





Abstract
Glioblastoma (GBM) represents the most aggressive and lethal disease of the central nervous system. Diagnosis is delayed following the occurrence of symptoms, and treatment is based on standardized approaches that are unable to cope with its heterogeneity, mutability, and invasiveness. The follow-up of patients relies on burdensome schedules for magnetic resonance imaging (MRI). However, to personalize treatment, biomarkers and liquid biopsy still represent unmet clinical needs. Extracellular vesicles (EVs) may be the key to revolutionize the entire process of care for patients with GBM. EVs can be collected noninvasively (eg, blood) and impressively possess multilayered information, which is constituted by their concentration and molecular cargo. EV-based liquid biopsy may facilitate GBM diagnosis and enable the implementation of personalized treatment, resulting in customized care for each patient and for each analyzed time point of the disease, thereby tackling the distinctive heterogeneity and mutability of GBM that confounds effective treatment. Herein, we discuss the limitations of current GBM treatment options and the rationale behind the need for personalized care. We also review the evidence supporting GBM-associated EVs as a promising tool capable of fulfilling the still unmet clinical need for effective and timely personalized care of patients with GBM.
Foreword
Glioblastoma (GBM) is the most severe and incurable disease of the central nervous system. Diagnosis is delayed following the occurrence of symptoms, and treatment is unable to cope with its heterogeneity, mutability, and invasiveness. Further, the patient follow-up is based on burdensome schedules for magnetic resonance imaging (MRI). Thus, the current GBM standard of care remains based on standardized processes that can only prolong survival by a few months and are associated with a poor prognosis. In this frame, a liquid biopsy that allows tumor diagnosis and constant characterization of its molecular landscape continues to represent a major unmet clinical need. Extracellular vesicles (EVs) may be the key to bridge this gap. They can be readily collected by noninvasive procedures (eg, peripheral blood sampling) and possess multilayered information that is provided by their concentration and cargo. This review discusses the evidence supporting GBM-associated EVs as diagnostic/prognostic biomarkers, providing the possibility of following treatment response and the capability to longitudinally monitor the dynamic molecular landscape of the tumor. Next, the main findings on the clinical relevance of EVs are also reviewed, and ultimately, potential future implications such as noninvasive GBM characterization and perspectives for personalized treatment are presented.
GBM
GBM is the most aggressive tumor among diffuse gliomas (WHO grade IV) accounting for 12–15% of all intracranial tumors, with a global incidence of 3.7/100 000 person-years.1 Despite aggressive treatment strategies, the prognosis remains dreadful achieving a 2-year and 5-year survival rate of 18% and 4%, respectively.1 Biologically, GBM holds two critical features that hamper the possibility of curing those afflicted even with experimental therapeutic approaches. The first is the extensive inter- and intra-tumoral heterogeneity that can mislead diagnosis and characterization, making it exceptionally challenging to target all tumor cells and all patients with the same drug.2 This obstacle is further strengthened by the temporal heterogeneity of the tumor, which bypasses the therapeutic insult by modifying its molecular set-up.2 The second feature is the highly invasive phenotype of the tumor that explains why: (i) surgery is not curative (ie, it is impossible to resect all tumor cells) and (ii) imaging techniques like MRI cannot fully describe the real tumor status and its changes3,4 as the 2–3-mm spatial resolution of MRI enables tumor cells to undergo more than 20 rounds of exponential growth before detection.5,6 Furthermore, MRI is also limited in specificity being unable to exclude with certainty other diagnoses or to differentiate real tumor progression from pseudoprogression, which represents 36% of cases with a form of progression.4,6 Notwithstanding these considerations, routine clinical practice is based on standardized and still suboptimal diagnostic and therapeutic protocols.7 For instance, once neurological symptoms have occurred, diagnosis and follow-up are routinely performed through imaging techniques, commonly MRI, regardless of its sensitivity and specificity.7 Similarly, histological and molecular characterizations are inherently limited, being impossible to achieve a comprehensive description of the whole molecular arrangement in such a heterogeneous tumor through surgical sampling. Furthermore, histological and molecular analyses are performed at a specific time point, conventionally at surgery, thus not reflecting GBM temporal heterogeneity and changes over time.2 To overcome this issue, the biopsy should be repeated several times during the disease course; however, this approach is neither practically nor ethically feasible when considering the potential cumulative surgical risk.5,6 The gold standard treatment for patients with GBM consists of maximum safe surgical resection followed by the Stupp protocol,8 which is based on a concomitant phase (1 month) of temozolomide (TMZ) and radiotherapy followed by repeated cycles of adjuvant TMZ. In addition, tumor-treating fields, based on low-intensity electric fields, may be given concurrently with adjuvant TMZ. Although first-diagnosis cases are homogeneously treated, a standard therapy for recurrent GBM is still not agreed upon by the neuro-oncological community. TMZ efficacy is limited, as it leads to relevant resistance mechanisms. Therefore, recurrent GBM is preferentially treated with second-line chemotherapies that have not already been employed. Based on National Comprehensive Cancer Network guidelines, surgical reresection is potentially indicated only in a limited subset of patients. Novel therapies, including tyrosine kinase inhibitors, checkpoint inhibitors such as nivolumab, immunotherapy, and angiogenesis inhibitors, are opening the door to an unprecedented personalization of treatment.9 However, this declination of precision medicine is only effectively possible once an accurate, reliable, repeatable, and noninvasive tool is available, capable of comprehensively characterizing tumor features. Additionally, these aforementioned considerations account for why the entire armamentarium of imaging, surgery, and chemoradiotherapy remains insufficient and unsuccessful in curing GBM.
Personalized Medicine in GBM
We are facing a significant paradox in GBM treatment. On the one hand, there is a heterogeneous and protean tumor capable of circumventing any treatment by continuously evolving; on the other hand, the diagnostic and therapeutic approaches are standardized and predetermined with inherent limitations. Over the years, considerable efforts have investigated immunotherapy and novel strategies to be introduced in the treatment pipeline.9 In reality, most of these novel treatments have achieved only partial results. In addition, although theoretically indicated, personalized medicine cannot be substantially implemented in GBM clinical practice. Despite any effort to characterize the surgical sample to identify targeted therapies against specific molecular alterations, the hypothetical treatment would be severely limited by both GBM heterogeneity and dynamicity.2 Thus, the key to defeating such a complex tumor is by taking a personalized approach that enables treating each case as dynamic and different from any other. In this sense, the effort to revolutionize treatment should begin from the implementation of a reliable liquid biopsy tool that allows the consistent and noninvasive assessment of tumor status/molecular setup and longitudinal monitoring for changes.2,10
Liquid Biopsy in GBM
GBM is the ideal candidate for a liquid biopsy-based therapeutic approach toward reducing risks of surgical intervention and maximizing treatment efficacy through true individualization of the therapy. In detail, similar to other tumors, GBM releases different molecules involved in intercellular signaling networks as well as metabolic and catabolic processes. These molecules can reach, through the circulation, the cerebrospinal fluid (CSF), from which they can be isolated and exploited for liquid biopsy. Several molecular substrates have been studied as potential diagnostic/prognostic biomarkers that can be isolated or obtained from body fluids.11 Circulating tumor cells (CTCs), cell-free nucleic acids (cfNAs, namely cfDNA, cfRNA, and microRNAs [miRNAs]), and proteins are considered of high potential as diagnostic biomarkers for either early-stage cancer detection/characterization or real-time monitoring of its progression. However, the development of a liquid biopsy platform for GBM must cope with unique challenges. First, the blood-brain barrier (BBB) reduces the amount of detectable tumor-derived material in the bloodstream. Second, the extensive intratumoral heterogeneity worsens difficulties related to accurately describing whole-tumor rearrangements. Under these premises, CTCs, cfNAs, and proteins are not the ideal solution. For CTCs, the small number of studies, the paucity of patients enrolled in each study, and the use of different approaches to isolate CTCs makes the results hardly comparable. In addition, the dearth of CTCs in the blood of patients with GBM (ie, only a single cell or a few cells are found in a 10-ml blood specimen12), coupled with their low stability in circulation, makes CTCs far from representing the whole-tumor molecular arrangement in a real clinical scenario where blood sampling is limited to a few milliliters. Concerning cfNAs, glioma is the tumor with the lowest frequency of detectable cfDNA.13,14 Moreover, cfDNA and cfRNA characterization is extremely demanding under both preanalytical and analytical phases.11 The analysis of a single molecular analyte at a time has its own challenges and limitations concerning specificity, sensitivity, technicality, and informative power. Optimally, concurrent detection of multiple markers would be needed to maximize the information potentially achievable.
Recently, EVs have attracted growing attention in the field of liquid biopsy. Indeed, they derive from whole-tumor cells and possess multilevel informative power, encompassing both their circulating concentration and their molecular cargo (eg, NAs and proteins). When packaged inside EVs, NAs and proteins are protected from degradation by circulating nucleases and proteases. Moreover, the stability of EVs under different conditions15,16 further supports their superiority to CTCs, cfNAs, and proteins. In addition, EVs freely overcome the BBB, even when it is intact, thus being able to reach the bloodstream.17 Several papers have reported enrichment of EVs in patients with GBM,18–21 demonstrating specificity for GBM pathology and not for other brain tumors.19 Furthermore, a continuous crosstalk exists between the GBM and the surrounding brain, and EVs are one of the main mediators of this interaction transferring their cargo between tumor cells and between tumor and nonmalignant cells like endothelial and immune cells, thereby also altering the reciprocal cell phenotype. That being said, it is also impossible to exclude the contribution of nonmalignant cells in plasma concentrations of EVs.
Extracellular Vesicles
EVs are a heterogeneous group of lipid-bilayer bound nanoparticles (30 nm to 10 μm in size) that are released by all cell types. Additionally, they can be isolated from many types of body fluids, spanning saliva, urine, plasma, serum, semen, bronchial lavage, milk, synovial fluid, tears, amniotic fluid, lymph, bile, and CSF,22 and are involved in both physiological and pathological processes.22,23 Furthermore, EVs play numerous roles promoting horizontal oncogene transfer and influencing cancer cell stemness, tumor growth, and progression as well as drug resistance, invasiveness, angiogenesis, immune response, and premetastatic niche preparation.24
Relying on the latest guidelines of the International Society for Extracellular Vesicles on the Minimal Information Required for Studies of Extracellular Vesicles (MISEV), EVs are defined as “particles naturally released from the cell that are delimited by a lipid bilayer and cannot replicate”.25 Three main subpopulations of EVs have been identified, namely, exosomes (EXOs), microvesicles (MVs), and large oncosomes (LOs).23–25 Here, we refer to EVs as the whole population of endosome-derived EXOs and plasma membrane-derived MVs and LOs. EXOs represent the subgroup of the smallest EVs ranging from 30 to 100 nm. They derive by inward budding of the plasma membrane to form early endosomes, whose membranes further invaginate generating intraluminal vesicles (ILVs). These endosomes are called multivesicular bodies (MVBs), and they eventually fuse with the plasma membrane, releasing the ILVs in the extracellular space in the form of EXOs.22–24,26 MVs possess a diameter ranging from 100 to 1000 nm and are derived from outward blebbing of the plasma membrane, which happens through a controlled but unknown mechanism.22,23,26,27 This process requires several contributions from cytoskeleton components, molecular motors, and fusion machinery, and thus, MVs are influenced by the cell’s physiological state and microenvironment.22–24,26 LOs are the subgroup of the largest EVs, ranging from 1 to 10 μm in size. They are specifically released via membrane blebs by tumor cells in advanced stages of disease.24,27,28 Notably, they are also secreted by glioma cells.24,28 LOs represent a relatively novel class of EV; little is known in their regard and their potential informative power has not yet been completely characterized. In virtue of their larger size, LOs are thought to carry a larger quantity of nucleic acids. Therefore, they are a rich source of cancer-specific markers.24,27,28
Currently, several approaches are available to isolate EVs from different body fluids. The more diffuse techniques include differential ultracentrifugation with modifications, density gradients, precipitation, filtration, size-exclusion chromatography, and immuno-isolation.25,29,30 Additional techniques and their combinations are currently under study or validation.25 The priority for research in EVs is standardization of the techniques required for isolation. The vast majority of techniques currently available are not optimized for concrete implementation in a clinical context, and each of these techniques has specific drawbacks and limitations, such as being time-consuming, expensive, technically demanding, low throughput, the presence of contaminants, low reproducibility, and poor isolation efficiency and purity.
The nomenclature of EVs represents another main unsolved issue. It is not always harmonized within the literature and the same terminology can refer to different subpopulations of vesicles, according to interpretations and technical variability during sample preparation and analysis.25 Although distinctive molecular profiles have been identified, a significant overlap exists between EXOs, MVs, and LOs and no specific markers allow to discriminate these different EV subtypes.22–24 Relying on origin, EV markers include tetraspanins (CD9, CD63, and CD81), endosomal sorting complexes required for transport proteins and their accessory proteins (Alix, TSG101, HSP70, and HSP90β),22,24,26 cytoskeletal proteins, and integrins. In the context of cargo, EVs contain proteins, nucleic acids (RNA and DNA), and lipids enclosed within a lipid bilayer that acts as a defense against enzymatic degradation.23–25 In this regard, different databases on EV content are available such as Exocarta (http://exocarta.org/) and Vesiclepedia (http://microvesicles.org/). It is still under debate whether these molecules are packed through specialized machinery or random assignment. Although some analytes are a common thread of EVs independent of the cell of origin, the presence of specific proteins and the enrichment of others in EVs, compared to parental cells, suggest a selective and controlled loading mechanism.22–24
GBM-Associated EVs
Human GBMs release an enormous number of EVs in vivo; a single GBM cell secretes approximately 10,000 EVs over a 48-h period (Figure 1A).31 Further, electron microscopy demonstrated the presence of several MVBs with EXOs inside human GBM tissue (Figure 1B). Notably, most of the vesicles isolated from the plasma of patients with GBM express CD9 which is a recognized marker of EVs (Figure 1C). Intriguingly, GBM-derived EVs differ from normal glial cell-derived EVs.32 A large body of literature reports a wide variety of signaling molecules, functional RNAs, and lipids that facilitate cell-cell and cell-stroma communication, supporting GBM progression, recurrence, and therapeutic resistance through the creation of a tumor-supporting microenvironment. In addition to directly stimulating GBM cell growth, survival, invasiveness, and drug resistance,31,33,34 GBM-derived EVs can also: (i) reprogram brain endothelial cells toward an enhanced and aberrant angiogenesis,35,36 (ii) modify the physiology of neighboring normal brain cells by transferring their oncogenic cargo,37,38 (iii) promote the immunosuppressive properties of microglia,39,40 (iv) stimulate monocytic cells to acquire tumor supportive behavior,41 and (v) inhibit T-cell-mediated immune response by influencing monocyte maturation.42 In this regard, the cargo of GBM-derived EVs includes several immunomodulating molecules (ie, transforming growth factor-beta, interleukin-10, and heat shock proteins)43,44 as well as programmed death ligand-1 (PD-L1),45 which leads to cancer immune evasion through the inhibition of T-cell function. Overall, GBM-associated EVs reflect the molecular asset of the tumor and its microenvironment.5,46–48 The expression level of several GBM-associated EV proteins and miRNAs mirrors that of glioma tissues and has been associated with GBM pathogenesis and progression.5,46–48 However, careful consideration must be given to their potential clinical utility. EV-related proteins and miRNAs, alone or as part of a signature, have been identified on small sample sizes with no independent validation, and a better understanding of their role requires further assessment. Considering the majority of published reports, the role of an EV’s cargo was mainly addressed in virtue of its ability to mirror specific features of the parental tumor, thus being of only theoretical value for diagnosis, prognosis, and tailored GBM treatment.

GBM-derived EVs: (A) Transmission electron micrograph of GBM tissue (green) releasing vesicles in the extracellular space. EXOs are shown by white arrows, whereas MVs and LOs are shown by black arrows (magnification × 25 000). (B) Transmission electron micrograph of GBM tissue containing MVBs (black arrows) with several EXOs inside (magnification × 14 500). (C) Transmission electron micrograph of EVs isolated from the plasma of a patient with GBM after immunogold labeling. The different EV subpopulations, EXOs (gray arrows) and MVs and LOs (black arrows) are classified by morphological features, similarly to that identified in GBM tissue and are stained with a mouse anti-CD9 primary antibody (1:10, BD Pharmingen), followed by a rabbit antimouse IgG bridge antibody (1:250, Dako) and protein A gold 10 nm (PAG10, 1:50).
GBM-Associated EVs: Sample Types for Liquid Biopsy
The two most suitable sources of GBM-associated EVs are blood and CSF. Interestingly, GBM EVs cross the BBB in both directions, thus being detectable in peripheral blood.31,49–51 The major drawback in dealing with blood stems from its complex composition as it collects the molecular contributions of almost every organ.49,52,53 Conversely, CSF is a confined compartment that is shielded from the blood by the BBB. It is less in volume and contains fewer EVs from other sources compared to blood54; however, CSF sampling is achievable only through lumbar puncture, which is invasive, painful, distressing, and carries the risk of infection and cerebral herniation, among others. These aspects make it inadvisable to repeat CSF sampling in a close timeframe, while supporting blood as the preferred liquid biopsy sample medium. Considering potential differences between CSF- and blood-derived EVs, few studies have directly addressed this issue, with controversial results. Recently, Jafari and coworkers evaluated the diagnostic accuracy of GBM-associated EVs.49 Interestingly, they did not observe any significant difference in sensitivity between serum and CSF EVs, although the specificity was slightly higher for CSF EVs (77% for serum vs. 89% for CSF). Accordingly, an independent analysis revealed that the levels of miR-21 in CSF-derived EVs mirrored that of serum EVs.55 However, mutant isocitrate dehydrogenase 1 (IDH1) mRNA was identified in CSF-derived EVs but not in serum-derived EVs collected from patients bearing mutant IDH1 gliomas.56 In addition, particular care must be taken to distinguish CSF collected during surgery, after opening the dura mater, and through lumbar puncture. Remarkably, CSF derived from these two compartments differs in its composition,57 potentially implying differences in the features of CSF-derived EVs. Akers and colleagues compared the diagnostic performance of a nine miRNA signature panel on CSF EVs from surgery (cisternal) to that of lumbar puncture,52 observing a sensitivity and specificity of 55% and 87%, respectively, in the first case compared to 13% and 100%, respectively, for lumbar puncture.52 This finding shows the importance of carefully translating CSF-based results into clinical practice according to the CSF collection site, lumbar or cisternal. Furthermore, the validation of the accuracy of liquid biopsy would require different controls, namely, healthy volunteers and patients affected by other conditions or tumors, to fully evaluate the area under the curve, calculate the positive and negative predictive values, and meaningfully determine the sensitivity and specificity. Although these types of comparisons are achievable for blood, they are inherently more challenging for CSF.54 Thus, based on current knowledge, CSF is not superior to blood in terms of informative power and lacks a fundamental aspect of liquid biopsy, which is having a noninvasive sampling procedure. These drawbacks significantly narrow CSF-based clinical applications, being ineligible for monitoring, follow-up, and continuous treatment tailoring. However, a few issues must be discussed when choosing blood as the sample medium to perform EV-based liquid biopsy. Concerning the choice between plasma and serum, the latter presents a higher concentration of platelet-derived EVs than plasma due to platelet activation and clot formation.58 In the case of plasma, the anticoagulant itself plays a role since EDTA and acid citrate dextrose inhibit platelet activation more efficiently than citrate.58 Consequently, to reduce the confounding factors potentially originating from platelet-released EVs, plasma is the preferred source of EVs for biomarker discovery, whereas serum is used to study cell-free miRNAs.
Clinical Significance of GBM-Associated EVs
From a clinical perspective, in addition to their pathophysiological commitment, EVs are appealing as potential liquid biopsy biomarkers in virtue of the possibility that they can be repeatedly collected in a noninvasive way and due to their multilayered informative power, which is constituted by their concentration and molecular cargo (ie, DNA, RNA, protein, lipid). By interrogating all these layers of information, a comprehensive overview of tumor status and molecular landscape is possible, thus facilitating diagnosis and characterization. Moreover, this information may help tailor personalized treatment for each patient: an accomplishment made possible by providing time points over the disease course and finally tackling the distinctive heterogeneity and mutability of GBM.2
Plasma Concentrations of EVs are a Reliable Biomarker of GBM Presence and Response to Treatment
The concentration of EVs in a fluid represents per se a reliable biomarker of GBM presence and status. To date, different groups have demonstrated that the number of EVs is increased in the peripheral blood of patients with GBM compared to healthy donors, thus suggesting that the plasma concentration of EVs can be indicative of GBM presence.18,19,21 We also demonstrated the specificity of this phenomenon. We found no measured increase in plasma concentrations of EVs in patients affected by brain metastasis or extraaxial brain tumors.19 In support of the specificity of the association between EVs and GBM cells, the concentration of EVs, although significantly reduced after surgical resection, increases again in patients facing a recurrence.19 Interestingly, plasma concentrations of EVs are significantly elevated in all patients with GBM regardless of the presence of specific GBM alterations, such as epidermal growth factor receptor (EGFR) amplification, phosphate and tensin homolog (PTEN) deletion, O6-methylguanine DNA methyltransferase (MGMT) expression, and IDH mutations. This finding indicates that the plasma concentration of EVs behaves as a biomarker sufficiently informative for diagnosis, follow response to therapy, and identify relapse independently from GBM molecular subtype/landscape.19 In this perspective, Sabbagh et al.20 recently coined the term “vesiclemia,” demonstrating the potential translation of plasma EV concentration in a clinical setting. The relevance of an EV concentration-based biomarker is even greater if contextualized in routine clinical practice, where EV counts may allow following of treatment response. Standard chemoradiation therapy (CRT) induces equivocal imaging findings, known as pseudoprogression, which are indistinguishable from real tumor progression. In this frame, the plasma concentration of EVs may effectively discriminate those patients facing tumor progression from those with pseudoprogression after CRT, thereby facilitating and anticipating treatment decisions toward improving patient survival.59 To maximize the accuracy of EV concentration as a biomarker, Shao and colleagues defined the “tumor progression index” (TPI)—a novel parameter that simultaneously reflects changes in both the number of EVs and molecular cargo relying on the analysis of EGFR, EGFRvIII, and IDH1R132H molecular markers. Changes in TPI during treatment allowed discrimination between CRT-responder and nonresponder.60 Interestingly, the possibility of following treatment response by assessing plasma EV concentrations has been demonstrated for both standard (TMZ) and experimental treatments (vaccination using antisurvivin).59,61 However, TMZ results remain controversial: some papers have demonstrated the enrichment of circulating EVs and a concomitant alteration of EV protein cargos, while others reported a decrease in the number of circulating EVs.18,60 Nevertheless, these findings provide proof of concept that the measurement of EV concentration represents an important source of diagnostic, prognostic, and predictive information to diagnose and monitor GBM response to treatment noninvasively in real time.
Molecular Cargo of EVs Allows Diagnosis and Prognosis Prediction and Potential Therapeutic Implications for Patients with GBM
Since 2000, to address the distinctive heterogeneity of GBM, scientists began classifying the disease into different subtypes relying on transcription profiles, genetic alterations, and DNA methylation. Concerning transcription-based classification, Phillips et al. in 2006 identified proneural, proliferative, and mesenchymal subtypes.62 Later, Verhaak and coworkers in 2010 confirmed and further furnished a more in-depth molecular grouping encompassing proneural, neural, classical, and mesenchymal subtypes.63 The proneural subtype is more common in younger patients owing to a better prognosis. Conversely, the mesenchymal subtype is characterized by the worst prognosis linked to its invasive growth and angiogenic features,64 whereas the neural subtype represents normal brain contamination.65 Worthy of note is the coexistence of these subtypes within the same tumor, thus making patient subclassification challenging.66 To increase the accuracy of GBM characterization, and improve the clinical/therapeutical approach to the disease, the WHO classification combined both molecular and histological features in a multilayered diagnosis protocol.67 As such, GBM is defined as a grade IV diffusely infiltrating glioma first stratified by IDH1 mutation status (IDH1R132H).67 Wild-type IDH1 GBMs share other genetic alterations like EGFR overexpression/amplification, PTEN deletion, and telomerase reverse transcriptase (TERT) promoter mutations.67 IDH1-mutant GBMs carry ATRX and TP53 mutations.67 Currently, routine practice is based on a single incomplete “snapshot” of the tumor obtained through tissue biopsy at a specific time point and location, thus being unable to describe whole-tumor heterogeneity and its changes over time. Conversely, EVs, deriving from all tumor cells, potentially allow the study of whole-GBM heterogeneity. A precise tumor molecular portrait will improve diagnosis and treatment. Of note, the specific molecular alterations currently used in GBM diagnosis, molecular classification, and subtyping (IDH, EGFR, EGFRvIII, PTEN, MGMT, and TP53) have also been identified in GBM-associated EVs (Table 1).17,43,47,56,60,68–74 IDH1 mutation is the first step in the new WHO Classification of Tumors of the Central Nervous System: its mutation rate is rare in primary GBM and more frequent (73%–85%) in secondary GBM. IDH1 mutation status allows differential diagnosis with other brain tumors, and it is considered a positive prognostic marker being associated with either better outcome metrics75 or increased radio- and chemosensitivity.76 Mutant and wild-type IDH1 DNA sequences and transcripts have been identified in EVs derived from blood and CSF of patients with glioma.17,56 The possibility to identify IDH1 mutation status, which is undetectable by conventional tissue biopsy, through an EV-based liquid biopsy highlights the great potential of EVs in describing the whole-tumor molecular setup.17 Therefore, the determination of IDH1 mutation status in EVs holds important implications for GBM diagnosis and subtyping. EGFR amplification and overexpression are two common genetic alterations in GBMs associated with the classical subtype.31,77 Moreover, the constitutively activated EGFRvIII variant, occurring in approximately one-third of GBMs, is specific for GBM cells, and thus, likely informative for prognosis, diagnosis, therapy monitoring, and targeted therapy. EGFR and EGFRvIII are expressed as oncoprotein or mRNA in GBM-associated EVs isolated from both CSF and blood.43,47,68,69 EGFR levels in EVs can differentiate patients with high-grade and low-grade glioma.69 Likewise, EGFRvIII-expressing EVs discriminate patients with GBM and healthy controls, potentially acting as a diagnostic biomarker.43,68 The disappearance of EGFRvIII-expressing EVs after tumor removal further reinforces the specificity of EVs for GBMs while supporting the possibility of following response to treatment and early detection of tumor relapse.68 In addition, EGFRvIII identification in those cases in which tissue biopsy excluded its presence demonstrates the superiority of EVs recapitulating the entire tumor heterogeneity.47,68 Notably, GBM-derived EVs transfer EGFRvIII from positive to negative cells, thereby promoting the acquisition of tumor-related features in recipient cells33 and accounting for the aforementioned GBM dynamicity. The sensitivity and specificity of EGFRvIII detection in EVs reached 61% and 98% in CSF,47 and 81.58% and 79.31% in serum, respectively.78 Interestingly, EGFRvIII-positive EXOs correlate with a poor patient outcome.78 Overall, these findings sustain a role for GBM-derived EVs as a mean for EGFRvIII-positive GBM diagnosis and tailored therapeutic approach (eg, introducing EGFR inhibitors or anti-EGFRvIII vaccinations). Gliomas are also potentially associated with the reduction or loss of PTEN expression. PTEN behaves as a tumor suppressor by inhibiting the PI3K/AKT/mTOR pathway, one of the main signaling cascades sustaining GBM progression. Therefore, its mutation fosters tumor proliferation. PTEN RNA is incorporated into EXOs from patients with GBM and its expression is associated with tumor grading.70 Thus, the identification of PTEN with other GBM alterations in circulating EVs may be a promising clinical tool for glioma classification. MGMT is the enzyme responsible for DNA repair. By neutralizing the effects of alkylating agents, MGMT plays a role in assessing sensitivity to alkylating chemotherapy, including TMZ.79 Methylation of the MGMT promoter occurs in approximately 50% of GBMs and affects the levels of functional MGMT protein, thus inhibiting proper DNA repair in response to alkylating chemotherapy.79 Therefore, MGMT status holds strong prognostic value. MGMT mRNA is present in serum-derived EVs from patients with GBM and its level mirrors that of the tumor.71 Notably, the MGMT level in EVs change during treatment, thus potentially allowing monitoring of treatment response and the derived molecular modifications.71 Moreover, the analysis of MGMT promoter methylation status, with genome-wide methylation profiling, is feasible in GBM-derived DNA-containing EVs and mirrors the methylation status of the corresponding tumor.80 In this scenario, the clinical significance of the EV cargo relies on the identification of multiple markers (Table 1). Shao and colleagues identified a molecular signature in GBM-associated EVs encompassing EGFR, EGFRvIII, and IDH1R132H, whose modulation is associated with TMZ and radiation therapy, indicating treatment efficacy.60 From a clinical perspective, the identification of these molecular traits will help in diagnosis and prognosis prediction (Table 1). Furthermore, unmethylated MGMT and elevated EGFR expression correlate with a short patient survival,81 whereas EGFR amplification and TERT promoter mutations indicate poor prognosis.82 In patients with good Karnofsky performance status and MGMT methylation, concurrent chemoradiation with adjuvant TMZ are the treatment of choice. Therapies targeting EGFRvIII83 or IDH84 are in clinical trials. There are also ongoing trials of anti-PD-L1 inhibitors targeting hypermutated GBM.85 GBM-related mutations and copy number variations are detectable in DNA-containing EVs and reflect whole-genomic alterations of the parental tumor.80 Therefore, BRAF/IDH1/IDH2 mutations, ERBB2/MET/EGFR/PDGFRA amplifications, mutations in DNA damage repair genes, and TGF-beta–mediated signaling are potentially exploitable for targeted therapeutic approaches. The possibility to identify further targetable and actionable molecular aberrations in EVs will open the door for a patient-tailored approach, also rescuing already approved drugs not currently used in GBM treatment. Additionally, this capacity may permit tracking of potentially targetable mutations in serial blood sampling, thus overcoming temporal intratumoral heterogeneity. Furthermore, the identification of these molecular markers in association with EVs may allow the accurate definition of patients with GBM subtype, determine their potential clinical aggressiveness, and provide a better indication of GBM progression and relapse (Table 1). In addition, numerous experimental investigations have addressed GBM-associated EVs as reservoirs of miRNAs (Table 1). Although the significance of miRNAs in GBM characterization has been widely described due to their abundance and stability, the effort to demonstrate a significant association with the course, history, or progression of the disease has yet to provide convincing evidence to translate the assessment of miRNAs in clinical practice. To our knowledge, no miRNA is specific for GBM and technical variability in measuring their expression levels under identical conditions challenges their clinical usefulness.53 Despite these clear drawbacks, we believe that some miRNAs are particularly worthy of mention. In particular, miR-21 levels in CSF-derived EVs reflect its overexpression in tumor cells, being, on average, ten-fold higher in patients with GBM than in controls. Moreover, it is associated with a worse prognosis. Interestingly, miR-21 is part of a signature that specifically distinguishes patients with GBM from healthy controls.55,86 Different miRNA signatures have been identified in EVs from GBM sera with the potential for GBM diagnosis, differentiation between GBM, lower-grade glioma and nonneoplastic brain lesions, and real-time monitoring of tumor burden and treatment response (Table 1).49,52,87–90 Of note, these panels of EV-encapsulated miRNAs maximize their sensitivity and specificity for GBM detection.
Summary of the Reviewed EV-Associated Biomarkers with Potential Clinical Implications
| Source | Isolation method | Identified molecules | Potential clinical Applications | Ref. |
|---|---|---|---|---|
| Mouse plasma | Differential ultracentrifugation | EGFRvIII | -Diagnosis | 23 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro(cell lines; GBM stem cells) | Differential ultracentrifugation | CLIC1 | -Treatment tailoring | 24 |
| In vitro(cell lines), Mouse plasma, Patient’s plasma | Differential ultracentrifugation | Proteins: CAV1, IL8, PDGFs, MMPs, PTX3, CD26, PAI1, Transcripts: ADM, LOX, IGFPB 3, BCL2, BNIP3, NDRG1, PLOD2, PAI1 | -Diagnosis | 25 |
| -Treatment tailoring/monitoring | ||||
| -Assessment of GBM oxygenation status | ||||
| -Angiogenesis inhibitors | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | miR-148a-3p, miR-9-5p, miR-16-2-3p, miR-182-5p, miR-9-3p, miR-22-3p, miR-186-5p, miR-378e | -Diagnosis | 26 |
| -Treatment tailoring/monitoring | ||||
| -Angiogenesis inhibitors | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | 97 mRNAs (RP, OXPHOS, glycolysis) | -Diagnosis | 28 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM cell line) | Differential ultracentrifugation | miR-21 | -Diagnosis | 29 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | miR-451, miR-21, miR-15b, miR-146a, miR-223 | -Diagnosis | 30 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | Proteins involved in leukocyte recruitment and focal adhesion mechanisms | -Treatment tailoring/monitoring | 31 |
| -Agents blocking CSF-1R | ||||
| Patients’ serum | Differential ultracentrifugation | EGFR, EGFRvIII, and TGF-β | -Diagnosis | 33 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro(cell lines) | Differential ultracentrifugation | HSPs 90, 70, 60, and 27 | -Diagnosis | 34 |
| -Treatment tailoring/monitoring | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | PD-L1 (protein and DNA) | -Diagnosis | 35 |
| -Treatment tailoring/monitoring | ||||
| Patients’ serum/plasma | -anti-PD1 receptor blocking antibody | |||
| -Tumor subtyping | ||||
| Patients CSF | Differential ultracentrifugation | wtEGFR RNA, EGFRvIII RNA | -Diagnosis | 37 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells), Mouse serum, Patients’ serum | Differential ultracentrifugation | Sema3A | -Diagnosis | 40 |
| -Treatment tailoring | ||||
| -Blocking Sema3A or its receptor Neuropilin1 (NRP1) | ||||
| Patients CSF | Differential ultracentrifugation | miR-21 | -Diagnosis/staging | 45 |
| -Treatment tailoring | ||||
| Patients CSF | Differential ultracentrifugation | mutant and wild-type IDH1 mRNA | -Diagnosis | 46 |
| -Assessment of IDH1 mut status | ||||
| In vitro (GBM stem cells) | -Size exclusion chromatography | -proteins related to ribosome, proteasome, cell adhesion, and extracellular matrix-receptor interaction. | -Diagnosis | 49 |
| -Treatment tailoring/monitoring | ||||
| -Differential ultracentrifugation | ||||
| -EVs released by GSCs upon TMZ challenge display a specific enrichment in molecules involved in cell adhesion. | ||||
| Patients’ plasma | Differential ultracentrifugation | members of the complement and coagulation cascade and regulators of iron metabolism (vWF, APCS, C4B, AMBP, APOD, AZGP1, C4BPB, Serpin3, FTL, C3, and APOE) | -Diagnosis | 51 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells, Cell lines), Mouse plasma | Differential ultracentrifugation | EGFR, PDGFRα, PDPN, EphA2, EGFRvIII, IDH1 R132H, HSP90, CD4124, MHCII. | -Diagnosis | 54 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Patients’ plasma | ||||
| Patients’ serum | Differential ultracentrifugation | Survivin | -Diagnosis | 55 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Mouse plasma, Patients’ serum | Differential ultracentrifugation | IDH1G395A DNA | -Diagnosis | 64 |
| -Assessment of IDH1 mut status | ||||
| Patients’ serum | Differential ultracentrifugation | miRNA21, EGFRvIII mRNA Proteins: angiogenin, FGFα, IL-6, IL-8, TIMP-1, VEGF, and TIMP-2 | -Diagnosis | 65 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM Cell lines), Patients’ serum | Differential ultracentrifugation | EGFR protein and NLGN3, PTTG1 mRNA | -Diagnostic and prognostic markers | 66 |
| Patients’ serum | - | PTEN RNA | -Diagnosis/staging | 67 |
| Patients serum | Microfluidic platform | MGMT and APNG RNA | -Diagnosis | 68 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Mouse plasma | Differential ultracentrifugation | DNM3, p65, CD117, PTEN and p53 mRNA and protein | -Diagnosis | 69 |
| In vitro (GBM Cell lines) | Density gradient ultracentrifugation | ANXA1, IGF2R, ITGB1, PDCD6IP, ACTR3, CALR, IPO5, MVP, PSMD2 proteins | -Diagnosis | 86 |
| -Markers for more aggressive disease | ||||
| In vitro (GBM stem cells, Cell lines) | Density gradient ultracentrifugation | EVs proteins content mirrors the phenotypic signature of the respective GBM cells | -Diagnosis | 63 |
| -GBM subtyping | ||||
| Patients serum | Total Exosome Isolation Kit | EGFRvIII RNA | -Diagnosis | 73 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells) | -Size exclusion chromatography | DNA methylome | -Diagnosis | 75 |
| -GBM subtyping | ||||
| -Differential ultracentrifugation | -Treatment tailoring/monitoring | |||
| Patients serum | Size exclusion chromatography | miR-182-5p, miR-328-3p, miR-339-5p, miR-340-5p, miR-485-3p, miR-486-5p, and miR-543 | -Diagnosis | 82 |
| Patients serum | Exoquick | 1 small noncoding RNA (RNU6-1) and 2 microRNAs (miR-320 and miR-574-3p) | -Diagnosis | 83 |
| Patients serum | Exoquick | miR-21, miR-222 and miR-124-3p | -Diagnosis | 85 |
| -Treatment tailoring/monitoring |
| Source | Isolation method | Identified molecules | Potential clinical Applications | Ref. |
|---|---|---|---|---|
| Mouse plasma | Differential ultracentrifugation | EGFRvIII | -Diagnosis | 23 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro(cell lines; GBM stem cells) | Differential ultracentrifugation | CLIC1 | -Treatment tailoring | 24 |
| In vitro(cell lines), Mouse plasma, Patient’s plasma | Differential ultracentrifugation | Proteins: CAV1, IL8, PDGFs, MMPs, PTX3, CD26, PAI1, Transcripts: ADM, LOX, IGFPB 3, BCL2, BNIP3, NDRG1, PLOD2, PAI1 | -Diagnosis | 25 |
| -Treatment tailoring/monitoring | ||||
| -Assessment of GBM oxygenation status | ||||
| -Angiogenesis inhibitors | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | miR-148a-3p, miR-9-5p, miR-16-2-3p, miR-182-5p, miR-9-3p, miR-22-3p, miR-186-5p, miR-378e | -Diagnosis | 26 |
| -Treatment tailoring/monitoring | ||||
| -Angiogenesis inhibitors | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | 97 mRNAs (RP, OXPHOS, glycolysis) | -Diagnosis | 28 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM cell line) | Differential ultracentrifugation | miR-21 | -Diagnosis | 29 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | miR-451, miR-21, miR-15b, miR-146a, miR-223 | -Diagnosis | 30 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | Proteins involved in leukocyte recruitment and focal adhesion mechanisms | -Treatment tailoring/monitoring | 31 |
| -Agents blocking CSF-1R | ||||
| Patients’ serum | Differential ultracentrifugation | EGFR, EGFRvIII, and TGF-β | -Diagnosis | 33 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro(cell lines) | Differential ultracentrifugation | HSPs 90, 70, 60, and 27 | -Diagnosis | 34 |
| -Treatment tailoring/monitoring | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | PD-L1 (protein and DNA) | -Diagnosis | 35 |
| -Treatment tailoring/monitoring | ||||
| Patients’ serum/plasma | -anti-PD1 receptor blocking antibody | |||
| -Tumor subtyping | ||||
| Patients CSF | Differential ultracentrifugation | wtEGFR RNA, EGFRvIII RNA | -Diagnosis | 37 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells), Mouse serum, Patients’ serum | Differential ultracentrifugation | Sema3A | -Diagnosis | 40 |
| -Treatment tailoring | ||||
| -Blocking Sema3A or its receptor Neuropilin1 (NRP1) | ||||
| Patients CSF | Differential ultracentrifugation | miR-21 | -Diagnosis/staging | 45 |
| -Treatment tailoring | ||||
| Patients CSF | Differential ultracentrifugation | mutant and wild-type IDH1 mRNA | -Diagnosis | 46 |
| -Assessment of IDH1 mut status | ||||
| In vitro (GBM stem cells) | -Size exclusion chromatography | -proteins related to ribosome, proteasome, cell adhesion, and extracellular matrix-receptor interaction. | -Diagnosis | 49 |
| -Treatment tailoring/monitoring | ||||
| -Differential ultracentrifugation | ||||
| -EVs released by GSCs upon TMZ challenge display a specific enrichment in molecules involved in cell adhesion. | ||||
| Patients’ plasma | Differential ultracentrifugation | members of the complement and coagulation cascade and regulators of iron metabolism (vWF, APCS, C4B, AMBP, APOD, AZGP1, C4BPB, Serpin3, FTL, C3, and APOE) | -Diagnosis | 51 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells, Cell lines), Mouse plasma | Differential ultracentrifugation | EGFR, PDGFRα, PDPN, EphA2, EGFRvIII, IDH1 R132H, HSP90, CD4124, MHCII. | -Diagnosis | 54 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Patients’ plasma | ||||
| Patients’ serum | Differential ultracentrifugation | Survivin | -Diagnosis | 55 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Mouse plasma, Patients’ serum | Differential ultracentrifugation | IDH1G395A DNA | -Diagnosis | 64 |
| -Assessment of IDH1 mut status | ||||
| Patients’ serum | Differential ultracentrifugation | miRNA21, EGFRvIII mRNA Proteins: angiogenin, FGFα, IL-6, IL-8, TIMP-1, VEGF, and TIMP-2 | -Diagnosis | 65 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM Cell lines), Patients’ serum | Differential ultracentrifugation | EGFR protein and NLGN3, PTTG1 mRNA | -Diagnostic and prognostic markers | 66 |
| Patients’ serum | - | PTEN RNA | -Diagnosis/staging | 67 |
| Patients serum | Microfluidic platform | MGMT and APNG RNA | -Diagnosis | 68 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Mouse plasma | Differential ultracentrifugation | DNM3, p65, CD117, PTEN and p53 mRNA and protein | -Diagnosis | 69 |
| In vitro (GBM Cell lines) | Density gradient ultracentrifugation | ANXA1, IGF2R, ITGB1, PDCD6IP, ACTR3, CALR, IPO5, MVP, PSMD2 proteins | -Diagnosis | 86 |
| -Markers for more aggressive disease | ||||
| In vitro (GBM stem cells, Cell lines) | Density gradient ultracentrifugation | EVs proteins content mirrors the phenotypic signature of the respective GBM cells | -Diagnosis | 63 |
| -GBM subtyping | ||||
| Patients serum | Total Exosome Isolation Kit | EGFRvIII RNA | -Diagnosis | 73 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells) | -Size exclusion chromatography | DNA methylome | -Diagnosis | 75 |
| -GBM subtyping | ||||
| -Differential ultracentrifugation | -Treatment tailoring/monitoring | |||
| Patients serum | Size exclusion chromatography | miR-182-5p, miR-328-3p, miR-339-5p, miR-340-5p, miR-485-3p, miR-486-5p, and miR-543 | -Diagnosis | 82 |
| Patients serum | Exoquick | 1 small noncoding RNA (RNU6-1) and 2 microRNAs (miR-320 and miR-574-3p) | -Diagnosis | 83 |
| Patients serum | Exoquick | miR-21, miR-222 and miR-124-3p | -Diagnosis | 85 |
| -Treatment tailoring/monitoring |
Summary of the Reviewed EV-Associated Biomarkers with Potential Clinical Implications
| Source | Isolation method | Identified molecules | Potential clinical Applications | Ref. |
|---|---|---|---|---|
| Mouse plasma | Differential ultracentrifugation | EGFRvIII | -Diagnosis | 23 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro(cell lines; GBM stem cells) | Differential ultracentrifugation | CLIC1 | -Treatment tailoring | 24 |
| In vitro(cell lines), Mouse plasma, Patient’s plasma | Differential ultracentrifugation | Proteins: CAV1, IL8, PDGFs, MMPs, PTX3, CD26, PAI1, Transcripts: ADM, LOX, IGFPB 3, BCL2, BNIP3, NDRG1, PLOD2, PAI1 | -Diagnosis | 25 |
| -Treatment tailoring/monitoring | ||||
| -Assessment of GBM oxygenation status | ||||
| -Angiogenesis inhibitors | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | miR-148a-3p, miR-9-5p, miR-16-2-3p, miR-182-5p, miR-9-3p, miR-22-3p, miR-186-5p, miR-378e | -Diagnosis | 26 |
| -Treatment tailoring/monitoring | ||||
| -Angiogenesis inhibitors | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | 97 mRNAs (RP, OXPHOS, glycolysis) | -Diagnosis | 28 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM cell line) | Differential ultracentrifugation | miR-21 | -Diagnosis | 29 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | miR-451, miR-21, miR-15b, miR-146a, miR-223 | -Diagnosis | 30 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | Proteins involved in leukocyte recruitment and focal adhesion mechanisms | -Treatment tailoring/monitoring | 31 |
| -Agents blocking CSF-1R | ||||
| Patients’ serum | Differential ultracentrifugation | EGFR, EGFRvIII, and TGF-β | -Diagnosis | 33 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro(cell lines) | Differential ultracentrifugation | HSPs 90, 70, 60, and 27 | -Diagnosis | 34 |
| -Treatment tailoring/monitoring | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | PD-L1 (protein and DNA) | -Diagnosis | 35 |
| -Treatment tailoring/monitoring | ||||
| Patients’ serum/plasma | -anti-PD1 receptor blocking antibody | |||
| -Tumor subtyping | ||||
| Patients CSF | Differential ultracentrifugation | wtEGFR RNA, EGFRvIII RNA | -Diagnosis | 37 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells), Mouse serum, Patients’ serum | Differential ultracentrifugation | Sema3A | -Diagnosis | 40 |
| -Treatment tailoring | ||||
| -Blocking Sema3A or its receptor Neuropilin1 (NRP1) | ||||
| Patients CSF | Differential ultracentrifugation | miR-21 | -Diagnosis/staging | 45 |
| -Treatment tailoring | ||||
| Patients CSF | Differential ultracentrifugation | mutant and wild-type IDH1 mRNA | -Diagnosis | 46 |
| -Assessment of IDH1 mut status | ||||
| In vitro (GBM stem cells) | -Size exclusion chromatography | -proteins related to ribosome, proteasome, cell adhesion, and extracellular matrix-receptor interaction. | -Diagnosis | 49 |
| -Treatment tailoring/monitoring | ||||
| -Differential ultracentrifugation | ||||
| -EVs released by GSCs upon TMZ challenge display a specific enrichment in molecules involved in cell adhesion. | ||||
| Patients’ plasma | Differential ultracentrifugation | members of the complement and coagulation cascade and regulators of iron metabolism (vWF, APCS, C4B, AMBP, APOD, AZGP1, C4BPB, Serpin3, FTL, C3, and APOE) | -Diagnosis | 51 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells, Cell lines), Mouse plasma | Differential ultracentrifugation | EGFR, PDGFRα, PDPN, EphA2, EGFRvIII, IDH1 R132H, HSP90, CD4124, MHCII. | -Diagnosis | 54 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Patients’ plasma | ||||
| Patients’ serum | Differential ultracentrifugation | Survivin | -Diagnosis | 55 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Mouse plasma, Patients’ serum | Differential ultracentrifugation | IDH1G395A DNA | -Diagnosis | 64 |
| -Assessment of IDH1 mut status | ||||
| Patients’ serum | Differential ultracentrifugation | miRNA21, EGFRvIII mRNA Proteins: angiogenin, FGFα, IL-6, IL-8, TIMP-1, VEGF, and TIMP-2 | -Diagnosis | 65 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM Cell lines), Patients’ serum | Differential ultracentrifugation | EGFR protein and NLGN3, PTTG1 mRNA | -Diagnostic and prognostic markers | 66 |
| Patients’ serum | - | PTEN RNA | -Diagnosis/staging | 67 |
| Patients serum | Microfluidic platform | MGMT and APNG RNA | -Diagnosis | 68 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Mouse plasma | Differential ultracentrifugation | DNM3, p65, CD117, PTEN and p53 mRNA and protein | -Diagnosis | 69 |
| In vitro (GBM Cell lines) | Density gradient ultracentrifugation | ANXA1, IGF2R, ITGB1, PDCD6IP, ACTR3, CALR, IPO5, MVP, PSMD2 proteins | -Diagnosis | 86 |
| -Markers for more aggressive disease | ||||
| In vitro (GBM stem cells, Cell lines) | Density gradient ultracentrifugation | EVs proteins content mirrors the phenotypic signature of the respective GBM cells | -Diagnosis | 63 |
| -GBM subtyping | ||||
| Patients serum | Total Exosome Isolation Kit | EGFRvIII RNA | -Diagnosis | 73 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells) | -Size exclusion chromatography | DNA methylome | -Diagnosis | 75 |
| -GBM subtyping | ||||
| -Differential ultracentrifugation | -Treatment tailoring/monitoring | |||
| Patients serum | Size exclusion chromatography | miR-182-5p, miR-328-3p, miR-339-5p, miR-340-5p, miR-485-3p, miR-486-5p, and miR-543 | -Diagnosis | 82 |
| Patients serum | Exoquick | 1 small noncoding RNA (RNU6-1) and 2 microRNAs (miR-320 and miR-574-3p) | -Diagnosis | 83 |
| Patients serum | Exoquick | miR-21, miR-222 and miR-124-3p | -Diagnosis | 85 |
| -Treatment tailoring/monitoring |
| Source | Isolation method | Identified molecules | Potential clinical Applications | Ref. |
|---|---|---|---|---|
| Mouse plasma | Differential ultracentrifugation | EGFRvIII | -Diagnosis | 23 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro(cell lines; GBM stem cells) | Differential ultracentrifugation | CLIC1 | -Treatment tailoring | 24 |
| In vitro(cell lines), Mouse plasma, Patient’s plasma | Differential ultracentrifugation | Proteins: CAV1, IL8, PDGFs, MMPs, PTX3, CD26, PAI1, Transcripts: ADM, LOX, IGFPB 3, BCL2, BNIP3, NDRG1, PLOD2, PAI1 | -Diagnosis | 25 |
| -Treatment tailoring/monitoring | ||||
| -Assessment of GBM oxygenation status | ||||
| -Angiogenesis inhibitors | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | miR-148a-3p, miR-9-5p, miR-16-2-3p, miR-182-5p, miR-9-3p, miR-22-3p, miR-186-5p, miR-378e | -Diagnosis | 26 |
| -Treatment tailoring/monitoring | ||||
| -Angiogenesis inhibitors | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | 97 mRNAs (RP, OXPHOS, glycolysis) | -Diagnosis | 28 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM cell line) | Differential ultracentrifugation | miR-21 | -Diagnosis | 29 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | miR-451, miR-21, miR-15b, miR-146a, miR-223 | -Diagnosis | 30 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | Proteins involved in leukocyte recruitment and focal adhesion mechanisms | -Treatment tailoring/monitoring | 31 |
| -Agents blocking CSF-1R | ||||
| Patients’ serum | Differential ultracentrifugation | EGFR, EGFRvIII, and TGF-β | -Diagnosis | 33 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro(cell lines) | Differential ultracentrifugation | HSPs 90, 70, 60, and 27 | -Diagnosis | 34 |
| -Treatment tailoring/monitoring | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells) | Differential ultracentrifugation | PD-L1 (protein and DNA) | -Diagnosis | 35 |
| -Treatment tailoring/monitoring | ||||
| Patients’ serum/plasma | -anti-PD1 receptor blocking antibody | |||
| -Tumor subtyping | ||||
| Patients CSF | Differential ultracentrifugation | wtEGFR RNA, EGFRvIII RNA | -Diagnosis | 37 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells), Mouse serum, Patients’ serum | Differential ultracentrifugation | Sema3A | -Diagnosis | 40 |
| -Treatment tailoring | ||||
| -Blocking Sema3A or its receptor Neuropilin1 (NRP1) | ||||
| Patients CSF | Differential ultracentrifugation | miR-21 | -Diagnosis/staging | 45 |
| -Treatment tailoring | ||||
| Patients CSF | Differential ultracentrifugation | mutant and wild-type IDH1 mRNA | -Diagnosis | 46 |
| -Assessment of IDH1 mut status | ||||
| In vitro (GBM stem cells) | -Size exclusion chromatography | -proteins related to ribosome, proteasome, cell adhesion, and extracellular matrix-receptor interaction. | -Diagnosis | 49 |
| -Treatment tailoring/monitoring | ||||
| -Differential ultracentrifugation | ||||
| -EVs released by GSCs upon TMZ challenge display a specific enrichment in molecules involved in cell adhesion. | ||||
| Patients’ plasma | Differential ultracentrifugation | members of the complement and coagulation cascade and regulators of iron metabolism (vWF, APCS, C4B, AMBP, APOD, AZGP1, C4BPB, Serpin3, FTL, C3, and APOE) | -Diagnosis | 51 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM stem cells, Cell lines), Mouse plasma | Differential ultracentrifugation | EGFR, PDGFRα, PDPN, EphA2, EGFRvIII, IDH1 R132H, HSP90, CD4124, MHCII. | -Diagnosis | 54 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Patients’ plasma | ||||
| Patients’ serum | Differential ultracentrifugation | Survivin | -Diagnosis | 55 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Mouse plasma, Patients’ serum | Differential ultracentrifugation | IDH1G395A DNA | -Diagnosis | 64 |
| -Assessment of IDH1 mut status | ||||
| Patients’ serum | Differential ultracentrifugation | miRNA21, EGFRvIII mRNA Proteins: angiogenin, FGFα, IL-6, IL-8, TIMP-1, VEGF, and TIMP-2 | -Diagnosis | 65 |
| -Treatment tailoring/monitoring | ||||
| In vitro (GBM Cell lines), Patients’ serum | Differential ultracentrifugation | EGFR protein and NLGN3, PTTG1 mRNA | -Diagnostic and prognostic markers | 66 |
| Patients’ serum | - | PTEN RNA | -Diagnosis/staging | 67 |
| Patients serum | Microfluidic platform | MGMT and APNG RNA | -Diagnosis | 68 |
| -Treatment tailoring/monitoring | ||||
| -Predict response to therapy | ||||
| Mouse plasma | Differential ultracentrifugation | DNM3, p65, CD117, PTEN and p53 mRNA and protein | -Diagnosis | 69 |
| In vitro (GBM Cell lines) | Density gradient ultracentrifugation | ANXA1, IGF2R, ITGB1, PDCD6IP, ACTR3, CALR, IPO5, MVP, PSMD2 proteins | -Diagnosis | 86 |
| -Markers for more aggressive disease | ||||
| In vitro (GBM stem cells, Cell lines) | Density gradient ultracentrifugation | EVs proteins content mirrors the phenotypic signature of the respective GBM cells | -Diagnosis | 63 |
| -GBM subtyping | ||||
| Patients serum | Total Exosome Isolation Kit | EGFRvIII RNA | -Diagnosis | 73 |
| -Treatment tailoring/monitoring | ||||
| -Anti-EGFRvIII treatment | ||||
| -Vaccine | ||||
| In vitro (GBM stem cells) | -Size exclusion chromatography | DNA methylome | -Diagnosis | 75 |
| -GBM subtyping | ||||
| -Differential ultracentrifugation | -Treatment tailoring/monitoring | |||
| Patients serum | Size exclusion chromatography | miR-182-5p, miR-328-3p, miR-339-5p, miR-340-5p, miR-485-3p, miR-486-5p, and miR-543 | -Diagnosis | 82 |
| Patients serum | Exoquick | 1 small noncoding RNA (RNU6-1) and 2 microRNAs (miR-320 and miR-574-3p) | -Diagnosis | 83 |
| Patients serum | Exoquick | miR-21, miR-222 and miR-124-3p | -Diagnosis | 85 |
| -Treatment tailoring/monitoring |
Discussion and Conclusions
To date, the lack of reliable and approved biomarkers significantly hinders glioma clinical practice. There is an urgent need for liquid biopsy medium and biomarkers allowing diagnosis, patient stratification, personalization of treatment, assessment of therapeutic responses, and identification of relapse. In this context, EVs represent an interesting candidate to answer this still unmet critical clinical need. EVs are released by GBM cells and are traceable in the blood and CSF, where they can be isolated and characterized. Notably, their informative power is much greater than that of other biomarkers because they can be read on different levels, exploiting both EV concentration and cargo. The concentration of EVs can act as biomarker information on tumor presence and status, differential diagnosis (ie, versus solitary brain metastasis), and monitoring of treatment response, whereas the molecular cargo provides a window into tumor biology allowing the study of multiple tumor molecules (eg, nucleic acids, proteins, lipids) at the same time. The possibility to study EVs at different levels makes them ideal candidates for developing a liquid biopsy approach to tailor the treatment and to finally cope with the protean and heterogeneous nature of GBM. In fact, a liquid biopsy approach based on EVs owns several advantages. First, EVs can be collected through minimally invasive procedures, such as blood sampling, whereas biopsy and surgery are inherently invasive and burdened with nonnegligible morbidity and mortality risk. Second, EVs are derived from all viable cells of the tumor, thus representing a more complete depiction of whole-tumor heterogeneity. Third, a liquid biopsy EV-based biomarker approach would allow patients to be followed in an outpatient setting, in contrast to the current approach based on hospitalization, multiple MRI scans, and surgery. Fourth, close monitoring of EV concentration and molecular cargo will allow better evaluation of the response to treatment obtaining a “real-time” whole-tumor picture of GBM molecular setup as needed during patient care. This latter advantage represents a substantial improvement compared to biopsy and surgery, which provide a partial image at a single time point of an ever-changing tumor. Despite these enormous advantages, several challenges must be addressed. First, extensive inter- and intratumoral heterogeneity inherent to GBM has dictated the absence of effective and reliable biomarkers capable of identifying GBM cells and GBM-derived EVs. Similarly, EVs derived from biological samples of patients are highly heterogeneous, encompassing different biological entities varying in size, cargo, and function. In this regard, the nomenclature and isolation techniques require standardization to allow reliable cross-study comparability. Additionally, the choice of sample type to collect EVs (blood vs. CSF) represents another challenge that can impact the procedure invasiveness, repeatability, and reliability of the results. Technically, sampling methods (eg, preprocessing and storage) and technologies for isolating EVs greatly influence the amount of starting material as well as its purity and yield, thus affecting subsequent analyses. The analytical phase of EV-based liquid biopsy testing also requires standardization to reduce the variability and clinical validation to allow implementation in clinical practice. Finally, the required technologies are still demanding in time and resources, being costly and requiring highly trained personnel.
In this era of precision medicine, the multilayered informative power of GBM-derived EVs would play a pivotal role by providing a complete, continuous picture of GBM presence and molecular assets without the risks and limitations to patients of conventional surgical biopsy. The combined assessment of EV number and cargo, which are known to be informative by themselves as demonstrated by the literature, can capture GBM features necessary for diagnosis, tumor-evolution/treatment-response monitoring, and the development of genotype-directed therapies based on actionable EV molecular aberrations (Figure 2). In the near future, tight cooperation between researchers, clinicians, and technologists will succeed in exploiting circulating GBM-associated EVs for improved care pathways and novel therapeutic tools for an effective patient-tailored approach.
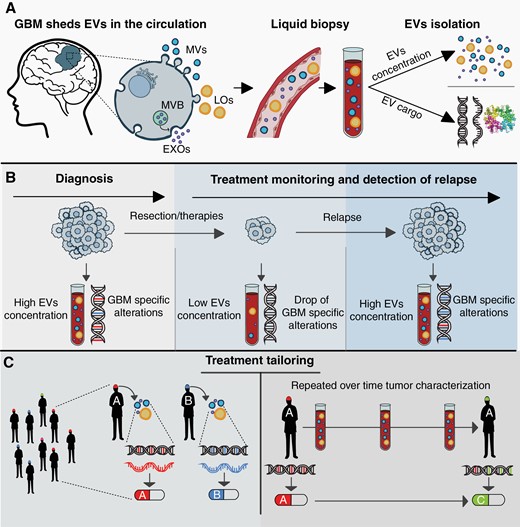
EVs: the key for precision medicine in GBM. (A) GBM-associated EVs (EXOs in purple, MVs in blue, and LOs in yellow) can be isolated from peripheral blood and used as a platform for liquid biopsy. EVs possess multilayered information constituted by their concentration and molecular cargo, spanning DNA and RNA to proteins and lipids. (B) The concentration and cargo of EVs indicate GBM presence allowing diagnosis, monitoring of tumor response to therapy, and identifying relapse. (C) An EV-based liquid biopsy approach may enable the characterization of the whole-GBM molecular set-up. Tumor characterization at baseline would allow treatment personalization (left panel), and serially, during treatment, it would allow treatment tailoring, thus maximizing its efficacy and tackling GBM molecular dynamicity (right panel).
Funding
This work has been supported by Fondazione Regionale per la Ricerca Biomedica (Regione Lombardia), Project ERAPERMED2020-104, GA 779282
Conflict of interest statement. The authors declare no conflicts of interest.
Authorship statement. M. Del Bene and D. Osti designed the review; M. Del Bene, D. Osti and S. Faletti wrote the manuscript; G. V. Beznoussenko performed electron microscopy; G. Pelicci and F. DiMeco supervised the review; all authors approved the submitted manuscript.
References
Author notes
These authors have contributed equally to this work.



{kind=link}
{kind=link}