





Abstract
Neurofibromatosis 2 (NF2) is an inherited disorder caused by bi-allelic inactivation of the NF2 tumor suppressor gene. NF2-associated tumors, including schwannoma and meningioma, are resistant to chemotherapy, often recurring despite surgery and/or radiation, and have generally shown cytostatic response to signal transduction pathway inhibitors, highlighting the need for improved cytotoxic therapies.
Leveraging data from our previous high-throughput drug screening in NF2 preclinical models, we identified a class of compounds targeting the ubiquitin–proteasome pathway (UPP), and undertook studies using candidate UPP inhibitors, ixazomib/MLN9708, pevonedistat/MLN4924, and TAK-243/MLN7243. Employing human primary and immortalized meningioma (MN) cell lines, CRISPR-modified Schwann cells (SCs), and mouse Nf2−/− SCs, we performed dose response testing, flow cytometry-based Annexin V and cell cycle analyses, and RNA-sequencing to identify potential underlying mechanisms of apoptosis. In vivo efficacy was also assessed in orthotopic NF2-deficient meningioma and schwannoma tumor models.
Testing of three UPP inhibitors demonstrated potent reduction in cell viability and induction of apoptosis for ixazomib or TAK-243, but not pevonedistat. In vitro analyses revealed that ixazomib or TAK-243 downregulates expression of c-KIT and PDGFRα, as well as the E3 ubiquitin ligase SKP2 while upregulating genes associated with endoplasmic reticulum stress-mediated activation of the unfolded protein response (UPR). In vivo treatment of mouse models revealed delayed tumor growth, suggesting a therapeutic potential.
This study demonstrates the efficacy of proteasomal pathway inhibitors in meningioma and schwannoma preclinical models and lays the groundwork for use of these drugs as a promising novel treatment strategy for NF2 patients.

Proteasome pathway inhibition induced apoptosis in NF2-null cell lines.
Treatment led to differential expression of proto-oncogenes and UPR-related genes.
Ixazomib treatment significantly reduced growth in preclinical mouse models of NF2.
Meningiomas and schwannomas are progressive, highly morbid, and resistant to traditional chemotherapies. Moreover, these tumors have shown, at best, cytostatic response to several implicated signaling pathway inhibitors, highlighting the need for improved cytotoxic therapies. Here, we identified ubiquitin–proteasome pathway inhibitors, TAK-243 and ixazomib, that induced apoptosis in NF2-related cell models. Treatment with these inhibitors in meningioma cell lines demonstrated decreased levels of several proto-oncogenes and upregulation of signature members of the unfolded protein response pathway. Importantly, in vivo treatment in NF2 mouse models revealed delayed tumor growth, suggesting a therapeutic potential for proteasomal pathway inhibition for NF2-associated and sporadic meningioma and schwannoma.
Neurofibromatosis 2 (NF2) is an autosomal dominant genetic disorder, caused by bi-allelic inactivating mutations in the NF2 tumor suppressor gene.1 NF2 is associated with central and peripheral nervous system tumors including schwannomas (especially vestibular schwannomas) and meningiomas, including intracranial and paraspinal locations.2NF2 loss also accounts for ~50% of sporadic meningiomas, the most common intracranial tumor in adults, and although the majority are histologically benign, they are often multifocal and tend to recur despite standard surgical or radiation therapies.3,4 We previously identified dysregulation of signaling pathways resulting from NF2 loss that has led to clinical trials for NF2, such as rapalogs and AZD2014 targeting mechanistic target of rapamycin (mTOR) signaling.5,6 However, treatment of meningioma and schwannoma preclinical models with these compounds have only shown cytostatic effects.7–9 Hence, there is a need to identify better noninvasive therapeutic strategies for NF2-associated and sporadic meningiomas and schwannomas.
As part of a large-scale collaborative in vitro drug screen, we recently investigated potential candidate compounds showing efficacy in NF2-related cell lines.10 Here, we leveraged our analyses of the high-throughput drug screen data and identified a distinct subset of drugs targeting the ubiquitin (Ub)-proteasome pathway (UPP) to be potential candidates leading to cytotoxicity in our NF2-deficient meningioma (MN) and Schwann cell (SC) lines. The UPP comprises a network of enzymes along with the multi-subunit 26S proteasome, which is responsible for regulating many biological processes including cellular protein homeostasis, to sustain an intricate balance between cell survival and apoptosis.11,12 Recently, UPP inhibitors have garnered much attention as candidate compounds to treat several types of cancers.13 Based on our analyses, we have tested three drugs targeting the UPP that are reported to have strong antitumor activity in preclinical and/or clinical studies, including the E1-Ub activating enzyme inhibitor TAK-243/MLN7243,14 the neuronal precursor cell-expressed developmentally down-regulated protein 8 (NEDD-8)-activating enzyme inhibitor pevonedistat/MLN492415 and the core 20S proteasome inhibitor ixazomib/MLN9708.16 We report here, for the first-time, cytotoxic effects in NF2-deficient MN and SC lines upon treatment with TAK-243 and ixazomib. To elucidate the mechanism, we performed global transcriptomic analysis in treated NF2-null MN cells, which demonstrated decreased expression of proto-oncogenes KIT, PDGFRA, and SKP2. In addition, we observed upregulation of several genes involved in the endoplasmic reticulum (ER) stress and activation of the unfolded protein response (UPR) pathway, which may be partially responsible for the observed cell death. Furthermore, the therapeutic potential of these drugs was evaluated in in vivo mouse models, where we found ixazomib to be effective in delaying tumor growth in both NF2-deficient meningioma and schwannoma.
Materials and Methods
Cell Culture and Treatment
Human meningiomas from surgical specimens used to generate primary cell cultures were collected following Massachusetts General Hospital Human Subjects protocols for tumor acquisition after obtaining informed consent. For further details of cell lines and reagents, proliferation assays, cell death assay, quantitative RT-PCR, immunoblotting, RNA sequencing, and cell cycle assays, see Supplementary Methods.
Cell Viability and Dose Response Testing
For immortalized MN lines Ben-Men-1 and MN1-LF, primary MN lines, and all SC lines; assays were carried out using the CellTiter-Glo cell viability kit (Promega) according to the manufacturer’s instructions. Relative luminescence units were measured using EnVision 2103 Multilabel Reader (Perkin Elmer). Treatment dosage details are outlined in the figure legends. For NF2-null AG-NF2-Men, AG-FP-Men, and KT21-MG cell lines, assays were performed using fluorescence-based resazurin (Sigma) assays and measured on a SpectraMax M2e plate reader (Molecular Devices). See Supplementary Methods for details.
Transcriptome/RNA Sequencing and Analysis
Ben-Men-1 and MN1-LF cells were treated for 24 h with 100 nM TAK-243, ixazomib, or DMSO followed by harvesting using TRIzol reagent (ThermoFisher Scientific) according to the manufacturer’s instructions. RNA sequencing libraries were made in sextuplets for treated cell lines, resulting in 36 total mRNA libraries, using Illumina’s TruSeq Stranded mRNA Library Preparation Kits (RS-122-2101, RS-122-2102, or RS-122-03). Differential expression analysis was performed separately for each MN line. For each pairwise comparison, genes in the top 5% by p-value were identified as differentially expressed genes (DEGs). Gene Ontology (GO) enrichment analysis was performed, testing enrichment of DEGs from each pairwise comparison and DEGs shared between Ben-Men-1 and MN1-LF lines upon TAK-243 or ixazomib treatments, for GO terms retrieved from MSigDB database (v. 7.4)17 using one-tailed Fisher’s exact test. See Supplementary Methods for details.
Treatment of In Vivo Mouse Models
All animal work was performed in accordance with Nationwide Children’s Hospital Institutional Animal Care and Use Committee or the Institutional Animal Care and Use Committee of Massachusetts General Hospital (MGH). Maximal tolerated dose (MTD) for TAK-243 (25 mg/kg) and ixazomib (7 mg/kg) was determined using previously reported dose ranges14,18,19 and confirmed in our model. To generate the orthotopic meningioma xenograft model, luciferase-expressing NF2-deficient KT21-MG1-Luc5D cells were injected intracranially into skull base of 8–12 week-old NSG mice (NOD.Cg-PrkdcscidIl2rgtmlWjl/SzJ; The Jackson Laboratory) as described previously.20 Injected mice were monitored for tumor growth by bioluminescence imaging (BLI) on Xenogen IVIS Spectrum imaging system (Perkin Elmer).21 The effects of treatment were measured by weekly BLI. See Supplementary Methods for details.
For schwannoma mouse models, Nf2−/− mouse SCs were implanted in the sciatic nerve of 8–12 weeks old immune-competent FVB/C57Bl6 mice, and patient-derived MGH-VS4 SCs were implanted in 8–12 weeks old immune-deficient nude mice.22,23 A total of 3 μl of tumor cell suspension (5 × 104 cells/mouse) was injected slowly (over 45–60 s) under the sciatic nerve sheath using a Hamilton syringe to prevent leakage. Treatment started when tumors reached 2–3 mm in diameter by caliper. Ixazomib (8 mg/kg) was administered by oral gavage every 3 days and continued until the study endpoint. Tumor size was measured every 3 days by caliper, and body weight was measured every 3 days as a sign of toxicity (defined as >15% body weight loss).
Results
Ubiquitin–Proteasome Pathway-related Drugs Induce Apoptosis in NF2-null Meningioma Cells
For NF2-associated therapies, majority of compounds identified to date have shown cytostatic effects in pre-clinical in vitro and in vivo models as well as in NF2-related clinical trials, with no sustained tumor shrinkage.7–10 We analyzed our recent high-throughput drug screening data set from NF2-deficient MN and SC lines10 (NF Data Portal Synapse ID: syn12087415; www.synapse.org) to search for potential cytotoxic drug candidates and identified a set of compounds targeting the ubiquitin–proteosome pathway (UPP). Based on this analysis, we chose three UPP inhibitors currently under clinical development, for further testing: pevonedistat, TAK-243, and ixazomib.
We initially performed drug treatment in 6 NF2-deficient human MN lines, including an immortalized Ben-Men-1 (WHO grade I) and 5 primary MN lines (two grade I, one grade I with atypical features, and two grade II), Cell viability experiments with these MN lines demonstrated potent effects for TAK-243 and ixazomib treatment compared to DMSO controls (Figure 1A, Table S1, Fig. S1A). For pevonedistat, all MN lines tested revealed minimal effects, with indeterminant IC50 and/or maximum response of >50% at the highest dose tested in many of the lines, and therefore we discontinued further testing (Figure 1A, Table S1). In addition, we also assessed the efficacy of TAK-243 and ixazomib in 5 NF2-expressing primary MN lines (two grade I, two grade II, and one grade III) and found the drug potency to be similar in these lines compared to NF2-deficient lines (Table S1, Fig. S1B, Fig. S2).
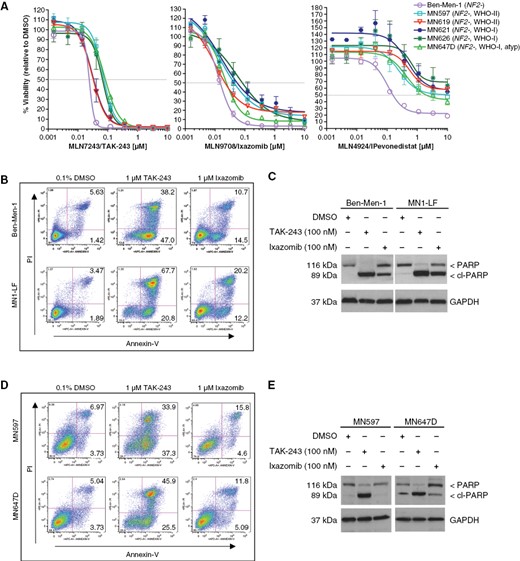
Treatment with ubiquitin-proteasome pathway inhibitors leads to reduced cell viability and apoptosis in NF2-deficient MN cell lines. (A) Dose-response curves (DRCs), representing percent cell viability relative to DMSO, are shown for human NF2-null MN lines, including immortalized Ben-Men-1 and 5 primary MN lines treated with TAK-243, ixazomib, or pevonedistat. Cell lines were treated with 9 dosage points (1.5 nM–10 μM, 1:3 serial dilution) for each drug representing 3 biological replicates (performed in triplicate) ± SEM. (B–E) Assessment of apoptosis was carried out by flow cytometry (B and D, numbers shown in upper and lower right quandrants for each plot represent percentage) and immunoblotting for cleaved-PARP (C and E). Treatment with TAK-243 or ixazomib (24 h) led to increased apoptosis in Ben-Men-1 and MN1-LF cells (B and C) and primary MN lines MN597 and MN647D (D and E).
We next assessed the ability of TAK-243 and ixazomib to induce apoptosis in two immortalized cell lines, Ben-Men-1 and MN1-LF, and two primary NF2-deficient MN lines, MN597 and MN647D. Flow cytometry using Annexin V/propidium iodide (PI) dual staining revealed increased apoptosis in all treated MN lines tested compared to DMSO controls (Figure 1B and D, Table S2). Additionally, immunoblotting confirmed cleavage of poly (ADP-ribose) polymerase (PARP), a standard apoptosis readout, in these MN lines (Figure 1C and E). Collectively, these results suggest that TAK-243 or ixazomib induces apoptotic cell death in the NF2-deficient MN cell lines.
Global Transcriptomic Analysis Reveals Downregulation of Several Proto-oncogenes Along With Activation of Unfolded Protein Response
To elucidate the molecular mechanism, we performed RNA-seq analysis in Ben-Men-1 and MN1-LF treated for 24 h with 100 nM of TAK-243 or ixazomib. Differential expression analysis of drug-treated lines compared with DMSO-treated controls revealed that either TAK-243 or ixazomib led to extensive gene expression changes (Figure 2A, Fig. S3). Direct comparison of the top 5% differentially expressed genes (DEGs) (assessed by P-value) following TAK-243 or ixazomib treatment revealed a total of 1253 or 1179 genes, respectively; among which 345 genes (for TAK-243) and 420 genes (for ixazomib) shared between Ben-Men-1 and MN1-LF, showed expression changes in the same direction (increased or decreased compared to DMSO controls) (Figure 2B).
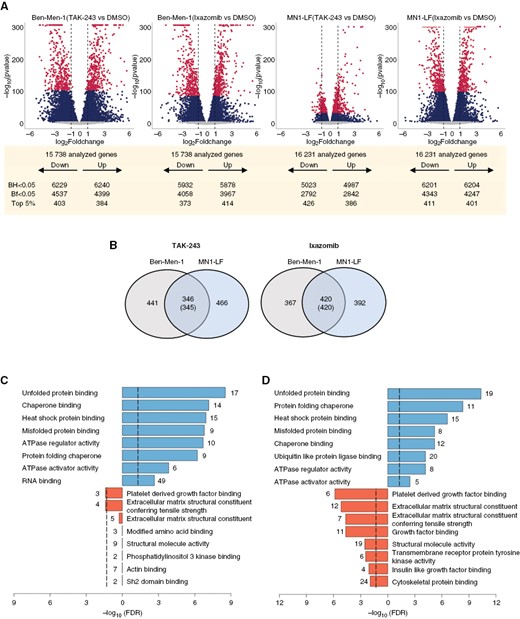
Transcriptome analysis reveals differentially expressed genes in NF2-deficient MN lines treated with UPP inhibitors. (A) Volcano plots of differentially expressed genes (DEGs) from drug-treated cell lines vs DMSO controls with the top 5% DEGs at Bonferroni-adjusted P-value (Bf) < .05 (maroon), the remaining DEGs at P-value (Bf) < .05 (dark blue), and the DEGs at P-value (Bf) > .05 (grey) shown. BH, Benjamini–Hochberg adjusted P-value. (B) Venn diagrams represent numbers of top 5% DEGs shared between Ben-Men-1 and MN1-LF upon treatment with TAK-243 (left) or ixazomib (right). Numbers within parenthesis denote DEGs in the same direction of dysregulation (up or down). (C and D) GO terms enriched for molecular function upon TAK-243 (C) or ixazomib (D) treatment that are upregulated (right-shifted from 0) or downregulated (left-shifted from 0) are shown, along with corresponding numbers of DEGs from the RNA-seq dataset.
Next, we categorized these shared DEG sets individually into upregulated or downregulated and performed Gene Ontology (GO) enrichment analysis separately on each category. This analysis revealed enrichment of downregulated shared DEGs for terms such as “platelet derived growth factor binding” upon ixazomib or TAK-243 treatment, whereas upregulated shared DEGs demonstrated a strong association with terms related to “protein folding chaperone”, “misfolded protein binding”, and “unfolded protein binding” upon treatment with TAK-243 or ixazomib (Figure 2C and D)
Validation of the Downregulated Gene Expression
We previously reported upregulation of c-KIT at both kinome and transcriptome levels in NF2-null CRISPR-modified arachnoid cells (ACs) and Ben-Men-1 cells.24,25 Further, increased expression of both c-KIT and platelet-derived growth factor receptor alpha (PDGFRα) receptors have been reported in NF2-related human schwannomas.26,27 Interestingly, RNA-seq analyses demonstrated decreased expression of both KIT and PDGFRA upon ixazomib treatment in Ben-Men-1 and MN1-LF (Table 1). To substantiate the transcriptomic analyses, we performed quantitative RT-PCR (qPCR) for KIT and PDGFRA. In Ben-Men-1 and MN1-LF cells, 24 h treatment with UPP inhibitors confirmed a significant decrease in KIT and PDGFRA expression, compared to DMSO controls (Figure 3A). To assess the effect of these UPP inhibitors at the protein level, we treated Ben-Men-1 and MN1-LF cells for 12 and 24 h with TAK-243 or ixazomib and performed immunoblotting. Results revealed reduced expression of c-KIT and PDGFR-α in both the cell lines (Figure 3B and Fig. S4A). Overall, these data suggest that, with regard to gene and protein expression of KIT and PDGFRα, Ben-Men-1 cells are more sensitive to TAK-243 treatment than MN1-LF cells, while ixazomib treatment led to similarly reduced expression in both cell lines.
Differentially Expressed Genes Regulated by Ixazomib or TAK-243 Treatment in Both Ben-Men-1 and MN1-LF Cells
| Drug | Symbol | Cell Line | Direction | Log2FC | Bonf. Adj. P Value |
|---|---|---|---|---|---|
| Ixazomib | PDGFRA | Ben-Men-1 | Downregulated | −2.024 | 1.29E-113 |
| MN1-LF | Downregulated | −2.372 | 4.27E-133 | ||
| KIT | Ben-Men-1 | Downregulated | −2.753 | 1.26E-161 | |
| MN1-LF | Downregulated | −2.937 | <1.00E-303a | ||
| SKP2 | Ben-Men-1 | Downregulated | −2.017 | 2.08E-90 | |
| MN1-LF | Downregulated | −1.953 | 1.62E-126 | ||
| TAK-243 | HSPA5 | Ben-Men-1 | Upregulated | 3.295 | <1.00E-303a |
| MN1-LF | Upregulated | 1.108 | 1.36E-114 | ||
| XBP1 | Ben-Men-1 | Upregulated | 1.399 | 5.35E-117 | |
| MN1-LF | Upregulated | 0.555 | 1.36E-29 | ||
| ATF3 | Ben-Men-1 | Upregulated | 3.423 | <1.00E-303a | |
| MN1-LF | Upregulated | 2.126 | 3.14E-157 | ||
| DDIT3 | Ben-Men-1 | Upregulated | 2.038 | 3.12E-104 | |
| MN1-LF | Upregulated | 0.871 | 6.06E-29 |
| Drug | Symbol | Cell Line | Direction | Log2FC | Bonf. Adj. P Value |
|---|---|---|---|---|---|
| Ixazomib | PDGFRA | Ben-Men-1 | Downregulated | −2.024 | 1.29E-113 |
| MN1-LF | Downregulated | −2.372 | 4.27E-133 | ||
| KIT | Ben-Men-1 | Downregulated | −2.753 | 1.26E-161 | |
| MN1-LF | Downregulated | −2.937 | <1.00E-303a | ||
| SKP2 | Ben-Men-1 | Downregulated | −2.017 | 2.08E-90 | |
| MN1-LF | Downregulated | −1.953 | 1.62E-126 | ||
| TAK-243 | HSPA5 | Ben-Men-1 | Upregulated | 3.295 | <1.00E-303a |
| MN1-LF | Upregulated | 1.108 | 1.36E-114 | ||
| XBP1 | Ben-Men-1 | Upregulated | 1.399 | 5.35E-117 | |
| MN1-LF | Upregulated | 0.555 | 1.36E-29 | ||
| ATF3 | Ben-Men-1 | Upregulated | 3.423 | <1.00E-303a | |
| MN1-LF | Upregulated | 2.126 | 3.14E-157 | ||
| DDIT3 | Ben-Men-1 | Upregulated | 2.038 | 3.12E-104 | |
| MN1-LF | Upregulated | 0.871 | 6.06E-29 |
Abbreviations: Bonf. Adj., Bonferroni adjusted; FC, fold change
aP value was below the calculatable threshold assessed by the edgeR Bioconductor software package.
Differentially Expressed Genes Regulated by Ixazomib or TAK-243 Treatment in Both Ben-Men-1 and MN1-LF Cells
| Drug | Symbol | Cell Line | Direction | Log2FC | Bonf. Adj. P Value |
|---|---|---|---|---|---|
| Ixazomib | PDGFRA | Ben-Men-1 | Downregulated | −2.024 | 1.29E-113 |
| MN1-LF | Downregulated | −2.372 | 4.27E-133 | ||
| KIT | Ben-Men-1 | Downregulated | −2.753 | 1.26E-161 | |
| MN1-LF | Downregulated | −2.937 | <1.00E-303a | ||
| SKP2 | Ben-Men-1 | Downregulated | −2.017 | 2.08E-90 | |
| MN1-LF | Downregulated | −1.953 | 1.62E-126 | ||
| TAK-243 | HSPA5 | Ben-Men-1 | Upregulated | 3.295 | <1.00E-303a |
| MN1-LF | Upregulated | 1.108 | 1.36E-114 | ||
| XBP1 | Ben-Men-1 | Upregulated | 1.399 | 5.35E-117 | |
| MN1-LF | Upregulated | 0.555 | 1.36E-29 | ||
| ATF3 | Ben-Men-1 | Upregulated | 3.423 | <1.00E-303a | |
| MN1-LF | Upregulated | 2.126 | 3.14E-157 | ||
| DDIT3 | Ben-Men-1 | Upregulated | 2.038 | 3.12E-104 | |
| MN1-LF | Upregulated | 0.871 | 6.06E-29 |
| Drug | Symbol | Cell Line | Direction | Log2FC | Bonf. Adj. P Value |
|---|---|---|---|---|---|
| Ixazomib | PDGFRA | Ben-Men-1 | Downregulated | −2.024 | 1.29E-113 |
| MN1-LF | Downregulated | −2.372 | 4.27E-133 | ||
| KIT | Ben-Men-1 | Downregulated | −2.753 | 1.26E-161 | |
| MN1-LF | Downregulated | −2.937 | <1.00E-303a | ||
| SKP2 | Ben-Men-1 | Downregulated | −2.017 | 2.08E-90 | |
| MN1-LF | Downregulated | −1.953 | 1.62E-126 | ||
| TAK-243 | HSPA5 | Ben-Men-1 | Upregulated | 3.295 | <1.00E-303a |
| MN1-LF | Upregulated | 1.108 | 1.36E-114 | ||
| XBP1 | Ben-Men-1 | Upregulated | 1.399 | 5.35E-117 | |
| MN1-LF | Upregulated | 0.555 | 1.36E-29 | ||
| ATF3 | Ben-Men-1 | Upregulated | 3.423 | <1.00E-303a | |
| MN1-LF | Upregulated | 2.126 | 3.14E-157 | ||
| DDIT3 | Ben-Men-1 | Upregulated | 2.038 | 3.12E-104 | |
| MN1-LF | Upregulated | 0.871 | 6.06E-29 |
Abbreviations: Bonf. Adj., Bonferroni adjusted; FC, fold change
aP value was below the calculatable threshold assessed by the edgeR Bioconductor software package.
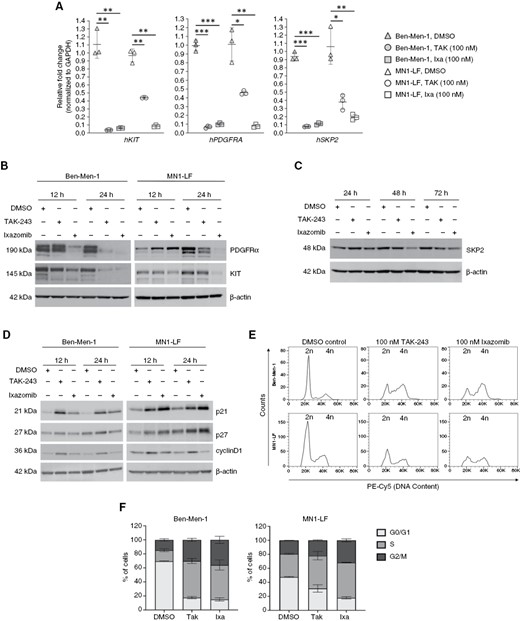
Treatment with proteasome pathway inhibitors leads to decreased expression of proto-oncogenes in NF2-null MN lines. (A) Quantification of qRT-PCR for KIT, PDGFRA, and SKP2 of Ben-Men-1 and MN1-LF cell lines demonstrated significant decrease in the expression levels of all three genes upon 24 h treatment with 100 nM TAK-243 or ixazomib. (B) Immunoblotting of Ben-Men-1 and MN1-LF cells treated with TAK-243 or ixazomib (12 and 24 h, 100 nM) for c-KIT and PDGFRα revealed decreased expression. β-actin serves as a loading control. (C) Immunoblotting of treated MN1-LF cells demonstrated decreased expression of SKP2 by 48 h or 72 h with 100 nM of TAK-243 or ixazomib, respectively. (D) Immunoblotting revealed increased p21, p27, and cyclinD1 in Ben-Men-1 and MN1-LF cells treated for 12 and 24 h with 100 nM TAK-243 or ixazomib. (E and F) Cell cycle data for Ben-Men-1 (n = 4) and MN1-LF (n = 2) treated for 24 h with TAK-243 or ixazomib are shown (F as a representative). Quantification for percentage of cells in cell cycle phases G0/G1, S, and G2/M is shown ± SEM (F). *P < .05, **P < .01, ***P < .001 (for A).
Ixazomib treatment of Ben-Men-1 and MN1-LF cells also led to decreased expression of the proto-oncogene S-Phase kinase-associated protein 2 (SKP2), a member of the SKP-Cullin-F-box Ub-ligase complex (SCFSKP2) shown to be upregulated in various tumor types (Table 1)28–30 Interestingly, in previous RNA-seq studies, we had observed SKP2 be increased in NF2-deficient Ben-Men-1 cells compared with NF2-expressing arachnoid cells (unpublished data). Similar to KIT and PDGFRA, confirmation by qPCR revealed decreased SKP2 in TAK-243 or ixazomib treated cell lines (Figure 3A). By immunoblotting, treatment in MN1-LF cells showed a decrease of SKP2 by 48 h with ixazomib treatment and by 72 h with TAK-243 treatment (Figure 3C). However, in Ben-Men-1 cells, we were unable to examine SKP2 protein levels at these longer timepoints due to potent cytotoxicity. Notably, SKP2 is an F-box substrate recognition member of the SCFSKP2, which functions to regulate cell cycle progression by directly targeting cyclin dependent kinase inhibitors (CKIs) such as p21 and p27 for degradation.31 Apart from p21 and p27, SKP2 also controls the degradation of cyclinD1 protein, which is also required for cell cycle progression.32 Immunoblotting revealed accumulation of CKIs p21 and p27, as well as cyclinD1 in both cell lines upon treatment with TAK-243 and ixazomib (Figure 3D, Fig. S4B and C). Moreover, cell cycle analyses of Ben-Men-1 and MN1-LF lines treated with TAK-243 or ixazomib demonstrated an increased percentage of cells in S and G2/M phases, while cells in G1 phase decreased correspondingly. (Figure 3E and F). Collectively, these data support the negative regulatory effects of UPP inhibitors on cell cycle progression in NF2-deficient MN cells.
Validation of the Upregulated Gene Expression
Inhibition of the UPP leads to impaired maintenance of protein homeostasis by blocking Ub-dependent protein degradation. As a result, accumulation of normally short-lived, damaged, or misfolded proteins leads to ER stress, and primes the cells for apoptosis.33,34 Upon ER stress, unfolded proteins bind to the molecular chaperone GRP78/BIP, which leads to its dissociation from the proximal membrane sensors PERK, IRE1α, and ATF6, thus initiating the three arms of the UPR signaling cascade. Analysis of the top 5% DEGs in Ben-Men-1 and MN1-LF cells revealed increased expression of several genes associated with ER stress and UPR pathways (Figure 2C and D, Table 1). Particularly, we found increased expression of HSPA5, encoding the upstream UPR chaperone GRP78/BIP, as well as DDIT3, encoding C/EBP Homologous Protein (CHOP) that is a downstream transcription factor regulating pro- and anti-apoptotic gene expression.35 We also identified XBP1 (X-box binding protein 1) and ATF3 (activating transcription factor 3) (Table 1), which are key players in the signaling axes of UPR.36,37 Immunoblotting confirmed increased expression of GRP78, XBP1s (spliced form) and ATF3 upon treatment with TAK-243 or ixazomib implying activation of the UPR signaling axes (Figure 4A–D).
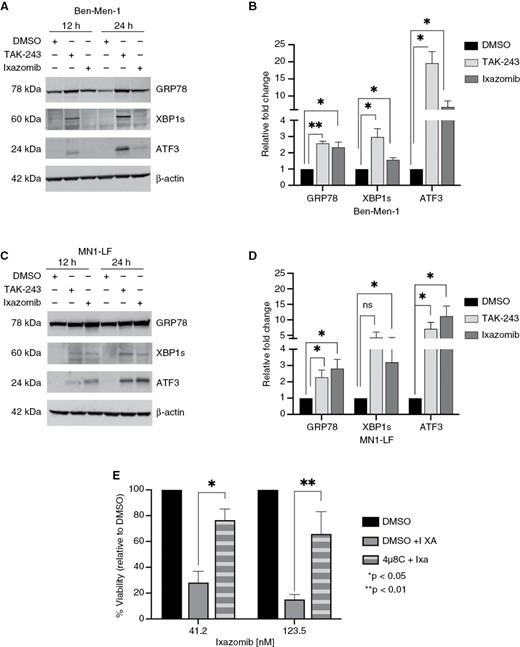
UPP inhibition leads to activation of the unfolded protein response in NF2-null MN cells. (A and C) Immunoblotting confirmed increased expression of unfolded protein response (UPR) markers GRP78, spliced XBP1 (XBP1s), and ATF3 in Ben-Men-1 (A) and MN1-LF (C) cell lines following 12 and 24 h treatment with 100 nM of TAK-243 or ixazomib. (B and D) Quantitation of 24 h treatment in Ben-Men-1 (B) and MN1-LF (D) using ImageJ/Fiji is shown representing the average of 3 replicates (DMSO, normalized to 1) ± SEM. (E) Cell viability assays, in MN1-LF cells pretreated for 24 h with 5 µM of IREα inhibitor 4µ8C, followed by 48 h treatment with ~41 nM or ~124 nM of ixazomib (4µ8C+Ixa) showed significant increase in percent viability compared to DMSO pretreatment (DMSO+Ixa). (DMSO alone, normalized to 100). Each bar represents average of 3 replicates ± SEM; *P < .05; **P < .01; ns, not significant.
To gain further insight into the role of UPR signaling in UPP inhibitor-induced cytotoxicity, we performed cell viability assays for TAK-243 or ixazomib in MN1-LF cells that were pretreated with PERK inhibitor GSK2606414, IRE1α inhibitor 4µ8C or ATF6 inhibitor nelfinavir. Cells pretreated for 24 h with 5 µM of 4µ8C (IRE1α inhibitor) were found to be significantly less sensitive to ixazomib compared to TAK-243 or DMSO pretreated cells (Figure 4E and data not shown). Conversely, cells pretreated with either PERK inhibitor or ATF6 inhibitor revealed no significant change in sensitivity to ixazomib or TAK-243 upon pretreatment (data not shown). These data suggest a role for IRE1α-driven UPR in the cytotoxic response of NF2-null meningioma cells to UPP inhibitor ixazomib. Taken together, these findings support the notion that UPP inhibition of NF2-deficient meningioma cells leads to reduced expression of proto-oncogenes KIT, PDGFRΑ, and SKP2 with concomitant induction of UPR transcriptional signatures.
In vivo Drug Testing in Meningioma Preclinical Models
Prior to generating the orthotopic mouse model, we evaluated the growth inhibitory activities of TAK-243 and ixazomib in three additional NF2-deficient cell lines in vitro, representing all three grades of meningioma: AG-NF2-Men (grade I), AG-FP-Men (grade II), and KT21-MG (grade III). The IC50 values of TAK-243 in AG-NF2-Men, AG-FP-Men, and KT21-MG1 cells were ~55, 113, and 100 nM, respectively. Ixazomib appeared more potent with IC50 values of ~25, 56, and 25 nM, respectively (Fig. S5A). Importantly, both ixazomib and TAK-243 showed high maximum inhibitory response. Next, KT21-MG1 cells were treated with 1×, 2×, and 3× IC50 doses of TAK-243 or ixazomib, and we observed enhanced cleavage of caspase-3 and PARP while decreasing total caspase 3 in a dose-dependent manner, as compared to GAPDH (Fig. S5B), confirming that TAK-243 and ixazomib induce apoptosis in these cells. Furthermore, treatment with either drug caused dose-dependent increase in γH2A.X, a DNA damage response marker, suggesting genotoxic stress. Additionally, UPP inhibitor treatment in KT21-MG cells also led to an increase in the expression of GRP78, ATF3, CHOP, and XBP1s further supporting the induction of UPR by these UPP inhibitors (Fig. S5C).
Although most meningiomas are benign, a significant subset, classified as atypical (WHO grade II) or anaplastic/malignant (WHO-grade III), display more aggressive clinical behavior, with rapid growth and increased recurrence rates, as well as high morbidity.3,38 To evaluate the anti-tumor efficacy of TAK-243 and ixazomib, we generated mice bearing intracranial WHO grade III KT21-MG1-Luc5D meningioma xenografts (Figure 5A). Bioluminescence imaging (BLI) of vehicle-treated mice showed that KT21-MG1-Luc5D meningiomas grew steadily over time. TAK-243 suppressed tumor growth by ~65% after six weeks of treatment, compared with the vehicle control (Figure 5B). Representative BLI images are shown in Fig. S6. Similarly, we observed that ixazomib suppressed meningioma xenograft growth by ~86% relative to the vehicle control after five-weeks of treatment (Figure 5B). Taken together, these results demonstrate that TAK-243 and ixazomib exhibit anti-tumor effects in NF2-deficient malignant meningiomas.
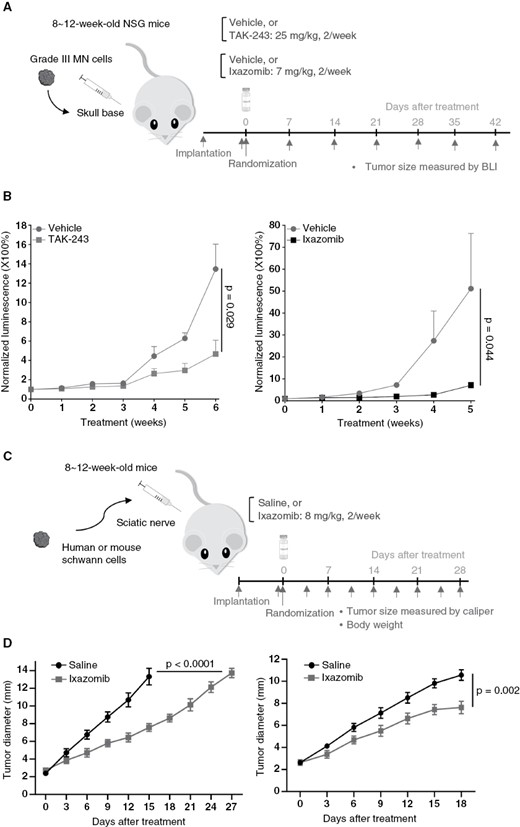
UPP inhibitor treatment leads to tumor shrinkage in mouse models of NF2-related meningioma and schwannoma. (A) SchematicFigure 5. Continuedrepresentation of drug treatment timeline for in vivo NF2-null Grade III meningioma mouse model. (B) Treatment with TAK-243 (left) or ixazomib (right) suppressed tumor growth in the KT21-MG1-Luc5D skull-base xenograft model. Relative tumor-emitted bioluminescent signals are calculated as the percentage of total photon flux during treatment relative to the pretreatment total flux, denoted as 1. Data are shown as mean ± SD for each treatment group. (C) Schematic representation of ixazomib treatment timeline for in vivo schwannoma mouse models. (D) Tumor growth curves of mouse Nf2−/− sciatic nerve model (left) and patient-derived VS4 sciatic nerve model (right). Schwannoma mouse model studies presented are representative of at least three independent experiments, n = 8 per group in each experiment and data presented as mean ± SEM. P-values are shown in the figure.
In vivo Drug Testing in Schwannoma Preclinical Models
We first performed dose response testing of the UPP-inhibitors in NF2-deficient Schwann cells including two human CRISPR-modified (SC-CRISPR) clones, S3-null and S7-null, as well as a mouse Nf2-mutant SC line (Nf2−/− SCs). In vitro drug treatment was performed using the same dosage points and timing as for meningioma cell lines. TAK-243 treatment demonstrated potent efficacy in human S3-null and S7-null SC-CRISPR, while mouse Nf2−/− SCs had greatly reduced sensitivity. Whereas ixazomib treatment revealed profound effects in all 3 SC lines (Fig. S1C, Table S1, Fig. S8A). For pevonedistat, human S3-null and S7-null SC-CRISPR cells exhibited relatively modest effects. (Table S1). Further, Annexin V/PI dual staining of SCs treated with ixazomib showed a 2-4-fold increase in apoptosis in human S3-null and S7-null as well as mouse Nf2−/− SCs. (Fig. S7). These data indicate ixazomib has superior effects in all 3 NF2-deficient SC lines compared to TAK-243 and pevonedistat, and we therefore proceeded with in vivo testing for ixazomib.
Based on in vitro results, we tested the efficacy of ixazomib in two schwannoma sciatic nerve models: (1) mouse Nf2−/− SCs and, (2) patient-derived MGH-VS4 vestibular schwannoma cells. Mouse Nf2−/− SCs or human MGH-VS4 cells were implanted in the sciatic nerve of mice and the tumor size was measured every 3 days. Mice were randomized into the treatment or control groups when the tumor reached 3 mm in diameter, and ixazomib or saline (control) was administered by oral gavage twice a week until experiment endpoints (Figure 5C). In both models, ixazomib treatment significantly delayed tumor growth (Figure 5D) without inducing significant systemic toxicity as evaluated by body weight loss (Fig. S8B). Taken together, our results establish the efficacy of ixazomib in in vitro and in vivo preclinical models of both meningiomas and schwannomas.
Discussion
Many of the drugs that have been tested in NF2 patients have shown cytostatic effects translating to tumor stabilization, but no consistent or durable shrinkage. Therefore, there is a clear need to identify drugs that show better efficacy. Our recent collaborative large-scale drug screening studies in preclinical models of NF2 led to the identification of novel single and combinatorial agents,10 with initiation of a clinical trial for an FDA-approved, multi-tyrosine kinase inhibitor, brigatinib (NCT04374305, www.ClinicalTrials.gov) for NF2. We took advantage of these data to identify agents that might have cytotoxic effects in NF2-deficient meningioma and schwannoma cell models and observed a subset of drugs targeting the UPP as potential candidates. Here, we investigated three distinct UPP inhibitors that are in clinical development or FDA-approved, including pevonedistat, ixazomib, and TAK-243, in NF2 preclinical models.
Of the three inhibitors, TAK-243 and ixazomib showed significant inhibition of proliferation in both human MN and SC cellular models. These two drugs also induced apoptosis in all the MN lines tested, which was not observed with other inhibitors such as rapamycin, AZD2014, INK-128, dasatinib, and brigatinib that we previously tested (our unpublished data). However, pevonedistat did not exhibit significant effects in inhibiting the proliferation of meningioma and schwannoma cells. This is consistent with an earlier study, which showed that while pevonedistat suppressed the E3 ubiquitin ligase CRL4DCAF1 and attenuated activation of YAP in NF2-mutant malignant mesothelioma models, it did not demonstrate significant preclinical activity, even when combined with chemotherapy in NF2-mutant in vitro and patient-derived xenograft models.39 We, therefore, did not include pevonedistat in further mechanistic and in vivo studies.
To further understand the mechanism of TAK-243 and ixazomib, we undertook transcriptome studies in MN lines treated with these drugs. Interestingly, both drugs significantly downregulated the expression of KIT and PDGFRA, which we and others reported to be upregulated in cells and tumors upon loss of NF2.10,24 Downregulation of c-KIT expression by ixazomib, and other inhibitors of the 20S subunit of the proteasome, was also observed in gastrointestinal stromal tumors.40 Further, our transcriptome analyses and subsequent validation also revealed the proto-oncogene SKP2, which we previously observed to be elevated in NF2-null cells (unpublished), is also downregulated by TAK-243 and ixazomib. More importantly, SKP2 has a pivotal role in regulating a plethora of survival pathways associated with cell cycle progression, cell growth, apoptosis, migration, and invasion.30,31 Our results here reveal accumulation of p21, p27, and cyclinD1 and cell cycle arrest in MN cells treated with TAK-243 or ixazomib, suggesting that these inhibitors act partly by downregulating SKP2 which can leads to cell cycle arrest, consistent with reports shown for other cell types.33,41 Interestingly, in T-cell and Hodgkin lymphoma, ixazomib induced cell death through the downregulation of CHK1 and MYC.42 However, in ixazomib treated MN cells, we did not observe altered expression of CHK1 or MYC genes, suggesting cell/tumor-dependent mechanisms controlling the drug effects.
Among the top 5% DEGs shared between TAK-243 and ixazomib, genes associated with ER stress and UPR were significantly upregulated in the two MN lines, and this was confirmed by immunoblotting. Our results are consistent with recent studies showing that TAK-243 or ixazomib is capable of inducing ER stress and UPR in glioblastoma models as well as other cancers.41,43–45 PERK, IRE1α, and ATF6 are three membrane sensors known to initiate the UPR signaling cascade.46 To gain further mechanistic insight, we pretreated MN cells with inhibitors for each of the three membrane bound sensor pathways, PERK/eIF2A, IRE1α/XBP1, and ATF6, known to initiate the UPR signaling cascade, followed by treatment with either TAK-243 or ixazomib. We observed that only the IRE1α inhibitor, 4µ8C, partially rescued the cytotoxic response of ixazomib, suggesting a potential role for the IRE1α/XBP1 axis of the UPR in triggering the apoptotic effects upon UPP inhibition. However, our results somewhat differ from those found in glioblastoma cells where both PERK and IREα were reported to play a role in TAK-243-induced cell death,33 again supporting a cell/tumor context-dependent process in meningioma.
Based on the efficacy of TAK-243 and ixazomib in NF2-related cell models, we tested the effects of these drugs in a xenograft model of malignant human WHO grade III meningioma and observed that both drugs were able to significantly suppress tumor growth, with ixazomib being more effective. Furthermore, ixazomib also showed efficacy in an allograft model of mouse schwannoma and a xenograft model of patient-derived schwannoma without any significant adverse effects. Taken together, our study demonstrates that UPP inhibition is effective in NF2-null cell and animal models and that the therapeutic potential of ixazomib is worth considering for NF2-associated tumors. Further, we believe that in NF2 patients, tumor formation is initiated by NF2 loss and many of the altered pathways downstream of NF2 loss could be shared among other meningiomas, which will explain the drug being effective in all meningiomas, regardless of the NF2 status. Ixazomib is an FDA-approved, oral second-generation proteasome inhibitor, which is extensively studied to treat multiple myeloma47 and other hematological malignancies.48 Although challenges remain for the use of ixazomib or other improved UPP inhibitors in solid cancers, our study highlights the therapeutic potential of proteasome inhibitors as an effective treatment strategy for NF2-associated and sporadic tumors.
Funding
This study was supported by National Institutes of Health (R01 NS113854 to V.R.), and Children’s Tumor Foundation Drug Discovery Initiative award (to V.R.), Department of Defense Investigator Initiated Research Award (W81XWH-20-1-0222 to L.X.), Clinical Research Award (W81XWH2210439 to L.X.), and American Cancer Society Mission Boost Award (MBGI-21-185-01-MBG to L.X.)
Acknowledgments
The authors thank Nicholas Redmond for technical help, and Takeda Development Center Americas, Inc. for kindly providing ixazomib, pevonedistat and TAK-243 for this study.
Conflict of interest statement
The authors declare that no conflicts of interest exist.
Authorship statement
Experimental design: S.B., J.L.O., R.L.B., L.X., L.S.C., V.R. Implementation: S.B., J.L.O., R.L.B., Z.Y., P.K., A.D.W., J.T.J., L.S.C. Analysis or interpretation of data: S.B., J.L.O., R.L.B., Z.Y., S.E., M.F., J.T.J., S.R.P., L.X., L.S.C., V.R. Manuscript writing/review: all authors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}