare examples of H3F3A G34R-mutated tumors with nonspecific histology were reclassified as HGG, which represents an emerging diagnostic category with a distinct clinical presentation in AYAs with hemispheric tumors.27 Negative genomic profiling results were also informative. For example, there were several young patients with a “silent” or “balanced” genome by NGS and CMA and posterior fossa ependymoma group A associated with reduced expression of H3K27me3 by IHC and poor prognosis.28
The WHO histological grading scheme is widely used for tumor grading across and within tumor entities. There are, however, instances where the molecular findings may not support the histological grading, as seen in Patent #95 with a histologically defined LGG. The presence of MYCN amplification, 2 TP53 mutations, and an IDH1 mutation was suggestive of a higher grade tumor. Notably, the patient is still alive 16 months after diagnosis.
Our data suggest that a significant proportion of CNS patients have a targetable somatic genomic variant (Supplementary Table 3). Multiple clinical trials have demonstrated the clinical efficacy of BRAF inhibitors for tumors with a BRAF V600E mutation. For children with KIAA1549–BRAF fusion-driven recurrent or refractory pediatric LGGs, monotherapy with a MEK inhibitor has been shown to be effective.29 Similarly, HTMB may suggest the potential use of immunotherapy treatments such as the checkpoint inhibitor pembrolizumab.18,30,31 However, the therapeutic implications of hypermutation in gliomas, particularly for those with an acquired temozolomide-related mismatch repair deficiency, require further study.19 Other alterations of therapeutic significance included fusions involving FGFR2, and QKI-RAF, MET amplification, as well as mutations in IDH1/2, NF1, FGFR1, TSC2, PIK3CA, PTEN, ALK, and PDGFRA. The demonstration of NTRK fusions may ultimately lead to the use of NTRK inhibitors, such as larotrectinib, as first-line therapy for such patients.
Our results demonstrate that the OncoKids NGS panel and CMA are complementary and thus provide a standard clinical approach for molecular diagnostics until the time that single gene or exon-level CNAs and LOH can be determined reliably with NGS. Specifically for non-AT/RT embryonal tumors, CMA appears to have greater utility than NGS. For example, MBs demonstrated chromosomal gains and losses consistent with subgroup 3/4 but were noninformative by NGS, and ETMRs showed characteristic high copy gain or amplification of the C19MC locus and trisomy 2 that may have been missed by NGS alone. Although several key diagnostic CNAs can be detected by fluorescence in situ hybridization, CMA provides comprehensive, genome-wide, high-resolution analysis.
Similarly, chromothripsis, a form of genomic instability characterized by numerous locally clustered rearrangements affecting one or more chromosomes, was detected by CMA in a chondroid chordoma as well as a diagnostic and recurrent PXA. In the routine clinical setting, this is much more easily detected by CMA than with NGS-based panel testing alone. Although chromothripsis is known to occur in a variety of tumor types, it has not been reported in association with PXA. Increased complexity of genomic abnormalities has been hypothesized to correlate with more aggressive behavior in PXA.32
The sensitivity of NGS or CMA depends on tumor content as well as underlying biology. For example, LGGs with KIAA1549–BRAF fusions were detected by both platforms and were concordant in the majority of cases. In several tumors with low tumor content, CMA did not reveal a copy number gain in 7q34 presumably because it was below the 10% limit of detection for the assay, whereas RNA analysis demonstrated a KIAA1549–BRAF fusion that was highly expressed. In contrast, CMA has a higher detection rate than NGS when the fusion is expressed at low levels, or when a complex rearrangement is implicated by the detection of chromosomal breakpoints that involve cancer-related genes, but the primers to detect the RNA fusion are not in the assay design.
Among the gliomas, in addition to the KIAA–BRAF fusions, NGS revealed Tier I or II sequence variants in the MAPK and citric acid cycle pathways, and CNAs revealed diagnostic or prognostic biomarkers, such as 1p/19q in oligodendroglioma, or homozygous loss of CDKN2A/B. NGS analysis of HGGs revealed diagnostic and prognostic Tier I or II mutations in H3F3A, HIST1H3B, ACVR1, TP53, NF1, PIK3CA, ATRX, PDGFRA, PTEN, MET, RB1, and PPM1D whereas CMA demonstrated key prognostic genomic markers including trisomy 7 and monosomy 10.
Our molecular approaches also facilitated the identification of 3 similar brainstem tumors harboring identical QKI–RAF1 fusions. The QKI–RAF1 fusion results in constitutive RAF1 kinase activity, leading to activation of the MAPK/ERK and PI3K/mTOR signaling pathways.33 Although rare, RAF1 rearrangements including the QKI–RAF1 fusion have been previously reported in pediatric LGGs.34–37QKI–RAF1 fusions appear to have a distinct drug response and have been associated with resistance to certain RAF inhibitors.33,38
Tumor profiling at recurrence demonstrated clonal evolution in several patients in this cohort,39 including the emergence of a potential drug resistance mutation (MAP2K1 p.Phe53Leu) in a patient with a BRAF V600E-mutant anaplastic astrocytoma treated with a targeted MEK inhibitor (Case #45). MAP2K (mitogen-activated protein kinase kinase 1, previously known as MEK1) is a dual-specificity kinase involved in the RAS–RAF–MEK–ERK pathway in cell proliferation and differentiation. Activating somatic mutations in MAP2K1 have been observed in a number of cancers including CNS tumors.40 This particular missense p.Phe53Leu variant does not lie within the catalytic domain, but has shown activation of MAP2K1 as indicated by increased Erk and Mek phosphorylation in cell culture.41 Preclinical studies demonstrate that this variant may show resistance to some MEK and BRAF inhibitors.42
Germline cancer predisposition was confirmed in 9 patients by follow-up targeted testing after tumor genomic profiling. The total number of patients with cancer predisposition was likely underestimated in this cohort due to (1) lack of blood samples for follow-up germline testing, (2) the presence of pathogenic variants in regions not covered by NGS, such as VHL, (3) exonic CNAs that were below the resolution of CMA but not detected by NGS, and (4) alterations in non-protein-coding regions of the genome not detected by this NGS panel. Cancer predisposition germline testing, even when family history is not remarkable, is important to avoid syndrome-related increased toxicity and critically to increase cancer surveillance in the proband and at-risk relatives. Ultimately, systematic monitoring of carriers of germline cancer predisposition variants may allow for the detection of cancers at their earliest and most curable stage, thereby improving patient outcomes.
Conclusions
Our results highlight the importance of molecular characterization of CNS tumors in pediatric and AYA patients as a basis for optimal personalized diagnosis and treatment at both diagnosis and in the setting of recurrent disease. The correlation of CNA and gene mutation profiles with histological features permits more accurate diagnoses and prognoses and supports the creation of personalized treatment plans. The testing algorithm employed in our clinical program also allows for the identification of a key subset of pediatric patients who require referral for germline testing. Overall, the OncoKids NGS panel was most informative for gliomas, whereas CMA had the greatest utility for ETMR and subgrouping choroid plexus tumors. Although these specific assays may be replaced by whole-genome DNA sequencing, RNA sequencing, and/or epigenetic profiling, it is important to recognize that subclonal alterations may be missed due to the lower depth of coverage with these genome-wide assays. Development of a rapid diagnostic strategy for integrated diagnosis and determination of prognosis will continue to be critical to the care of patients with CNS tumors.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Acknowledgments
We thank Jennifer Han, Dolores Estrine, Cindy Fong, and Vandana Mehta for their technical assistance.
Authorship Statement. J.J.: Overall responsibility for the execution of the project; data collection, analysis, and interpretation; writing the manuscript. K.K.: Data collection and contributed to writing the manuscript. M.C.H.: OncoKids data analysis, interpretation, and contributed to writing the manuscript. G.D.: Clinical data collection, interpretation, and contributed to writing the manuscript. T.B.D.: Clinical data collection, interpretation, and contributed to writing the manuscript. A.E.E.: Clinical data collection, interpretation, and contributed to writing the manuscript. D.E.: Pathology data collection, interpretation, and contributed to writing the manuscript. K.H.: Pathology data interpretation and contributed to writing the manuscript. A.S.M.: Clinical data collection, interpretation, and contributed to writing the manuscript. A.J.M.: Pathology data interpretation and contributed to writing the manuscript. N.J.R.: Clinical data collection, interpretation, and contributed to writing the manuscript. R.J.S.: OncoKids data analysis, interpretation, and contributed to writing the manuscript. H.N.T.: Clinical data collection, interpretation, and contributed to writing the manuscript. A.R.J.: Overall execution of the study, pathology data interpretation, and contributed to writing the manuscript. J.A.C.: Data collection, interpretation, and contributed to writing the manuscript. J.A.B.: Overall execution of the study, data interpretation, and contributed to writing the manuscript.
Conflict of interest statement. All authors declare no conflict of interests.

{kind=link}
{kind=link}
{kind=link}
{kind=link}