





Abstract
Pediatric high-grade glioma (pHGG) is largely incurable and accounts for most brain tumor-related deaths in children. Radiation is a standard therapy, yet the benefit from this treatment modality is transient, and most children succumb to disease within 2 years. Recent large-scale genomic studies suggest that pHGG has alterations in DNA damage response (DDR) pathways that induce resistance to DNA damaging agents. The aim of this study was to evaluate the therapeutic potential and molecular consequences of combining radiation with selective DDR inhibition in pHGG.
We conducted an unbiased screen in pHGG cells that combined radiation with clinical candidates targeting the DDR and identified the ATM inhibitor AZD1390. Subsequently, we profiled AZD1390 + radiation in an extensive panel of early passage pHGG cell lines, mechanistically characterized response to the combination in vitro in sensitive and resistant cells and evaluated the combination in vivo using TP53 wild-type and TP53 mutant orthotopic xenografts.
AZD1390 significantly potentiated radiation across molecular subgroups of pHGG by increasing mutagenic nonhomologous end joining and augmenting genomic instability. In contrast to previous reports, ATM inhibition significantly improved the efficacy of radiation in both TP53 wild-type and TP53 mutant isogenic cell lines and distinct orthotopic xenograft models. Furthermore, we identified a novel mechanism of resistance to AZD1390 + radiation that was marked by an attenuated ATM pathway response which dampened sensitivity to ATM inhibition and induced synthetic lethality with ATR inhibition.
Our study supports the clinical evaluation of AZD1390 in combination with radiation in pediatric patients with HGG.
AZD1390 potentiated radiation in pHGG cells representing distinct molecular subgroups.
AZD1390 + radiation increased survival in TP53 wild-type and TP53 mutant xenografts.
ATR inhibition was synthetically lethal to pHGG cells resistant to the combination.
Using a multi-modal screening approach involving a unique collection of molecularly annotated, early passage pHGG models, we identified the ATM inhibitor AZD1390 as an exceptional radio-sensitizing agent. While AZD1390 is currently being evaluated in adult brain tumors, there are significant differences between adult HGG and pHGG tumors with respect to oncogenic drivers and response to DNA damaging therapy that motivate a comprehensive evaluation of the drug in pHGG models. Notably, we showed for the first time that AZD1390 + radiation—using clinically relevant doses and schedules—significantly prolonged survival in vivo in both TP53 wild-type and TP53 mutant orthotopic xenograft models of pHGG. We also identified a novel mechanism of resistance to AZD1390 + radiation that was characterized by synthetic lethality with ATR inhibition. Our findings provide a strong rationale for the clinical evaluation of AZD1390 + radiation in pHGG and identify potential biomarkers that may govern the efficacy of this novel therapeutic approach.
Pediatric high-grade glioma (pHGG) is among the most aggressive malignant brain tumors in children, with few long-term survivors.1 Radiation remains a mainstay of treatment for pHGG, yet palliation of symptoms is temporary and the impact on overall survival is limited. Minimal progress has been made toward improving the prognosis of this disease, despite numerous clinical trials exploring alternative radiotherapy approaches2 and the addition of conventional chemotherapeutics and/or molecularly targeted therapy to irradiation.3,4
A dysregulated DNA damage response (DDR) has emerged as a common driver of resistance to conventional DNA damaging therapies in both adult HGG and pHGG,5–7 and has spurred the clinical development of several central nervous system (CNS)-penetrant drugs targeting the DDR in adults (eg, NCT03423628 and NCT04555577). Crucially, the strategies used to enhance radiation therapy for adult gliomas, most conspicuously using concomitant and adjuvant temozolomide,8 have failed to improve outcomes for children with HGG.9,10 This observation no doubt reflects critical biologic differences between adult HGG and pHGG. It is upon this background that we evaluated the therapeutic potential and molecular consequences of selective DDR inhibition in pHGG by leveraging a unique, recently reported panel of early-passage, patient-derived models that recapitulate the heterogeneity of the disease and vary by histone H3 mutation status and associated co-occurring mutations.11
Materials and Methods
Cell Culture
The etiology, source, and cell culture conditions for the cell lines used in this study have been reported previously.11 Briefly, cells were plated in Corning® 3471 Ultra-low Attachment plates in media used for neural stem cells and glial progenitor cells consisting of a 1:1 mixture of Neurobasal™ without phenol red (ThermoFisher, 12348017) supplemented with 2% of B27 without vitamin A (ThermoFisher, 12587010) and 1% of N2 (ThermoFisher, 17502048); and Knock-Out DMEM/F12 (ThermoFisher, 12660012) supplemented with 2% of Stempro® neural supplement (ThermoFisher, A1050801), 20 ng/mL of human recombinant EGF (PeproTech, AF-100-15), 20 ng/mL of human recombinant bFGF (PeproTech, 100-18B), 10 ng/mL of human recombinant PDGF-AA (Cell Guidance Systems, GFH16AF-100) and PDGF-BB (Cell Guidance Systems, GFH18AF-100), 1% of Glutamax (ThermoFisher, 35050061), 1% of sodium pyruvate, 1% of NEAA, 10 mM of HEPES, 2 μg/mL of heparin, and 1× Primocin (InvivoGen, ant-pm-1). The cell lines were maintained on human ESC-qualified Geltrex (ThermoFisher, A1413302) artificial extracellular matrix-coated tissue culture surface at 37 °C, 5% CO2, and 5% of O2. Cell identity was verified using short tandem repeat fingerprinting, and cells in culture were routinely checked for mycoplasma contamination.
In vitro Assays and Genetic Engineering
For Western blot analysis, images were acquired and quantified using an Odyssey CLx (LI-COR Biosciences). RNA was extracted with QIAshredder (Qiagen, 79656) and a RNeasy Mini Kit (Qiagen, 74106), and quantified using the TaqMan Fast Advanced Master Mix (ThermoFisher, 4444554). Antibodies and primers are listed in Supplementary Table S6A. Cell viability was assessed using CellTiter-Glo® (Promega, G7570); cell death was monitored using Sytox® Green (ThermoFisher, S7020) or annexin V-APC/DAPI staining with flow cytometry; senescence assays were performed using Cell Signaling Technology (9860) or Abcam (ab228562) kits. CFAs were quantified using GelCount (Oxford Optronix). Radiation was administered using orthovoltage x-rays (300 kVp) at a dose rate of 0.445 cGy/MU by using a Gulmay D3300 (Gulmay Medical). The alkaline comet assay was performed using CometAssay® (Trevigen) and analyzed with the Lionheart™ FX Automated Microscope (BioTek) using TriTek CometScore 2.0.0.38. Gene editing using CRISPR-Cas9 was performed by the Center for Advanced Genome Engineering at St. Jude (see Supplementary Table S6B for gRNA sequences). Phosphoproteomics and proteomics were performed as described previously12 by the Center for Proteomics and Metabolomics at St. Jude.
Patient-Derived OX Studies
SJ-DIPG7 and SJ-DIPG37 OXs were established as previously reported.11 Mice were purchased from Charles River Laboratories and were maintained in the Animal Resource Center at St. Jude. All animal studies were conducted according to protocols approved by the St. Jude Institutional Animal Care and Use Committee and in accordance with the NIH guidelines for the care and use of laboratory animals. For in vivo survival analysis, 28 animals with cranial implants were randomized to the control and treatment groups and were treated for 7 days. The median tumor bioluminescence signals at enrollment were 6.03 × 106 photon/s and 1.57 × 106 photon/s, respectively, for the SJ-DIPG7 and SJ-DIPG37 survival studies, and treatments were started 4 days after enrollment (postimplantation day 17 for DIPG7 and day 25 for DIPG37). Tumor growth was tracked weekly by BLI using a PerkinElmer IVIS. Treatment failure was monitored by using BLI to reveal patterns of tumor progression along the entire neuroaxis.
Radiation delivery.
A small animal radiation research platform (SARRP) was used to deliver image-guided, arc-based (−90° to 90°) fractionated radiation to encompass the whole brain.13 The SARRP incorporates computerized tomography (CT) imaging and rotating X-ray source with adjustable collimator to deliver conformal radiation dose, minimizing exposure to nontargeted tissues and organs. High resolution, low imaging dose, on board CT imaging and 3D reconstruction, and MuriPlan software simplifies targeted dose delivery and radiation treatment planning.
Statistical Analysis
The number of independent biological replicates is indicated in parentheses in figure legends. Comparisons between 2 groups were performed using the paired or unpaired Student’s t-test unless otherwise indicated. Comparisons between >2 groups were performed using one-way or two-way ANOVA followed by Tukey’s or Sidak’s multiple comparisons test, respectively. For in vivo experiments, mice were randomly assigned to each experimental group by a biostatistician. Survival data were analyzed using the Mantel-Cox log-rank test, by first running a global log-rank test on whether the survival curves were different from each other, and then comparing individual treatment arms posthoc using the log-rank test. P values from the t-test, multiple comparison test, and log-rank test are reported as follows: ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. For ANOVA analysis, the F-statistic and P value for main effects are reported in the manuscript, and the P value is annotated in figures as follows: ns, not significant, #P < 0.05, ##P < 0.01, ###P < 0.001, and ####P < 0.0001. All statistical analyses were performed with Prism (GraphPad v. 9).
Data and Materials Availability
Proteomics and phosphoproteomics data have been deposited with the ProteomeXchange Consortium via the Proteomics Identifications Database (PRIDE) under PXD028285 (30395289 and 31686107). Next-generation sequencing data have been deposited in the European Genome-Phenome Archive (EGA) under accession numbers EGAS00001005159 (whole-genome), EGAS00001005160 (whole-exome), and EGAS00001005161 (RNA sequencing).
Results
Unbiased Drug Screening in DIPG Cells Identified AZD1390 as an Exceptional Radiosensitizer
To identify radiosensitizers of pHGG, we first treated SU-DIPGXIII cells with 24 CNS-penetrant FDA-approved drugs or clinical candidates targeting the DDR, evaluated at 10 concentrations in combination with radiation at 0–8 Gy (Figure 1A–B). Cell viability was measured 7 days after treatment with the CellTiter-Glo (CTG) assay, and combination experiments were analyzed using the BRAID response surface model,14 which identifies synergy when κ > 0, additivity when κ = 0, and antagonism when κ < 0. The ATM inhibitor (ATMi) AZD1390 emerged as the compound most synergistic with radiation in our panel and showed little cytotoxicity on its own. We confirmed the strong radiosensitizing activity of AZD1390 in this cell line by using the colony-forming assay (CFA) (Figure 1C). The ratio of the mean inactivation dose (Dbar), a well-validated measure of intrinsic radiosensitivity, with the drug to that with vehicle alone (plotted in red) was 2.08 at 4 nM drug and 3.37 at 100 nM. By comparison, 3 drugs (AZD1775, temozolomide, CUDC907) promoted as radiosensitizing agents for pHGG15–17 demonstrated weaker potentiation of radiation when tested at noncytotoxic drug concentrations (Figure 1D–E; Supplementary Figure S1A).
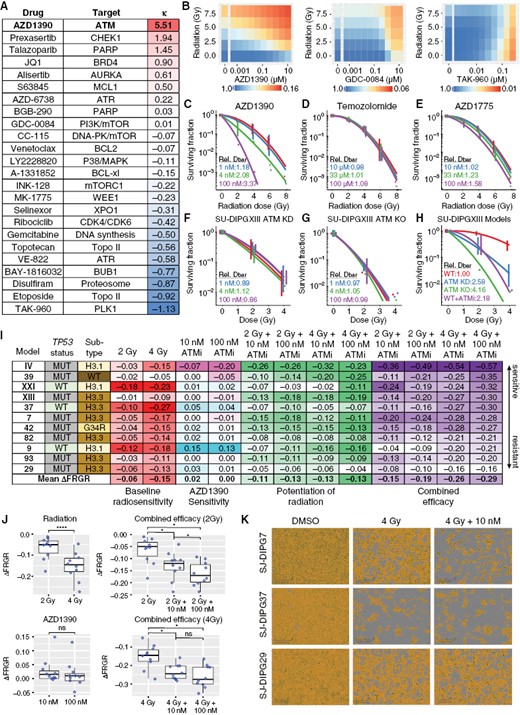
AZD1390 radiosensitized cells representing distinct molecular subgroups of pHGG. (A) BRAID response surface model analysis of 24 CNS-penetrant FDA-approved drugs or clinical candidates targeting the DDR in combination with radiation in SU-DIPGXIII cells (n = 3). (B) Exemplar BRAID response surface models from (A). (C-E) Linear quadratic (LQ) fits of colony-forming assays (CFAs) testing the combination of radiation with AZD1390 (C) or with 2 drugs (D-E) currently used or in clinical development for pHGG (n = 3). Dbar, the mean inactivation dose, was averaged over biological replicates and divided by the Dbar for the vehicle (red). (F-G) LQ fits from CFA experiments in SU-DIPGXIII ATM kinase dead (KD) or knockout (KO) cells treated with increasing doses of AZD1390 (n = 3). No further radiosensitization was observed in ATM KD and KO cells after treatment with up to 100 nM AZD1390, confirming that the drug potentiates radiation solely through ATM inhibition within this concentration range. (H) Overlay of the LQ fits from CFA experiments in SU-DIPGXIII WT cells (± 100 nM AZD1390), SU-DIPGXIII ATM KD cells, or SU-DIPGXIII ATM KO cells (n = 3). Wild-type SU-DIPGXIII cells treated with 100 nM AZD1390 showed radiation sensitivity comparable to that of ATM KD cells (Dbar relative to vehicle = 2.18 vs. 2.58, respectively) but less than that of ATM KO cells (Dbar relative to vehicle = 4.16). (I) Summary of the average change in FRGR calculated from the confluence assays of 11 pHGG cell lines for 14 days (n ≥ 3). (J) Statistical analysis of the distributions of ΔFRGR values reported in (I). Data are presented as Tukey box-and-whisker plots and were analyzed by paired Student’s t-test (when comparing 2 groups) or one-way within-subjects ANOVA modeling the effect of drug treatment on ΔFRGR (when comparing >2 groups). (K) Representative microscopy images from the confluence assay of SJ-DIPG7, SJ-DIPGXIII, and SJ-DIPG37 cells. Scale bar: 300 µm.
To verify the target specificity of AZD1390, we established an ATM knockout (KO) cell line and a kinase-dead (KD, ATMD2870A/N2875K) cell line derived from SU-DIPGXIII cells by using the CRISPR/Cas9 system. No further radiosensitization was observed in ATM KD and KO cells after treatment with up to 100 nM AZD1390 (Figure 1F–G), confirming that the drug potentiates radiation solely through ATM inhibition within this concentration range. Wild-type SU-DIPGXIII cells treated with 100 nM AZD1390 showed radiation sensitivity comparable to that of ATM KD cells (Dbar relative to vehicle = 2.18 vs. 2.58, respectively) but less than that of ATM KO cells (Dbar relative to vehicle = 4.16) (Figure 1H).
AZD1390 Radiosensitized pHGG Cells Representing Distinct Molecular Subgroups
To determine the extent to which AZD1390 potentiated radiation in pHGG, we expanded our studies to include 10 additional cell lines representing the 4 primary histone H3 subgroups (H3 wild-type, H3.3K27M, H3.1K27M, and H3.3G34R) and harboring common associated recurrent mutations (Supplementary Table S1).11 The CFA confirmed strong radio-sensitization by AZD1390 in SJ-DIPG7 cells (Supplementary Figure S1B). As the other pHGG cell lines in the panel failed to form colonies, we developed a long-term confluence-based assay to assess radio-sensitization and combined efficacy in vitro and analyzed these experiments using the fitted relative growth rate (FRGR), a robust measure of the average growth rate observed in the confluence assay that correlates well with the CFA (Supplementary Figure S1C–E). The change in FRGR (ΔFRGR) is negative for treatments that reduce cell growth.
ΔFRGR after treatment with single-agent AZD1390 at 10 nM and 100 nM, with radiation alone at 2 Gy and 4 Gy, and with the 4 combinations of these 2 agents were quantified in 11 pHGG cell lines by using the confluence assay (Figure 1I; Supplementary Table S2). Cell lines were rank ordered according to combined efficacy at 4 Gy + 100 nM AZD1390 and were annotated by TP53 status and histone subtype, as previous studies suggested that these 2 genetic features affected the cellular response to radiation in pHGG.18,19 As expected, the average ΔFRGR for single-agent radiation became increasingly negative as the radiation dose increased (P < 0.0001) (Figure 1J). When single-agent AZD1390 responses were examined, the average ΔFRGR across all cell lines was close to zero and there was no significant difference in ΔFRGR between 10 nM and 100 nM drugs. To evaluate the combination of radiation and AZD1390, we quantified both the overall combined efficacy (ΔFRGReff), defined as the change in FRGR relative to that with no treatment and the potentiation of radiation (ΔFRGRpot), defined as the change in FRGR induced by the drug at a specified dose of radiation after accounting for the effect of the drug alone. The addition of 10 nM or 100 nM AZD1390 to 2 Gy resulted in a significant decrease in average ΔFRGR (P = 0.040 and P = 0.022, respectively), and a significant decrease when combined with 4 Gy (P = 0.022 and P = 0.012, respectively).
Notably, AZD1390 consistently potentiated radiation in 2 models of cortical pHGG (H3 wild-type SJ-HGG39 and H3.3G34R SJ-HGG42). In contrast, AZD1390 failed to substantially potentiate radiation at any drug concentration or radiation dose in TP53mut SJ-DIPG29 cells. Representative images from these confluence assay experiments enable the responses in TP53mut SJ-DIPG7 and TP53wt SJ-DIPG37 to be compared to those in SJ-DIPG29 (Figure 1K).
AZD1390 + Radiation Increases Genomic Instability and Decreases Viability in Sensitive pHGG Models
We confirmed the in vitro pharmacodynamics (PD) of AZD1390 in SJ-DIPG7 and SU-DIPGXIII cells, 2 models with sensitivity to combination therapy, using Western blot and a cell-based reporter system that simultaneously quantifies the extent of homologous recombination (HR) repair and mutagenic NHEJ (mNHEJ) repair20 (Supplementary Figure S2A–B). Irradiation alone increased the percentage of γH2A.X-intense cells after 1 h, but co-treatment with AZD1390 prevented H2A.X phosphorylation immediately after irradiation (main effect F1,16 = 134.6; P < 0.0001 in SJ-DIPG7 cells; main effect F1,16 = 53.40; P < 0.0001 in SU-DIPGXIII cells) (Figure 2A). Yet, the combination increased γH2A.X signal intensity after 6 days (main effect F1,16 = 6.543; P = 0.021 in SJ-DIPG7 cells; main effect F1,16 = 14.69; P = 0.002 in SU-DIPGXIII cells) (Figure 2A; Supplementary Figure S2C). These results suggest that AZD1390 exacerbated DNA damage in the long term by inhibiting the DDR and impeding repair. Consistent with this hypothesis, the Olive tail moment,21 a measure of physical DNA damage assessed using the alkaline comet assay, was unchanged with the drug alone, increased with radiation, and was further potentiated by AZD1390 (P = 0.003 in SJ-DIPG7 cells; P < 0.0001 in SU-DIPGXIII cells) (Figure 2B–C). Persistent genotoxic stress also induces the formation of micronuclei.22 AZD1390 significantly increased the average number of micronuclei per cell 6 days after exposure to radiation in SJ-DIPG7 cells (main effect F1,16 = 34.36; P < 0.0001) and trended toward statistical significance in SU-DIPGXIII (main effect F1,16 = 4.322; P = 0.054) (Figure 2D–E).
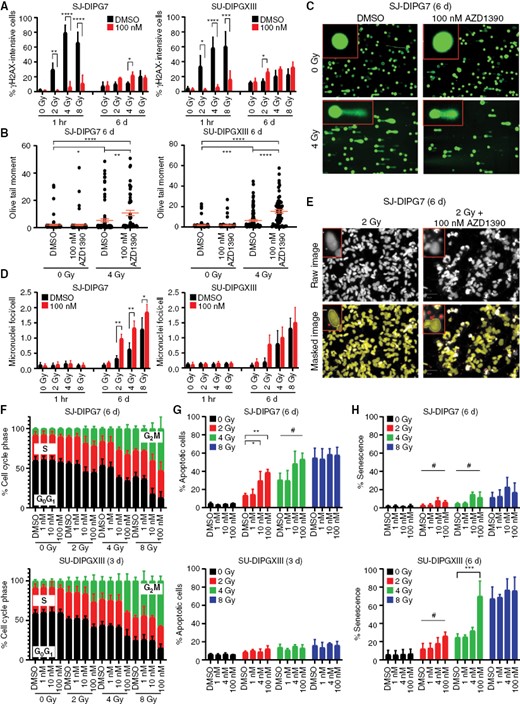
AZD1390 increased radiation-induced genomic instability, cell cycle arrest, and apoptosis or senescence. (A) Percentage of γH2A.X-intense SJ-DIPG7 or SU-DIPGXIII cells 1 h and 6 days after treatment with 100 nM AZD1390 + radiation (n = 3). Data are presented as the mean ± SD. Two-way ANOVA was performed to analyze the effect of radiation dose and drug treatment on the percentage of γH2A.X-intense cells. (B) Olive tail moment from comet assay of SJ-DIPG7 or SU-DIPGXIII cells 6 days after treatment with 100 nM AZD1390 + 4 Gy (n = 3, ≥50 cells/replicate). Data are presented as the mean ± SEM. One-way ANOVA was performed to analyze the effect of treatment (radiation + drug) on the Olive tail moment. (C) Exemplar comet assay images of SU-DIPG7 cells from (B). (D) Quantification of micronuclei in SJ-DIPG7 and SU-DIPGXIII cells after treatment with radiation and AZD1390 (n = 3). Data are presented as the mean ± SD. Two-way ANOVA was performed to analyze the effect of radiation dose and drug treatment on micronuclei foci count. (E) Illustration of the quantification of micronuclei in SJ-DIPG7 cells after treatment with radiation and AZD1390. Microcopy images were masked to isolate the nuclei (yellow) from the micronuclei (red) present within each cell. Scale bar: 50 µm. (F-H) Cell cycle distribution (F), percentage of apoptotic cells (G), and percentage of senescent cells (H) as assessed by assays in SJ-DIPG7 or SU-DIPGXIII cells after treatment with radiation and AZD1390 (n ≥ 2). Data are presented as the mean ± SD. One-way ANOVA was performed at each radiation dose to analyze the effect of drug treatment on cell fate outcome.
Cell cycle progression and apoptosis were assessed using flow cytometry and annexin V/DAPI staining. AZD1390 enhanced radiation-induced G2/M accumulation in both SJ-DIPG7 cells and SU-DIPGXIII cells (Figure 2F). Interestingly, radiation-induced apoptotic cell death in SJ-DIPG7 cells, and this effect was enhanced by AZD1390 at 2 Gy (main effect F3,8 = 12.43; P = 0.002) and 4 Gy (main effect F3,8 = 4.407; P = 0.042) (Figure 2G). In contrast, SU-DIPXIII cells showed little apoptosis with radiation or combination therapy. Conversely, senescence was the primary outcome in SU-DIPXIII cells exposed to radiation, and this response was significantly potentiated by 100 nM AZD1390 at 4 Gy (P = 0.001) (Figure 2H; Supplementary Figure S2D). Taken together, these studies suggest that in sensitive models, AZD1390 potentiates the mechanism by which radiation adversely affects cell viability, driving apoptosis in SJ-DIPG7 cells and senescence in SU-DIPGXIII cells.
In Vitro, TP53 Alteration Did Not Change the Combined Efficacy of AZD1390 + Radiation
A reanalysis of the confluence assay of 11 pHGG models reported in Figure 1I based on TP53 status highlights the opposing behavior of ATMi in TP53wt cells (Figure 3A), in which intact p53 pathway signaling was confirmed by measuring sensitivity to MDM2 inhibition and p21 expression at baseline and after treatment with radiation (Supplementary Figure S3A-B). DNA damage induced by radiation activates the p53 pathway through ATM and commits TP53wt cells to cell cycle arrest and/or apoptosis, while ATM inhibition attenuates p53 pathway activation by reducing the stability of p53. In contrast, inhibiting ATM also impairs the DDR and induces genotoxicity in the long term. Consistent with this mechanism, we observed that pHGG TP53wt cells were significantly more radiosensitive than were TP53mut cells when treated with radiation alone at doses of 2 Gy (P = 0.0009) and 4 Gy (P = 0.0005). When treated with drug alone, TP53wt status had a significant positive effect on ΔFRGR (main effect F1,18 = 8.780; P = 0.008), consistent with ATM inhibition promoting growth in these cells. AZD1390 potentiated the effect of radiation (ΔFRGRpot) in both groups, but did so better in TP53mut cells (main effect F1,36 = 5.857; P = 0.021). Yet, mutation status did not have a significant effect on combined efficacy (ΔFRGReff), which became increasingly negative in both groups, indicating greater overall efficacy with increasing drug and radiation doses.
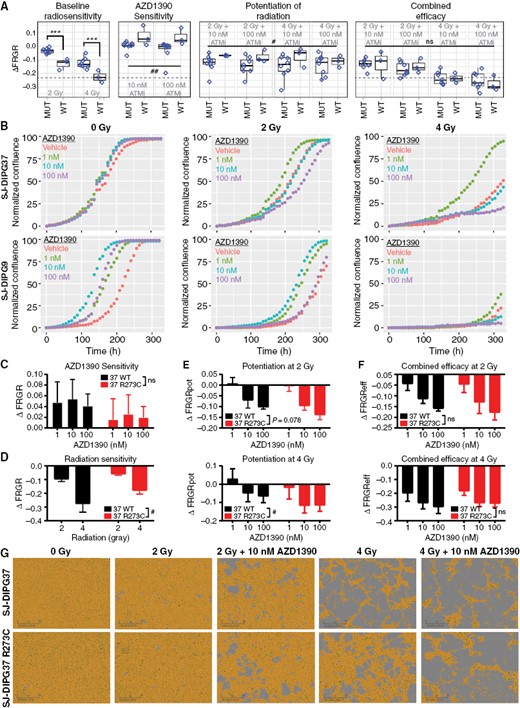
In vitro, TP53 alteration did not change the combined efficacy of AZD1390 + radiation. (A) Statistical analysis of the distributions of ΔFRGR values reported in Figure 1I as a function of TP53 status. Data are presented as Tukey box-and-whisker plots and were analyzed by two-way ANOVA modeling the effect of treatment (radiation, drug, or radiation + drug) and TP53 status on ΔFRGR. The dotted line equals the median ΔFRGR of TP53wt pHGG cell lines treated with 4 Gy of radiation. (B) Representative confluence assay curves for SJ-DIPG37 and SJ-DIPG9 cells, both TP53wt, treated with radiation and AZD1390 (n = 3). (C) ΔFRGR calculated from the confluence assay of SJ-DIPG37 TP53wt cells and isogenic SJ-DIPG37 TP53 R273C mutant cells treated with AZD1390 alone (n = 3). Data are presented as the mean ± SD. Two-way ANOVA was performed to analyze the effect of drug treatment and TP53 status on ΔFRGR. (D) ΔFRGR calculated from the confluence assay of SJ-DIPG37 TP53wt cells and SJ-DIPG37 TP53 R273C mutant cells treated with radiation alone (n = 3). Data are presented as the mean ± SD. Two-way ANOVA was performed to analyze the effect of radiation and TP53 status on ΔFRGR. (E) Potentiation of radiation, ΔFRGRpot, calculated from the confluence assay of SJ-DIPG37 TP53wt cells and SJ-DIPG37 TP53 R273C mutant cells treated with radiation and AZD1390 (n = 3). Data are presented as the mean ± SD. Two-way ANOVA was performed at each radiation dose to analyze the effect of drug and TP53 status on ΔFRGRpot. (F) Reanalysis of the experiment in (E) using combined efficacy, ΔFRGReff, as the dependent variable. (G) Representative microscopy images from week 2 of the confluence assay from (C-F) of SJ-DIPG37 TP53wt and SJ-DIPG37 TP53 R273C mutant cells. Scale bar: 300 µm.
To further explore this phenomenon, we extended our live-cell microscopy studies to investigate a lower dose of AZD1390 (1 nM) in SJ-DIPG9 and SJ-DIPG37, 2 TP53wt pHGG cell lines that showed substantial augmented growth upon treatment with single-agent ATMi (Figures 3B, 1I). In the absence of radiation, AZD1390 at concentrations from 1 nM to 100 nM increased growth relative to that with vehicle control (red) in both cell lines. However, this proliferative effect diminished at higher drug concentrations with increasing radiation dose, such that at 4 Gy, only the 1 nM condition (green) resulted in enhanced growth relative to that with vehicle control, whereas treatment with ≥10 nM AZD1390 reduced cell growth relative to that with vehicle control. These data demonstrate how the “protective” effect of AZD1390 in TP53wt cells diminished as a function of increasing drug concentration and/or increasing radiation dose.
Next, after confirming the expected PD effect of AZD1390 + radiation in SJ-DIPG37 cells (Supplementary Figure S3C), we engineered a TP53 R273C mutation into those cells by using the CRISPR/Cas9 system and compared the parental and mutant cell models using the confluence assay. When treated with drug alone, the average ΔFRGR was lower in TP53mut SJ-DIPG37 cells at every concentration tested, although the result did not reach statistical significance. However, TP53mut SJ-DIPG37 cells were less sensitive to radiation alone (main effect F1,8 = 10.87; P = 0.011). When treated with both radiation and drug, TP53mut SJ-DIPG37 cells trended towards higher potentiation at 2 Gy (main effect F1,12 = 3.720; P = 0.078), and were significantly more potentiated at 4 Gy (main effect F1,12 = 6.181; P = 0.029). Consistent with the results from our panel of pHGG models, the combined efficacy of AZD1390 + radiation was comparable in isogenic TP53wt and TP53mut SJ-DIPG37 cells. Exemplar images from the confluence assay illustrate the higher baseline radiation sensitivity of SJ-DIPG37 TP53wt cells, especially with 4 Gy (Figure 3C). Although the TP53 R273C mutation reduced cellular sensitivity to radiation, it also radiosensitized the cells to a greater extent in the presence of ATMi; therefore, the net effects of the combination in TP53wt and TP53mut cells were negative and similar in magnitude. We observed similar behavior in TP53wt and TP53mut SJ-DIPG9 cells (Supplementary Figure S3D–E), although in this case we were unable to isolate single clones containing the TP53 mutation and instead screened a pool of SJ-DIPG9 cells bearing >90% of the desired mutation. Collectively, our data indicate that AZD1390 is an effective radiosensitizer for both TP53wt and TP53mut pHGG cell lines and that TP53 status alone does not correlate with response to this combination therapy.
AZD1390 + Radiation Prolonged Survival in Both TP53wt and TP53mut Orthotopic Xenografts
To test the efficacy of AZD1390 + radiation in vivo, we orthotopically implanted TP53mut SJ-DIPG7 cells or TP53wt SJ-DIPG37 cells in the brain cortices of female CD-1 Nude mice. We first confirmed that oral administration of AZD1390 1 h before a single fraction of 2.5 Gy substantially reduced the phosphorylation of both ATM (Ser1981) and RAD50 (Ser635) in the tumors of mice bearing SJ-DIPG7 orthotopic xenografts (OXs) (Figure 4A). To assess the tolerability and efficacy of AZD1390 in combination with radiation, we administered vehicle control, radiation, AZD1390, or AZD1390 + radiation to mice harboring SJ-DIPG7 or SJ-DIPG37 OXs. Based on clinical radiation regimens for HGG and our in vitro studies demonstrating that AZD1390 was better at potentiating low-dose radiation, we administered 2 Gy/fraction over consecutive days. In previous work, we demonstrated that a cumulative dose of 10–20 Gy (2 Gy/fraction/day) was well tolerated and resulted in transient tumor growth inhibition with significant improvement in the overall survival of mice with SJ-DIPG7 or SJ-DIPG37 OXs.23 Consequently, we decided to treat mice for 7 days to achieve a cumulative dose of 14 Gy: radiation was delivered in daily fractions of 2 Gy for 5 days/week for 7 days, and AZD1390 was administered via oral gavage 1 h before radiation for 7 days (Figure 4B).
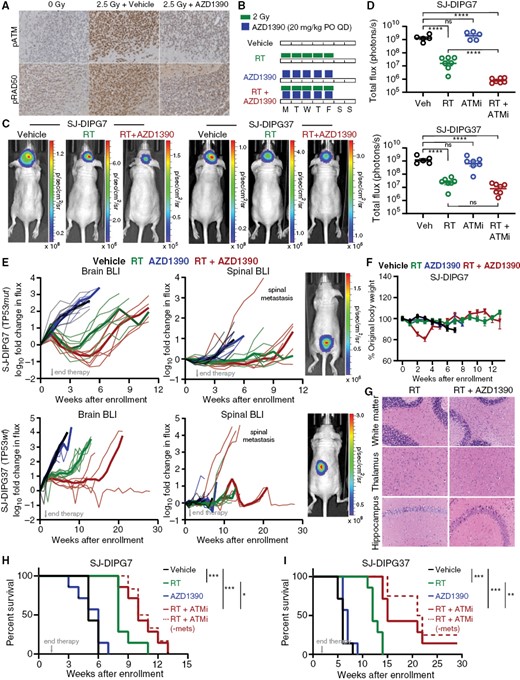
AZD1390 + radiation prolonged survival in both TP53wt and TP53mut OXs. (A) Immunohistochemical staining for p-ATM and pRAD50 in mice bearing SJ-DIPG7 OXs treated as indicated. (B) Dose and schedule for in vivo efficacy studies. (C) Representative bioluminescence images of mice bearing SJ-DIPG7 or SJ-DIPG37 OXs 3 weeks after treatment completion. (D) BLI intensity in SJ-DIPG7 OXs or SJ-DIPG37 OXs 3 weeks after treatment completion. Data are presented as the mean ± SEM. One-way ANOVA was performed to analyze the effect of treatment on total flux. (E) In vivo growth of SJ-DIPG7 OXs (top) and SJ-DIPG37 OXs (bottom) in the brain (left) after enrollment. BLI intensities at each timepoint were normalized to the BLI intensity at enrollment for each animal. Pre-euthanasia bioluminescence images of spinal metastases are shown at right. (F) Body weight as a percentage of weight at enrollment for SJ-DIPG7 study animals. Data are presented as the mean ± SEM. (G) H&E staining of the brain regions of mice after treatment with radiation and/or AZD1390. (H-I). Survival analysis of mice harboring SJ-DIPG7 OXs (H) or SJ-DIPG37 OXs (I) treated as indicated (n = 7 per treatment arm). Data were analyzed using the log-rank test. The dotted line represents survival after excluding mice euthanized because of hind-quarter paralysis.
In both the SJ-DIPG7 and SJ-DIPG37 OX studies, tumor burden as assessed by bioluminescence imaging (BLI) 3 weeks after the end of therapy showed a significant reduction in tumor growth in the radiation alone arms (P < 0.0001 for both models), but no significant difference in the AZD1390 alone arms (Figure 4C–E; Supplementary Table S3). However, AZD1390 + radiation reduced tumor burden relative to radiation alone in SJ-DIPG7 OXs (P < 0.0001), but not in SJ-DIPG37 OXs. At its nadir approximately 3 to 6 weeks post-treatment, the median brain tumor BLI signal from SJ-DIPG7 OXs was 25 times (IQR, 15–38 times) lower in the combination arm compared to the radiation alone arm and was lower than the median signal at enrollment. In contrast, AZD1390 + radiation was more cytostatic in SJ-DIPG37 OXs and we did not observe tumor shrinkage below the level at enrollment. However, combination therapy yielded a more sustained response and delayed tumor recurrence in SJ-DIPG37 OXs compared to SJ-DIPG7 OXs (Figure 4E). These in vivo findings are consistent with the tendency of SJ-DIPG7 cells to undergo cell death and the tendency of SJ-DIPG37 cells to become senescent after exposure to radiation (Figure S4A). One animal in the SJ-DIPG7 study and 3 mice in the SJ-DIPG37 study, all from the AZD1390 + radiation arm, were euthanized because of hind-quarter paralysis before they exhibited symptoms associated with brain tumor. BLI revealed signs of tumor metastasis to the spinal cord in these animals (Figure 4E). Despite the increase in BLI signal in the spinal cord, the tumor burden in the brains of these mice was comparable to that in the brains of the other animals in the treatment group, suggesting some degree of local tumor control (Supplementary Table S3).
We also evaluated the study animals for signs of toxicity and observed weight loss and lethargy in animals in the combination arm during the second week of treatment (Figure 4F; Supplementary Figure S4B). However, these animals regained their body weight by day 10 after the end of therapy. Assessment of hematologic toxicity in study animals revealed transient leukopenia and lymphocytopenia in both treatment arms with radiation, but these blood cell counts recovered within 4 weeks after treatment, consistent with the clinical observations of patients undergoing radiation treatment (Supplementary Figure S4C). No change in other blood cell counts or hemoglobin concentration was noted in the study animals. To understand further the pathology underlying the observed weight loss and lethargy, we performed whole-body necropsies of mice harboring SJ-DIPG7 OXs treated for 10 days with vehicle or with AZD1390 + radiation (cumulative dose, 20 Gy). Histopathologic examination revealed moderate to marked stomatitis, glossitis, and esophageal re-epithelialization in animals treated with the combination (Supplementary Figure S4D). Critically, detailed evaluation of major brain regions showed normal tissue morphology and no noticeable signs of neurotoxicity were detected in these animals (Figure 4G).
Strikingly, adding AZD1390 to radiation significantly improved overall survival of mice in both studies. In the SJ-DIPG7 study, mice that received AZD1390 + radiation showed a significant improvement in overall survival compared to mice treated with radiation alone (P = 0.042), and median survival increased from 8 weeks to 10 weeks (Figure 4H). In the SJ-DIPG37 study, treatment with the combination also significantly increased overall survival compared with radiation alone (P = 0.001), and median survival increased from 12 weeks to 15 weeks with one mouse in this arm surviving till the end of study with no evidence of tumor progression (Figure 4I).
SJ-DIPG29 Showed Attenuated ATM Pathway Activation and Concomitant Synthetic Lethality With ATR Inhibition
Pharmacodynamic assessment of SJ-DIPG29, the pHGG model in our panel most resistant to treatment with AZD1390 + radiation (Figure 1I), indicated that these cells showed substantially lower levels of total ATM and total CHK2 when compared with the 2 exemplar sensitive cell lines (Supplementary Figure S5A). However, the change in the ratio of phosphorylated to total ATM and CHK2 was not statistically different among the 3 cell lines, and treatment with 1–100 nM AZD1390 decreased the levels of p-ATM, p-KAP1, and p-CHK2 in SJ-DIPG29 cells after activation by 2 Gy, indicating that the residual ATM and CHK2 in SJ-DIPG29 remained functional (Supplementary Figure S5B–C). Remarkably, ATMi failed to potentiate radiation-induced cytotoxicity (Supplementary Figure S5D–H). To better understand how SJ-DIPG29 responded to radiation despite an attenuated ATM pathway response, we quantified the change in the proteome and phosphoproteome in SJ-DIPG29 and sensitive SJ-DIPG7 and SU-DIPGXIII cells 2 h after exposure to 4 Gy (Figure S6A–B; Supplementary Table S4). Ingenuity Pathway Analysis24 of phosphoproteome changes identified ATM signaling as one of the most differentially phosphorylated pathways in both groups, although the magnitude of the response in SJ-DIPG29 was significantly smaller (main effect F1,22 = 88.00; P < 0.0001) (Figure 5A; Supplementary Table S5). Kinase Substrate Enrichment Analysis (KSEA)25 found enrichment of a common set of kinase targets in both groups: activation of ATM, CHK2, and ATR and deactivation of CDK1 and CDK2 (Figure 5B). However, CDK3, MAPK9 (JNK2), PBK, VRK1, and MAPK15 (ERK7) were selectively repressed in SJ-DIPG29, whereas RET, MAP2K1 (MEK1), CAMK2B, and PRKCA were selectively activated in SJ-DIPG29.
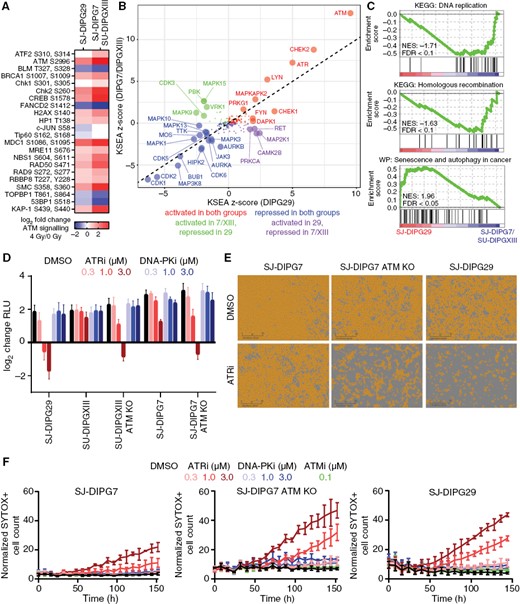
SJ-DIPG29 showed attenuated ATM pathway activation and concomitant synthetic lethality with ATR inhibition. (A) Log2 change in phosphoprotein abundance in SU-DIPGXIII/SJ-DIPG7 and SJ-DIPG29 ATM signaling pathways after irradiation. (B) KSEA z-scores from postirradiation phosphoproteome change in SJ-DIPG7/SU-DIPGXIII vs. SJ-DIPG29. Kinases are color-coded by response in SJ-DIPG7/SU-DIPGXIII vs. SJ-DIPG29 and enlarged if P < 0.05 (FDR-adjusted). (C) Differentially regulated pathways in the baseline proteomes of SJ-DIPG7/SU-DIPGXIII vs. SJ-DIPG29 identified by GSEA. (D) Log2 change (CTG RLU) in DIPG cells treated with the ATR inhibitor AZD6738 or the DNA-PK inhibitor AZD7648 (n = 3). Data are presented as the mean ± SD. (E) Representative microscopy images from week 2 of the confluence assay of SJ-DIPG7, SJ-DIPG7 ATM KO, and SJ-DIPG29 cells treated with 1 µM AZD6738. Scale bar: 300 µm. (F) Results of SYTOX Green cell death assay of SJ-DIPG7, SJ-DIPG7 ATM KO, and SJ-DIPG29 cells treated with the ATR inhibitor AZD6738 or the DNA-PK inhibitor AZD7648 (n = 3). The y-axis reflects the number of SYTOX Green-positive cells divided by the percent confluence. Data are presented as the mean ± SD.
Gene Set Enrichment Analysis (GSEA)26 using proteome data identified the KEGG “DNA replication” and “HR” pathways as deficient in SJ-DIPG29 cells, whereas the WikiPathways “Senescence and Autophagy in Cancer” pathway was enriched (Figure 5C; Supplementary Figure S6C). Interestingly, autophagy can contribute to radioresistance, especially in glioma stem cells.27,28 Consistent with altered autophagy, we confirmed elevated expression of RNASEL, ATG7, and SMAD4 in SJ-DIPG29 and increased expression of HMGA1 in SJ-DIPG7/SU-DIPGXIII by Western blot analysis (Supplementary Figure S6D). Furthermore, expression of LC3B (MAP1LC3B), a widely used marker for autophagosomes,29 was substantially higher in SJ-DIPG29 cells than in SJ-DIPG7/SU-DIPXIII cells.
Both proteomics studies suggested that SJ-DIPG29 compensated for an attenuated ATM pathway response after exposure to genotoxic stress—and subsequent impairment of HR repair—by modulating pro-survival responses. We reasoned that if SJ-DIPG29 was functionally ATM deficient, the tumor might be sensitive to ATR inhibition alone, a known synthetic lethality that has recently been exploited in clinical trials.30 To test this hypothesis, SJ-DIPG29, SU-DIPGXIII WT and ATM KO, and SJ-DIPG7 WT and ATM KO cells were treated with increasing concentrations of the ATR inhibitor AZD6738. Single-agent treatment with AZD6738 was most cytotoxic to SJ-DIPG29 cells—even more so than to the ATM KO cell lines (Figure 5D). We confirmed these results using the confluence assay and a live/dead cell assay (Figure 5E–F). In summary, SJ-DIPG29 presents a novel model of resistance to AZD1390 + radiation, which mechanistically involves decreased dependence on the ATM pathway and concomitant synthetic lethality with ATR inhibition.
Discussion
Inherent resistance to DNA damaging therapies is a hallmark of HGG,7 and yet both radiographic and clinical responses, albeit transient, are often observed with these therapies, and they remain a mainstay of treatment beyond surgical resection. Notably, alterations in DNA repair genes are common in pHGG, including mutations in a diverse set of genes involved in HR. Here, we report that AZD1390 induced remarkable potentiation of radiation in genetically distinct pHGG cell lines in vitro and significantly increased survival relative to that with radiation alone in DIPG OXs, irrespective of TP53 status.
Deland et al. showed that genetic knockout of ATM in a mouse model of brainstem glioma lacking histone H3 mutation significantly improved median survival after hypofractionated radiation treatment (10 Gy × 3) in TP53-deficient mice, as compared with TP53wt mice.31 However, ATM kinase inhibition does not phenocopy the loss of ATM protein,32 and it is unclear how the combination of AZD1390 and conventionally fractionated radiation would perform in the diverse genetic backgrounds present in human pHGG. Consistent with work by others,33–35 Durant et al. concluded that ATM inhibition potentiated radiation to a greater extent in TP53-deficient glioma cells than in TP53wt glioma cells, prompting a discussion about whether TP53 status should be used for patient stratification in future clinical trials.36 Indeed, in our in vitro pHGG cell line panel study, AZD1390 was more effective at sensitizing TP53mut cells to radiation, and protected TP53wt cells from radiation at low doses of drug and radiation. However, as our live cell microscopy experiments illustrated, the proliferative effect of ATMi was reversed as the dose of drug and radiation was increased. Importantly, when combined in vivo daily with fractionated radiation totaling a cumulative dose of 14 Gy, the addition of AZD1390 yielded a more durable response in TP53wt SJ-DIPG37 OXs than in TP53mut SJ-DIPG7 OXs. Interestingly, SJ-DIPG37 cells harbor an activating mutation in PPM1D (Supplementary Table S1A), a phosphatase frequently altered in pHGG that can reverse the ATM-mediated phosphorylation of p53.37 These findings underscore the limitations of using TP53 status alone as a biomarker for predicting response to ATM inhibition in combination with radiation, and they argue against its use as a criterion for clinical trial participation at present.
We uncovered a novel mechanism of resistance to AZD1390 + radiation in SJ-DIPG29 cells, which showed marked reduction in ATM pathway activation after treatment with radiation. While proteomics analysis identified elevated autophagy and altered kinase signaling as potential drivers of radioresistance in these cells, the ATM deficiency presents in SJ-DIPG29 cells induced synthetic lethality with ATR inhibition. Our work supports evaluating components of the ATM pathway, including downstream targets CHK2 and KAP1, as potential biomarkers for response to AZD1390 + radiation combination therapy.
Importantly, we observed no neurotoxicity in study animals treated with combination therapy, and we found no difference in the histopathology of normal brains from mice treated with radiation or the combination. However, we did observe weight loss in mice in the combination arm that was reversed after therapy ended. Further investigation of these mice revealed acute inflammation of the oral mucosa and oropharynx that resembled radiation-induced oral mucositis in patients receiving radiotherapy. The damage in the epithelial compartment was consistent with the potentiation of radiation delivered to the upper aerodigestive tract as part of the whole-brain irradiation procedure employed in our efficacy studies. This toxicity may be mitigated by more conformal and localized delivery of radiation, which is the current clinical standard of care. In conclusion, our study supports the clinical evaluation of AZD1390 in combination with radiation in pediatric patients with HGG and has revealed a novel mechanism of resistance to combination therapy that may be targeted with clinically actionable ATR inhibitors.
Acknowledgments
We are grateful to the patients and families who donated tissue to support pHGG research. We thank Keith A. Laycock, PhD, ELS, for scientific editing of the manuscript; Dr Richard Ashmun and the Flow Cytometry and Cell Sorting Shared Resource (SJCRH); and Divyabharathi Chepyala and Lei Yang from the Analytical Technologies Center of the Chemical Biology and Therapeutics Department (SJCRH) for drug formulation. Bioluminescence imaging, orthotopic xenograft implants, and chemotherapy dosing were performed by the Center for In Vivo Imaging and Therapeutics (SJCRH), which is supported in part by NIH/NCI grant R50CA211481. Cytogenetics was performed by the Cytogenetic Shared Resource (SJCRH), and WGS/WES and RNA-sequencing data were generated by the Hartwell Center for Bioinformatics (SJCRH). The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest
Suzanne J. Baker owns stock in Astra Zeneca. The other authors declare no potential conflicts of interest.
Funding
National Brain Tumor Society Defeat Pediatric Brain Tumors Research Collaborative (SJB, AAS, CLT); National Institute of Health, National Cancer Institute Cancer Center Support Grant P30 CA021765 (AAS, CLT); National Institutes of Health, National Cancer Institute Project Grant P01CA096832 (SJB); American Lebanese Syrian Associated Charities (ALSAC) (AAS, CLT).
Author Contributions
Conceptualization: JX, TK, AAS, CLT; Methodology: JX, TK, BB, NT, ES, XZ, CH, AOT, BF, AAS, CLT; Investigation: JX, TK, BB, ES, XZ, CH, BH, KX, HW, AH, YL, JR, HT, YP; Visualization: JX, TK, NT, ES; Funding acquisition: SJB, AAS, CLT; Project administration: AAS, CLT; Supervision: ZR, GW, JP, SM, SJB, AAS, CLT; Writing—original draft: JX, TK; Writing—review and editing: AAS, CLT.
References
Author notes
These authors contributed equally.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}