





Abstract
Group 3 medulloblastoma (MB) is often accompanied by MYC amplification. PLK1 is an oncogenic kinase that controls cell cycle and proliferation and has been preclinically validated as a cancer therapeutic target. Onvansertib (PCM-075) is a novel, orally available PLK1 inhibitor, which shows tumor growth inhibition in various types of cancer. We aim to explore the effect of onvansertib on MYC-driven medulloblastoma as a monotherapy or in combination with radiation.
Crisper-Cas9 screen was used to discover essential genes for MB tumor growth. Microarray and immunohistochemistry on pediatric patient samples were performed to examine the expression of PLK1. The effect of onvansertib in vitro was measure by cell viability, colony-forming assays, extreme limiting dilution assay, and RNA-Seq. ALDH activity, cell-cycle distribution, and apoptosis were analyzed by flow cytometry. DNA damage was assessed by immunofluorescence staining. Medulloblastoma xenografts were generated to explore the monotherapy or radio-sensitizing effect.
PLK1 is overexpressed in Group 3 MB. The IC50 concentrations of onvansertib in Group 3 MB cell lines were in a low nanomolar range. Onvansertib reduced colony formation, cell proliferation, stem cell renewal and induced G2/M arrest in vitro. Moreover, onvansertib in combination with radiation increased DNA damage and apoptosis compared with radiation treatment alone. The combination radiotherapy resulted in marked tumor regression in xenografts.
These findings demonstrate the efficacy of a novel PLK1 inhibitor onvansertib in vitro and in xenografts of Group 3 MB, which suggests onvansertib is an effective strategy as monotherapy or in combination with radiotherapy in MB.
Novel oral inhibitor of PLK1, Onvansertib, suppresses tumor growth, increases DNA damage, and apoptosis in MYC-driven medulloblastoma.
Onvansertib efficiently sensitizes medulloblastoma xenografts to radiation.
Medulloblastoma is the most common malignant childhood brain tumor. Maximal safe resection, chemotherapy, and craniospinal irradiation remain the first line of treatment. Current treatments cure only a subset of patients and result in long-term morbidity. Patients that express high levels of MYC have a very poor prognosis: 5-year survival is less than 30%, emphasizing the urgency need for more effective therapies. In this preclinical study, we used CRISPR-CAS9 screen to identify that PLK1 is required for tumorigenesis of MYC-driven medulloblastoma. PLK1 is overexpressed in Group 3 MB and reveals a poor prognosis. We demonstrate the therapeutic potential of a novel orally available PLK1 inhibitor, onvansertib in Group 3 medulloblastoma. Onvansertib significantly enhances the impact of radiation in vitro and in animal studies. Our results justify developing a clinical trial to determine the safety and efficacy of combining onvansertib with radiation in patients with MYC-driven medulloblastoma.
Brain tumors are now the leading cause of mortality from cancer in children.1 Medulloblastoma (MB), the most common malignant brain tumor in childhood, is a diverse and heterogeneous disease. Transcriptional profiling identified four major MB subgroups, and among these groups, patients of Group 3 with high MYC expression show the worst prognosis.2 However, novel therapy for Group 3 medulloblastoma is lacking.
Progression of the cell cycle is tightly regulated by coordinated control of phosphorylation and proteolytic events.3 Polo-like kinase-1 (PLK1) belongs to a family of serine-threonine kinases and plays an important role in cell cycle progression through its effects on chromosome segregation, spindle assembly, and cytokinesis.4 PLK1 is activated by phosphorylation and promotes mitotic entry via regulation of cyclin B, the regulatory subunit of CDK1, the phosphatase CDC25, and the kinase Wee1.5 Moreover, PLK1 is critical for the recovery from the DNA damage checkpoint-mediated arrest at G2/M after the DNA damage has been successfully repaired.6–9
PLK1 is highly expressed in rapidly dividing normal cells and is overexpressed in many types of cancer.10 PLK1 inhibition with shRNA or chemical inhibitors decreases tumor growth both in vitro and in vivo.11–14 In pediatric solid tumors including brain tumors, PLK1 expression is often associated with adverse prognosis, and inhibition of PLK1 leads to apoptosis in vitro and in tumor xenografts.15,16 Considering the essential role of PLK1 in the cell-cycle and DNA damage that are relevant to cancer progression, PLK1 is being considered as a “druggable target” in a variety of cancers.17 Several PLK1 inhibitors have been developed to be used as monotherapy and in combination with existing chemotherapies to treat cancer.5,18 BI2536 was found to sensitize N-MYC amplified neuroblastoma cells for vinca alkaloid-induced apoptosis.19 BI2536 also prevents medulloblastoma cell proliferation, self-renewal, cell-cycle progression, and induced apoptosis.12,20 BI6727 was shown to be effective in preclinical models of neuroblastoma, aggressive B cell lymphomas, and small cell lung carcinomas.21–23 Other PLK1 inhibitors including TAK-960 and GW843682 inhibit the proliferation of a wide range of human solid tumor in vitro and in vivo.24,25 While several compounds that target PLK1 were tested in clinical trials following preclinical success, most have been discontinued largely due to nonspecific off target toxicity.5
Onvansertib is a novel, PLK1 specific inhibitor.26 Interestingly, this inhibitor is a pyrazoloquinazoline that can be administered orally rather than via intravenous administration like other PLK1 inhibitors. It shows tumor growth inhibition in hematologic tumors, osteosarcoma, ovarian carcinoma, breast cancer, and colon adenocarcinoma cells.27–29 Clinical trials involving onvansertib are underway in patients with acute myeloid leukemia (NCT03303339), prostate cancer (NCT03414034), and colorectal cancer (NCT03829410). Recently onvansertib was shown to be well tolerated in a phase 1b study of relapsed acute myeloid leukemia patients.30 However, the effects of onvansertib on brain tumor including medulloblastoma as monotherapy or in combination with other therapeutics remains to be determined.
In this study, we investigated the first orally available PLK1-specific inhibitor, onvansertib, in medulloblastoma. A CRISPR-Cas9 screen established PLK1 as a critical mediator in MYC-driven medulloblastoma. We demonstrated that onvansertib suppressed medulloblastoma tumor development and neurosphere formation. We found onvansertib induced G2/M arrest, increased apoptosis, and DNA damage. Onvansertib markedly sensitized medulloblastoma to radiotherapy in vitro and in vivo. Our findings suggest onvansertib in combination with radiotherapy may improve the therapeutic outcome of patients with MYC-driven medulloblastoma.
Materials and Methods
CRISPR-Cas9 Screen
The CU Druggable Library (CUDL) consists of 9761 sgRNAs from 1140 genes targeted by 1194 FDA-approved drugs involved in metabolism, protein modifications, signal transduction, and kinases and druggable genes of interest from multiple research labs in University of Colorado’s Anschutz Medical Campus31 (Supplementary Figure 1A). The oligo pool for the sgRNA library was synthesized on a custom chip and cloned into lentiCRISPRv2 (#52961, Addgene) and lentiGUIDE-puro (#52963, Addgene). The library was transduced into D341, D425, and D458 cells in triplicate at a multiplicity of infection (MOI) of ~0.3. Uninfected cells were removed with puromycin selection, and the population was cultured for a total of 18 days, after which genomic DNA was harvested, the sgRNA cassette retrieved by PCR, and the abundance of sgRNAs quantitated by Illumina sequencing. The sequencing reads were aligned to the shRNA library using Bowtie and the ambiguous reads were excluded from the analysis. Resulting read counts per sgRNA were analyzed using R-based package MAGeCK (1.10.0) (Supplementary Tables 1 and 2), which ranks sgRNAs based on P-values calculated from the negative binomial model and uses a modified robust ranking aggregation (RRA) algorithm to identify positively or negatively selected sgRNA and genes. The sample-to-sample distances were analyzed by hierarchical clustering (Supplementary Figure 1B).
Extreme Limiting Dilution Assay
The cells were treated with indicated concentrations of onvansertib then seeded into 96-well ultra-low-attachment plate in neurosphere media at increasing numbers from 1 cell/well to 250 cells/well. Cells were seeded from n = 5 wells (250 cells/well, 100 cells/well), n = 10 wells (10–50 cells/well), or n = 30 wells (1 cell/well) per condition. Cells were allowed to grow for 14 days, and the number of wells containing neurosphere was counted under microscopy.
Animal Studies
Female athymic Nude Foxn1nu and female NOD-scid gamma mice aged 4–8 weeks were utilized for orthotopic xenograft studies. D458 cells were collected and resuspended as a single cell suspension of 20000 cells/3 μl. MED411 cells were injected at 200000 cells/3 μl. Intracranial injection of cells into outbred athymic Nude Foxn1nu mice (The Jackson Laboratory, strain 07850) was done at 1.5 mm lateral and 2 mm posterior of lambda at 400 nanoliters/minute. Mice were monitored for tumor growth daily and euthanized when 15% weight loss was reached. To monitor tumor growth in D458 xenografts mice, mice were injected intraperitoneally with 10 ul/g of 15 mg/mL D-luciferin potassium salt solution (Gold Biotechnology) and imaged using the Xenogen IVIS 200 In Vivo Imaging System (PerkinElmer). Tumor bioluminescence was analyzed using Living Image 2.60.1 software (PerkinElmer).
Study Approval
All patients provided written informed consent for molecular studies of their tumor and the protocol was approved by the ethics committee of University of Colorado and Children's Hospital Colorado (COMIRBs #95–500). All animal procedures were performed in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals and were approved by the University of Colorado Anschutz Campus Institutional Animal Care and Use Committee.
Results
CRISPR-Cas9 Screen Establishes PLK1 as a Critical Mediator in MYC-Driven Medulloblastoma
To systematically identify genes that are therapeutic vulnerabilities in MYC amplified medulloblastoma, we performed a CRISPR-CAS9 screen targeting 1140 genes that were identified as druggable. D341, D425, and D458 medulloblastoma cells were transduced with 9761 sgRNAs and collected lysate before and after selection puromycin (Figure 1A). The abundance of sgRNAs was quantitated by sequencing and analyzed by MAGeCK.32 KEGG pathway and GO enrichment analysis identified cell cycle and DNA replication as top signaling hubs for MB cell growth (Figure 1B and C). MAGeCK revealed multiple genes that negatively impact the overall cellular fitness, thus identifying the targets for an effective treatment. Both robust rank aggregation score and fold change demonstrated PLK1 was one of the top negatively selected genes in our screen (Figure 1D–G).
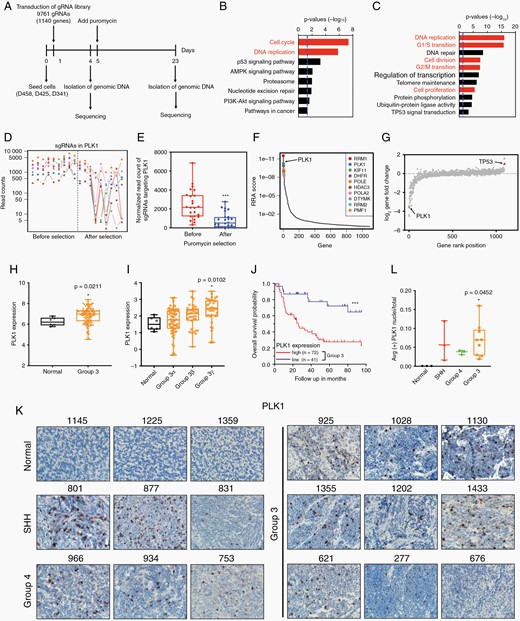
CRISPR-Cas9 screen establishes PLK1 as a critical mediator in MYC-driven medulloblastoma. (A) Schematic of the CRISPR-Cas9 screen performed on D425, D458, and D341 cell lines. (B, C) KEGG and GO enrichment analysis of significantly negative selected genes. (D) The read counts of sgRNAs targeting PLK1. The columns are cell lines. Each dot represents a sgRNA in PLK1. (E) Average read counts of sgRNAs targeting PLK1 before and after puromycin selection. (F) The distribution of top 10 negative robust rank aggregation score. (G) Log fold change of gene expression. PLK1 was identified as one of top negative selective genes as indicated by the green triangle. TP53 was labeled with red as positive control. (H) Microarray analysis of PLK1 in 134 Group 3 patients' samples versus 6 normal cerebellum samples. Dataset of MB was obtained from Cavalli et al. 2017, Cancer Cell. (I) Microarray analysis of PLK1 expression in Group 3 MB. n = 6 in normal, n = 61 in Group 3α, n = 35 in Group 3β, n = 38 in Group 3γ. (J) Kaplan–Meier plots indicating overall survival in relation to PLK1 expression in Group 3 MB patients. (K) Immunohistochemical staining of PLK1 in normal cerebellum and medulloblastoma pediatric patient tissues (Group 3, Group 4, SHH) at 40× magnification. (L) Quantification of IHC.
We therefore investigated PLK1 expression in DepMap which can be used for discovering essential genes in tumor models.33 PLK1 was found as a critical mediator in 55 central nervous system cell lines, including 8 MB cell lines (Supplementary Figure 1C). To further elucidate the expression of PLK1 in medulloblastoma, we performed microarray-based gene expression analysis in 11 Group 3 MB patients and 6 normal cerebella samples collected by our institution. We also examined the expression of PLK1 in a cohort of 763 recently described MB samples.2 Gene expression array data generated from two platforms was normalized and merged to generate a combined series. Group 3 samples were found to express significantly higher PLK1 compared to normal cerebellum, especially in high c-MYC expressed Group 3γ subtype (Figure 1H and I; Supplementary Figure 1D). Kaplan–Meier survival curves revealed that low PLK1 expression considerably correlated with better overall survival in Group 3 (Figure 1J).
We then examined PLK1 and Ki67 expression in MB subtypes by immunohistochemistry staining (Figure 1K and L). The MYC expression was markedly upregulated in selected group 3 samples (Supplementary Figure 1E). Ki67 displayed aggressive tumor cell proliferation and growth in MB (Supplementary Figure 1F, G). PLK1 protein level was notably increased in tumor tissues, even PLK1 gene expression was not significantly changed in selected samples. These data demonstrated high expression of PLK1 is associated with high malignancy and poor prognosis in Group 3 medulloblastoma cases.
PLK1 Inhibition With Onvansertib Suppresses Tumor Growth in Group 3 Medulloblastoma
Onvansertib is a novel orally administration PLK1 specific inhibitor.26 To examine the effect of onvansertib on medulloblastoma, we first performed cell viability assay. We found onvansertib markedly inhibited survival of MB cell lines with varying potency. The half-maximal inhibitory concentration varied from 4.90 to 6 nM in Group 3 cells and up to 27.94 nM in SHH cells. Nontransformed normal human astrocytes (NHA) demonstrated significantly higher resistance to onvansertib with the IC50 concentration at 131.60 nM (Figure 2A). Treatment of onvansertib revealed marked time- and dose-dependent sensitivity suggesting PLK1 function is essential for MB cell survival (Supplementary Figure 2A, B). Onvansertib treatment showed decreased levels of p-CDC25C, which is a downstream target of PLK1, indicating inhibition of PLK1 activity (Figure 2B; Supplementary Figure 2C). Moreover, methylcellulose colony-forming assay revealed that PLK1 inhibition with onvansertib dramatically suppressed adhesion independent colony formation and tumor cell growth in Group 3 MB cells (Figure 2C).
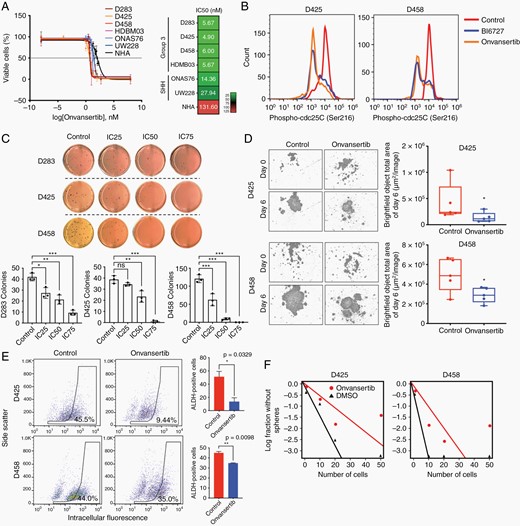
PLK1 inhibition with onvansertib suppresses tumor growth in Group 3 medulloblastoma. (A) Log IC50 determination of onvansertib. Color scale indicates low (green) to high (red) concentrations. (B) The expression of phospho-CDC25C (Ser216) determined by flow cytometry. (C) Methylcellulose assay in D283, D425, and D458 treated with DMSO, IC25, IC50, and IC75 for 2 weeks, respectively. (D) Representative images of neurosphere growth assay in MB cells treated with DMSO or onvansertib. (E) Identification of brain tumor initiating cell fraction in MB cells by ALDH expression demonstrates a decrease in ALDH+ fraction following onvansertib treatment. (F) Neurosphere formation efficacy by extreme limiting dilution assay (DMSO versus onvansertib treatment P = .035 in D425, P = .023 in D458).
Medulloblastoma cells consist of undifferentiated stem- or progenitor-like tumor initiating cells.34 The population of cancer stem cells is thought to be responsible for cancer recurrence in medulloblastoma.35 Following the treatment of onvansertib we assessed cancer stem cell self-renewal and proliferation through a neurosphere growth assay. PLK1 inhibition with onvansertib suppressed neurosphere growth, whereas control cells have ability to developed new neurospheres (Figure 2D; Supplementary Figure 2D). Onvansertib likewise reduced capacity for self-renewal as determined by both aldehyde dehydrogenase activity, a marker of the brain tumor initiating cell fraction with a given cell population (Figure 2E), and neurosphere formation efficacy (Figure 2F).
Onvansertib Induces G2/M Arrest and Suppresses Proliferation
To study the effect of PLK1 inhibition in medulloblastoma, we performed whole transcriptome analyses in onvansertib treated D425 and D458 cells. The RNA-seq results demonstrated a big overlap of differential expressed genes between D425 and D458 treated with onvansertib (Supplementary Figure 3A). Similarly, there are more than 50% overlap hallmark gene sets in two MB cell lines (Supplementary Figure 3B). The gene sets of G2M checkpoint, mitotic spindle, MYC targets, P53 pathway, and apoptosis were significantly enriched in both cell lines (Figure 3A). Onvansertib treatment declined the enrichment score of cell cycle checkpoints and transition gene sets (Figure 3B; Supplementary Figure 3C). Consistent with the gene expression data, we identified that onvansertib induced G2/M arrest of cell cycle (Figure 3C; Supplementary Figure 3D, E). Further, onvansertib led to a marked decrease in cyclin E protein which is required for the transition from G1 to S phase, and to an increase in cyclin B protein which is necessary for the progression of the cells into and out of M phase (Figure 3D).
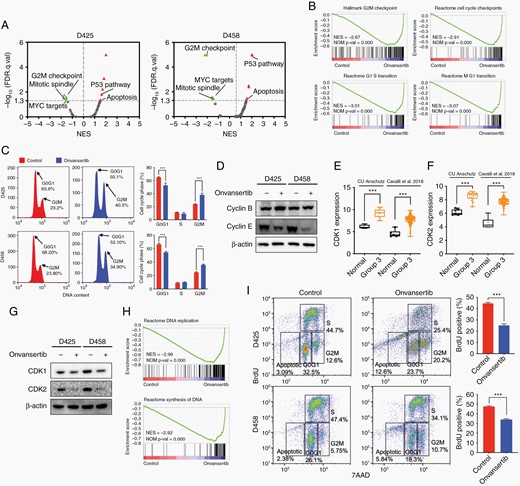
Onvansertib induces G2/M arrest and suppresses proliferation. (A) GSEA hallmark gene sets of onvansertib treated D425 and D458 cells. (B) GSEA of cell cycle checkpoints and transition. (C) Flow cytometry analysis shows increased G2/M arrest that was seen consistently in MB cells treated with onvansertib for 24 h. (D) Cyclin B and cyclin E protein levels in onvansertib versus DMSO treated MB cells. (E, F) CDK1 and CDK2 expression in 11 Group 3 patient samples from our dataset and 134 Group 3 patient samples from Cavalli dataset. (G) CDK1 and CDK2 protein levels in onvansertib versus DMSO treated MB cells. (H) GSEA of DNA replication and synthesis. (I) Flow cytometry profile showing BrdU incorporation into nuclei as a function of DNA content. Four boxes are shown, representing nuclei in G1, S, G2/M, and apoptotic, respectively.
PLK1 drives the transformation of G2/M phase by regulating the activity of the CDK1/Cyclin B protein complex, while CDK2 is required for the transition from G1 to S phase.36,37 We analyzed our data and public datasets for CDK1 and CDK2 expression in Group 3 MB. Significant elevation of CDK1 and CDK2 mRNA expression was observed in Group 3 MB (Figure 3E and F). Inhibition of PLK1 with onvansertib reduced the protein level of CDK1 and CDK2 in MB cells (Figure 3G). GSEA also revealed that expression signatures related to DNA replication and synthesis were attenuated by onvansertib (Figure 3H; Supplementary Figure 3F). Consistent with this observation, onvansertib treated MB cells reduced active DNA synthesis as determined by bromodexoyuridine (Brdu) incorporation measured by flow cytometry (Figure 3I). Taken together, these results indicated that PLK1 is an important factor for promoting cell progression, and inhibition of PLK1 with onvansertib induces G2/M arrest and suppresses the proliferation in Group 3 medulloblastoma.
Onvansertib Suppresses MYC Expression and Transcriptional Activity
MYC is a key driver of tumorigenesis in Group 3 MB.2 Onvansertib treatment caused a substantial downregulation of MYC target genes in MB cells (Figure 4A and B). Flow cytometry analysis showed both BI6727 and onvansertib led to notable loss of c-MYC protein abundance in MB cells (Figure 4C). Further, treatment with onvansertib exhibited a time-dependent loss of c-MYC protein abundance (Figure 4D). To reproduce these results genetically, we performed PLK1 knockdown in MB cells. Onvansertib affected almost 50% differential expressed genes that were regulated by shRNA mediated PLK1 depletion (Supplementary Figure 4A). Comparably, 6 of 9 hallmark gene sets in PLK1 depletion cells were altered by onvansertib treatment, indicating the inhibition of PLK1 activity (Supplementary Figure 4B). PLK1 knockdown suppressed proliferation of medulloblastoma cells and led to similar marked reductions in PLK1 and c-MYC mRNA and protein levels (Figure 4E; Supplementary Figure 4C–E). Thus, these results suggested that PLK1 inhibition by onvansertib suppresses MYC expression and transcriptional activity in medulloblastoma.
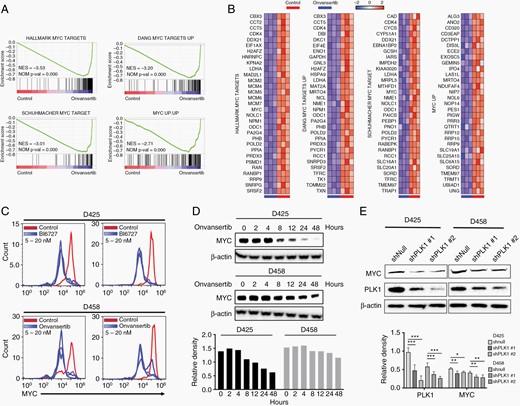
Onvansertib suppresses MYC expression and transcriptional activity. (A) GSEA of MYC targets gene sets. (B) Leading edge analysis of MYC targets gene sets. (C) Flow cytometry analysis c-MYC protein levels upon various concentration of either BI6727 or onvansertib treatment (5 nM, 10 nM, or 20 nM). (D) Time-course analysis of c-MYC protein level with treatment of onvansertib. (E) PLK1 and c-MYC protein levels in MB cells with PLK1 knockdown. The protein was collected after transfection with shPLK1 for 72 h.
Onvansertib Increases Apoptosis and Attenuates Tumor Growth In Vivo
We previously demonstrated that BI2536 increased apoptosis in Daoy and ONS-76 cells.12 Similarly, GSEA revealed alteration of the expression of apoptosis gene set with treatment of onvansertib (Figure 5A). To verify this result, we evaluated caspase 3/7 activity in onvansertib treated MB cells by live cell imaging. As shown in Figure 5B, the administration of onvansertib led to caspase 3/7 activation over time. Correspondingly, we found significantly increased caspase 3 activity and elevated level of cleaved PARP (Supplementary Figure 5A, B). In addition, the Annexin V positive population was markedly enhanced in onvansertib treated cells, indicating an increased total percentage of apoptosis (Figure 5C).
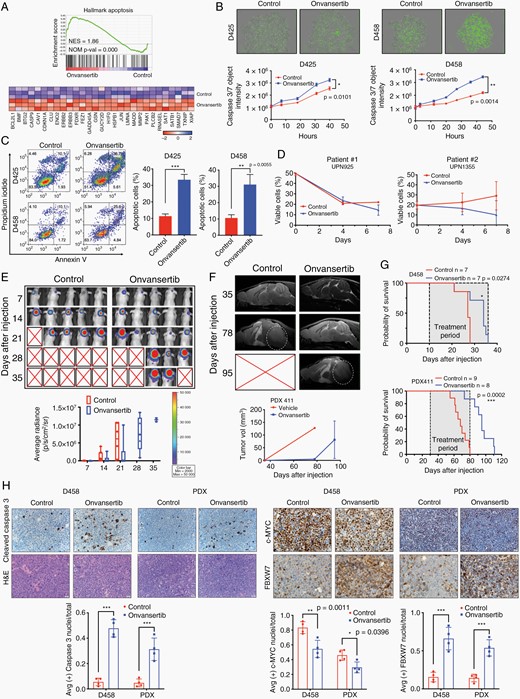
Onvansertib increases apoptosis and attenuates tumor growth in vivo. (A) GSEA of apoptosis pathway. (B) Real-time detection of apoptosis in Incucyte Nuclight Green labeled MB cells treated with DMSO or onvansertib. Apoptotic cells are quantified using the caspase 3/7 Incucyte Live-Cell Analysis System. (C) Annexin V apoptosis analysis of MB cells. (D) Cell viable analysis in short term cultured primary cancer cells from two patients. (E) Representative images of bioluminescence imaging after injection were shown. (F) Representative MRI in PDX xenografts mice. White circles indicate tumor. (G) Kaplan–Meier survival curve of mice treated with vehicle or onvansertib. (H) Representative immunohistochemistry staining for H&E, cleaved caspase 3, c-MYC, and FBXW7. Images taken at 40× magnification.
We next examined the impact of onvansertib on two patient-derived short-term cultures from Group 3 MB patients. In both cases, onvansertib reduced viable cells further establishing the efficacy of onvansertib in Group3 medulloblastoma in vitro (Figure 5D). We then employed an onvansertib treatment in an orthotopic mouse model of medulloblastoma growth. Bioluminescent D458 or patient-derived xenograft (PDX) cells were injected into murine cerebellum to generate orthotopic xenografts.38 Tumor bearing mice were treated three times weekly with onvansertib at 100 mg/kg or vehicle by gavage. For the D458 xenografts model, initial IVIS signals showed similar tumor size in both groups, while tumor growth continued to slow in treated mice (Figure 5E). For the PDX model, using a patient analogous MRI, we found the vehicle-treated mice displayed early signs of edema and visible tumor in the ventricular space at 78 days in PDX models. In contrast, treatment group strongly attenuated MB tumor growth (Figure 5F). The onvansertib treated cohorts survived markedly longer than the vehicle-treated cohorts in both models (Figure 5G). Most notably, onvansertib dramatically improved the median survival of PDX model from 68 to 95 days.
Immunohistochemistry was performed at endpoint of each group. Compared to vehicle treated tumors, onvansertib treated tumors exhibited higher cleaved caspase 3 expression (Figure 5H). Notably, consistent with in vitro data, onvansertib substantially depleted c-MYC expression concomitant with more intense expression of FBXW7. In addition, hematology analyses were also assessed before mice were sacrificed. The oral administration of onvansertib did not induce significant acute hematological toxicity as measured by stable white blood cells, neutrophils, and lymphocyte counts (Supplementary Figure 5C). Taken together, these results indicated that PLK1 inhibition with onvansertib increases apoptosis and suppresses tumor growth in vitro and in vivo.
Onvansertib Enhances Radiation Sensitivity of Medulloblastoma
PLK1 targets various key factors in the DNA damage response.6 GSEA revealed considerably diminished gene expression of DNA damage gene sets (Figure 6A; Supplementary Figure 6A). Onvansertib induced phosphorylation of γH2AX, a marker of DNA damage, as determined by flow cytometry (Figure 6B; Supplementary Figure 6B). RAD51, an important homologous recombination gene that assists in the repair of double strand DNA breaks, also declined in the treatment of onvansertib (Supplementary Figure 6C, D).
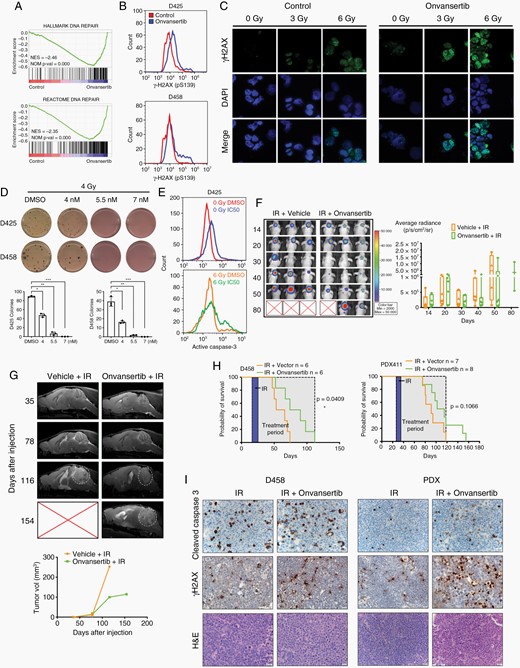
Onvansertib enhances radiation sensitivity of medulloblastoma. (A) GSEA of DNA repair gene sets. (B) Flow cytometry analysis of P-γH2AX (pS139) in MB cells. (C) Immunofluorescence of P-γH2AX (green) and DAPI (blue) at 40×. D425 cells were treated with onvansertib for 48 h and then exposed to 0 Gy, 3 Gy, or 6 Gy radiation. (D) Methylcellulose assay in MB cells treated with 4 Gy radiation in combination with onvansertib treatment. (E) Flow cytometry analysis of apoptotic population measured by cleaved caspase 3. D425 cells were treated with onvansertib for 48 h and then exposed to 0 Gy or 6 Gy radiation. (F) Nude mice xenograft injected with D458 cells were treated with radiation alone or radiation with onvansertib. Representative images of bioluminescence imaging after injection were shown. (G) Representative MRI in PDX mice. White circles indicate tumor. (H) Kaplan–Meier survival curve of xenografts models. (I) IHC analyses of cleaved caspase 3 and P-γH2AX protein expression in xenografts mice. Original magnification, ×40.
Radiation remains a key component of MB therapy and acts by inducing DNA damage. To test if onvansertib and radiation treatment synergistically increase DNA damage, MB cells were treated with vehicle or onvansertib followed by 3 Gy or 6 Gy radiation. In comparison to the vehicle control, onvansertib alone increased P-γH2AX signal in MB cells. Radiation treatment resulted in the accumulation of P-γH2AX which was drastically enhanced by onvansertib in both cell lines (Figure 6C; Supplementary Figure 6E, F). Further, elevated P-γH2AX expression was also observed in vivo (Supplementary Figure 6G). Radiation combined with onvansertib reduced colony formation compared with radiation alone (Figure 6D). Radiation also caused notably increased active caspase 3 activity, while the combination of onvansertib with radiation markedly amplified the active caspase-3 activity compared with radiation alone (Figure 6E). Thus, the combination of onvansertib with radiation resulted in higher DNA damage and apoptosis compared to either treatment alone.
To test potential efficacy in vivo, we further explored the combined treatment of onvansertib and radiation using orthotopic cerebellar tumor xenograft models. For D458 xenograft mice, visible tumors were identified by IVIS imaging on day 11 postinjection. Following tumor verification, mice were irradiated focally using a CT guided IR system at 1.5 Gy fractions for 5 days to simulate a patient analogous fractionation schedule. Tumors size as measured by IVIS signal was visibly reduced within one week after radiation commenced. Combination treatment led to sustained tumor regression in D458 xenograft model (Figure 6F).
To further validate our in vivo findings, the patient-derived xenograft that is more reflective of human patients was used to test responses to combination treatment. MRI revealed onvansertib and IR delayed tumor recurrence compared to IR treatment alone (Figure 6G). The combination treatment improved overall survival compared to radiation treatment alone in both xenograft models (Figure 6H). Further, the expression of cleaved caspase 3 and P-γH2AX was markedly elevated in combination treated models (Figure 6I). The PDX model is highly sensitive to IR so the added impact of onvansertib is modest compared to the D458 cell line model. Nevertheless, these results suggested onvansertib can extend the latency of tumor growth beyond treatment with radiation alone and may be an effective treatment for combination therapy.
Discussion
MYC-driven Group 3 medulloblastoma is an aggressive pediatric brain tumor that is refractory to intensive multimodal therapy. While specifically targeting MYC amplification has remained problematic due to the absence of druggable domains, the potential to indirectly target MYC has shown promise. Here, we performed a CRISPR-Cas9 screen to identify specific targets in MYC-amplified medulloblastoma that have a potential chemical inhibitor available. PLK1 was identified as a top hit which mediating medulloblastoma growth.
PLK1 has been known as a promising target for cancer therapy, however nonspecific PLK1 inhibitors have shown high toxicity profiles and limited activity in clinic so far.5 Pan-PLK inhibitors result in inhibition of PLK2 and PLK3, both mediators of DNA damage and oxidative stress that have tumor suppressor functions.39 Additionally, long half-life of PLK inhibitors such as Volasertib (~5 days) limits the ability to adequately manage treatment-induced toxicities such as myelosuppression. Onvansertib with its high specificity for PLK1, its bioavailability, and its short half-life (~24 h) enables flexible dosing schedules and has the potential to mitigate toxicities observed with previous nonspecific PLK1 inhibitors. Good oral bioavailability in rodent and nonrodent species and proven antitumor activity in different preclinical models make onvansertib as a promising agent for clinical trials. A recent Phase1b study in AML showed that onvansertib was well tolerated and demonstrated antileukemic activity with target engagement and decreased mutant ctDNA following treatment.30
MYC is a key driver of tumorigenesis in medulloblastoma, suggesting that therapeutic efforts aimed at inhibiting its expression or activity should have an important clinical relevance.40 In this study, we found inhibition of PLK1 with onvansertib led to a reduction of c-MYC. Mechanistically, we recently reported that PLK1 inhibition decreases c-MYC through suppressing FBXW7 auto poly-ubiquitination which is consistent with previous studies in neuroblastoma.23,41 The positive, reciprocal activation circuit between PLK1 and c-MYC reinforces MYC-dependent oncogenic programs and promotes aggressive progression of MB. In detail, PLK1 sustains MYC protein levels in MB by enhancing MYC protein stability through kinase cascade.22,41 PLK1 also phosphorylates FBXW7 and promotes its autoubiquitylation and destruction results in stabilization of c-MYC.23 Meanwhile, MYC activates PLK1 transcription and forms positive feed-forward regulatory loop amplifies MYC-regulated oncogenic programs.23,41 Comparably, in this study, we observed diminished c-MYC and increased FBXW7 in onvansertib treated xenograft models. These data support the notion that a PLK1/FBXW7/MYC signaling circuit underlies tumorigenesis and validate PLK1 inhibitors as potential effective therapeutics for MYC-overexpressing cancers.
The inhibition of PLK1 is considered a promising approach for targeting cell cycle. We found onvansertib caused a reduction of cyclin E and an increase of cyclin B. This observation is consistent with our knowledge of the temporal expression of cyclins during the cell cycle. Specifically, E-type cyclins are upregulated during the G1–S phase transition and levels subsequently decline, whereas B-type cyclins are upregulated at the G2–M transition then decrease sharply upon entry to mitosis.42,43 Onvansertib causes the loss of CDK1, abolishes cell cycle progression, and results in inhibition of cell proliferation. Further, the effects of onvansertib on MB cell survival were linked to apoptosis, with markedly increase of cleaved PARP, cleaved caspase-3, Annexin-V, and reductions in MCL-1. There are studies reported that FBXW7 regulates apoptosis by targeting MCL-1 for ubiquitylation and destruction.44,45 Deletion of FBXW7 attenuates PLK1 inhibitor induced MCL-1 degradation.22 Thus, PLK1 signaling controls the cellular apoptosis by regulating MCL-1 protein stability through FBXW7.
PLK1 depletion was shown to cause postmitotic DNA damage and senescence.46,47 BI2536 significantly induced DNA damage and impaired the DNA damage repair pathway in DDP-treated cells both in vitro and in vivo.48 Consistent with the role of PLK1 in DNA damage, our results showed onvansertib enhanced IR-induced DNA damage, which was accompanied by γH2AX induction, indicative of increased DNA damage. Onvansertib in combination with radiotherapy resulted in complete tumor regression of medulloblastoma xenografts. Thus, our study suggests that onvansertib is an effective strategy for radiation sensitization of Group 3 MB.
No preclinical study or clinical trial of onvansertib has yet been performed in any pediatric cancer. This is the first report to evaluate the therapeutic potential of onvansertib in medulloblastoma. Our preclinical study paves the way for testing onvansertib in combination with radiation for the treatment of medulloblastoma and we are now developing a first pediatrics clinical trial for onvansertib targeting pediatric tumors.
Funding
This work was funded by the Morgan Adams Foundation (S.V., R.V.), the Cancer League of Colorado (R.V.) and NIH RO1NS086956 (R.V.).
Conflict of interest statement. M. Ridinger and M. Erlander are employed by Cardiff Oncology Inc. All other authors report no conflict of interest relevant to this manuscript.
Authorship statement. D.W., S.V., and R.V. conceptualized and prepared the manuscript. D.W., B.V., S.F., A.P., K.M., I.B., K.S., M.J., and I.A. performed the experiments. N.F. and A.D. provided tumor specimens. M.E. and M.R. provided scientific inputs.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}