





Abstract
H3K27M-mutant diffuse midline glioma (DMG) is a lethal brain tumor that usually occurs in children. Despite advances in our understanding of its underlying biology, efficacious therapies are severely lacking.
We screened a library of drugs either FDA-approved or in clinical trial using a library of patient-derived H3K27M-mutant DMG cell lines with cell viability as the outcome. Results were validated for clinical relevance and mechanistic importance using patient specimens from biopsy and autopsy, patient-derived cell lines, inhibition by gene knockdown and small molecule inhibitors, and patient-derived xenografts.
Kinase inhibitors were highly toxic to H3K27M-mutant DMG cells. Within this class, STAT3 inhibitors demonstrated robust cytotoxic activity in vitro. Mechanistic analyses revealed one form of activated STAT3, phospho-tyrosine- 705 STAT3 (pSTAT3), was selectively upregulated in H3K27M-mutant cell lines and clinical specimens. STAT3 inhibition by CRISPR/Cas9 knockout, shRNA or small molecule inhibition reduced cell viability in vitro, and partially restored expression of the polycomb repressive mark H3K27me3, which is classically lost in H3K27M-mutant DMG. Putative STAT3-regulated genes were enriched in an H3K27M-knockout DMG cell line, indicating relative gain of STAT3 signaling in K27M-mutant cells. Treatment of patient-derived intracranial xenografts with WP1066, a STAT3 pathway inhibitor currently in clinical use for pediatric brain tumors, resulted in stasis of tumor growth, and increased overall survival. Finally, pSTAT3(Y705) was detected in circulating plasma extracellular vesicles of patients with H3K27M-mutant DMG.
STAT3 is a biologically relevant therapeutic target in H3K27M-mutant DMG. STAT3 inhibition should be considered in future clinical trials.
STAT3(Y705) is activated in H3K27M-mutant DMG.
STAT3 inhibition results in stasis of intracranial patient-derived H3K27M-mutant xenografts.
STAT3 is detectable in the plasma of H3K27M-mutant DMG patients.
H3K27M-mutant diffuse midline glioma (DMG) is a rare but deadly tumor that most often occurs in childhood; most children die within one year of diagnosis. While our understanding of the epigenetic basis of this tumor has expanded rapidly, this has not yet translated into efficacious, targeted therapy. We screened a library of drugs either FDA-approved for clinical use or in clinical trial and identified STAT3 inhibition as a potentially efficacious therapeutic strategy in H3K27M-mutant DMG. CRISPR/Cas9-mediated knockout of STAT3 resulted in growth stasis in vitro, and a small molecule inhibitor of STAT3 currently in clinical trial halted tumor growth and prolonged survival in orthotopic xenograft models. While JAK/STAT signaling is ubiquitous in many cancers, we found STAT3 is significantly activated in H3K27M-mutant DMG and is detectable in the plasma of patients harboring these tumors, indicating STAT3 is not only a therapeutic target but also a potential biomarker for this tumor.
H3K27M-mutant diffuse midline glioma (DMG), formerly known as diffuse intrinsic pontine glioma (DIPG), is a lethal CNS cancer with a dismal prognosis.1 While some cases occur in young adults, most occur in children, and the median overall survival is less than one year from diagnosis.2 The current mainstay of therapy is palliative external beam radiotherapy, and there are no long-term survivors. While clinical trials are underway using a range of different therapeutic approaches including small molecular inhibitors, immunotherapy, and convection-enhanced delivery, no treatment has demonstrated a benefit to overall or progression-free survival in this tumor.3 The extremely poor prognosis and lack of efficacious, targeted therapy demand a novel approach to its treatment.
Basic research over the past decade has dramatically expanded our understanding of the genetic and epigenetic underpinnings of this tumor. Most midline gliomas of childhood harbor missense H3F3A or HIST1H3B mutations resulting in a lysine-to-methionine substitution at the 27th position (K27M),4,5 resulting in a net loss of H3K27 trimethylation (H3K27me3) and a reprogramming of the cell’s epigenetic landscape.6 While numerous mechanistic studies have cemented the importance of the H3K27M mutation in the pathogenesis of DMG, the field has not yet been able to leverage this knowledge into improved outcomes for patients.
The purpose of this study was to identify molecular pathways that are both pathologically relevant and therapeutically targetable in H3K27M-mutant DMG. We screened a panel of drugs currently in clinical use for efficacy against a library of H3K27M-mutant and H3-wildtype patient-derived cell lines and identified STAT3 as a druggable target in H3K27M-mutant DMG. Treatment with a STAT3 inhibitor resulted in growth stasis and increased survival in intracranial H3K27M-mutant patient-derived xenografts. Moreover, STAT3 was detectable in the extracellular vesicles from plasma of patients with H3K27M-mutant DMG but not normal donors. Our results illustrate a previously underappreciated role for STAT3 and outline its potential role as a therapeutic target and biomarker for this tumor.
Materials and Methods
Cell Lines and Culture
For all human tissue studies and patient-derived cell lines, informed consent and Institutional Review Board (IRB) approval were obtained. Details regarding cell lines can be found in Supplementary Table 1. All cell lines were validated by short tandem repeat (STR) DNA fingerprinting annually and tested for Mycoplasma every 3 months. All H3K27M-mutant cell lines were validated for K27M-mutant histone expression using Western blot and Sanger sequencing over the same interval. All patient-derived tumor cell lines were maintained in cell-line appropriate media, the details of which are provided in Supplementary Table 2.
Drug Screen
A panel of 120 drugs, either FDA-approved or in clinical trial was obtained from Selleckchem, (Houston, TX). All drugs in this study were dissolved in 100% dimethylsulfoxide (DMSO, Sigma-Aldrich) and stored as 10 mM stock. For the preliminary drug screen, cells were plated with culture media in 96-well clear bottom black microplates (Corning Costar) at a density of 5 × 103 cells per well and cultured overnight at 37° C with 5% CO2. The next day, cells were treated with either vehicle (DMSO) or one three drug concentrations (0.1 μM, 1 μM, and 10 μM in 0.5% DMSO) in triplicate for 72 h. The drugs were given to the cells at a final concentration of 0.5% DMSO. Cell viability was measured using the CellTiter-Blue Cell Viability Assay (Promega) according to the manufacturer’s instructions and an Infinite M200 Pro microplate reader (Tecan). The potency (IC50) of each drug was calculated by nonlinear least-squares curve-fitting using Prism 6 (GraphPad). Selected drugs were further assessed using a 13-point concentration range with 2-fold dilutions in triplicate to confirm key findings from the drug screen.
Clinical Specimen Gene Expression Analysis
Normalized microarray and overall survival data were obtained from PedcBioPortal (pedcbioportal.org). Microarray analysis was performed using R and Bioconductor. MAS5.0-P/A normalized datasets of childhood DIPG (N = 27; GSE265767) were compared to normal prefrontal cortex (N = 44; GSE135648–11). Volcano plots illustrate genes with FDR < 0.05 using MATLAB 2016a. STAT3-related genes were identified using BioGRID and STRING.
Protein Expression, Construct Transduction, and Cell Viability Experiments
Details regarding the Western blot protocol, lentiviral transduction, gene editing, cell viability, and clonogenic assays are presented in the Supplementary Methods.
RNA Extraction and Sequencing (RNA-Seq)
Whole RNA was extracted using RNeasy Plus micro kit (QIAGEN). RNA expression experiments were performed in triplicate. RNA quality, library preparation, and sequencing were performed by Novogene (Beijing, China). Input RNA sample integrity, purity, and quantitation were validated using NanoPhotometer spectrophotometer (IMPLEN), 1% agarose gel electrophoresis, and Bioanalyzer 2100 (Agilent Technologies). One microgram of RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext UltraTM RNA Library Prep Kit for Illumina (New England Biolabs). Following library preparation, cDNA fragments of 150–200 bp length were size-selected using AMPure XP magnetic beads (Beckman Coulter). Final library quality was assessed using an Agilent Bioanalyzer 2100. The clustering of the index-coded samples was performed on an Illumina Novaseq 6000 sequencer according to the manufacturer’s instructions. After cluster generation, the libraries were sequenced on the same machine and paired-end reads were generated.
Following quality control using FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), reads were trimmed using fastx trimmer (http://hannonlab.cshl.edu/fastx_toolkit/). Trimmed reads were mapped to hg38 (patch 13) (http://genome.ucsc.edu/ENCODE/releaseLog.html) and counted using RNA STAR v2.5.12 Differential gene expression analysis with batch normalization was performed using edgeR (v3.32.1)13 with a false discovery rate (FDR) cutoff of <0.05 defining differential expression between conditions. Pathway and gene set enrichment were performed using Gene Ontology (geneontology.org), Kyoto Encyclopedia of Genes and Genomes (KEGG; https://www.genome.jp/kegg/pathway.html), and Gene Set Enrichment Analyses (GSEA).14,15 Volcano plots were created using VolcaNoseR.16
Chromatin Immunoprecipitation with High-throughput Sequencing (ChIP-Seq)
For ChIP-Seq studies, DIPGXVII cells were treated with 2 μM WP1066 or vehicle (0.5% DMSO) for 24 h. Cells were then fixed in 1% formaldehyde for 20 min, and fixation was quenched with 125 mM glycine. Nuclei were subsequently extracted and lysed as described previously.17 Chromatin was sonicated using a Diagenode Bioruptor (Diagenode, Denville, NJ) to an average fragment length of 300–500bp. Immunoprecipitation was performed using 2 μg of target-specific antibody (Diagnode) overnight at 4° C followed by a two-hour incubation with protein A/G-magnetic beads (Pierce Biotechnology, Thermo Scientific). DNA was subsequently obtained using phenol–chloroform extraction and ethanol precipitation. Library preparation was performed using the Diagenode Microplex v2 kit followed by size selection using sparQ magnetic beads (QuantaBio). Paired-end 50-bp length DNA libraries were sequenced using an Illumina HiSeq4000. Following quality control, trimmed reads were mapped to hg38 (patch 13) using bowTie2 v1.2.2.18 Peak calling was performed using the broad peaks function of DeepTools MACS219 using a q-value cutoff of 0.1. Read pileups were visualized using DeepTools bamCoverage and plotHeatmap functions.20
Patient-derived Xenografts
Diffuse midline glioma cell lines (DIPGXIII, PED17, SF8628, SF8628-B23) were transduced with a lentiviral vector expressing green fluorescent protein (GFP) and luciferase (gift from Dr. Michelle Monje, Stanford) as previously described.21 All animals were handled in accordance with IACUC guidelines and the Mayo Clinic Institutional Committee for Animal Research approved all the experiments. Tumor engraftment using cultured cells was performed as previously described.21 Briefly, cells were placed in single-cell suspension and injected stereotactically into the pons of 6 to 7-week-old female Hsd:athymic nude Foxn1nu mice (Envigo) or RNU nude rats (Charles River) using 300 000 cells in 3 μL of sterile PBS through a 0.5 mm burr hole (coordinates: 1 mm inferior to the lambdoid suture, 1 mm lateral to the mid-sagittal plane, and 4 mm in depth) using a 26-gauge (2 mm, point style AS) Hamilton syringe (Bonaduz, GR, Switzerland). As all cell lines used for xenografting in this study were derived from female patients, female mice were used for all PDXs to preserve the tumor microenvironment as much as possible. In vivo tumor growth was monitored using IVIS bioluminescence imaging acquired using the IVIS-2000 Imaging System (Xenogen Corporation) and analyzed by LivingImage 4.3 software. Tumor growth was assessed between groups using a two-tailed t test.
For drug treatment studies, after tumor presence was confirmed by bioluminescence imaging, animals were randomized to control and treatment groups such that each group had equivalent distribution of tumor sizes at the beginning of the study. Administration of WP1066 or DMSO was performed 6 weeks post tumor implantation with oral gavage 3 times per week (M, W, F) at 20 mg/kg. WP1066 was dissolved in a mixture of DMSO: PEG300 (20:80) for administration. Animals were monitored daily and euthanized at indication of progressive neurologic deficit or if found in a moribund condition. For PED17 orthotopic xenografts, administration of WP1066 or DMSO was performed six weeks post tumor implantation with oral gavage three times per week (M, W, F) at 20 mg/kg. For DIPGXIIIp xenografts, WP1066 treatment was performed 5 days post tumor implantation via oral gavage five times per week (5 days on, 2 days off) at 40 mg/kg. Treatment was continued until the study endpoint was reached.
For studies utilizing an ALZET pump (Durect), animals were engrafted with DIPGXIIIp cells, and the pump was implanted eight days following engraftment. 200 uM WP1066 was dissolved in 1% DMSO in PBS followed by 5–10 min of sonication using a bath sonicator. The 7-day ALZET pump (Model 2001) was connected to an ALZET cannula (ALZET brain infusion kit 1), filled, and primed with WP1066 or vehicle (1% DMSO in PBS) prior to installation. The pump was then inserted into a subcutaneous pocket created between the scapulae. The ALZET cannula was installed 4 mm below the pedestal through the same burr hole as tumor engraftment. Dental glue (Ivoclar Vivadent) was used to affix the ALZET cannula onto the skull. Pumps were removed on Day 8 post pump implant per manufacturer’s recommendation.
Immunohistochemical Staining
Animals were euthanized with cervical dislocation and brains were removed, fixed in 4% paraformaldehyde at room temperature overnight. The brains were then embedded in paraffin and sections in the coronal plane using a microtome. Hematoxylin and eosin (H&E) stains were performed according to standard procedures. For immunohistochemistry, 5-μm paraffin sections were used for staining were dewaxed in xylene and rehydrated in ethanol. Antigen retrieval was performed by steaming slides in either citrate buffer (10 mM tri-sodium citrate, 0.05% Tween 20, pH 6.0) or in Tris buffer (100 mM Tris–HCl, 5% urea w/v, pH 9.0, pY-STAT3). Sections were then washed in Tris-buffered saline (TBS), blocked with 10% normal goat serum (NGS) in TBS for 60 min at room temperature. Primary antibodies were then diluted in TBS with 2% NGS and 0.5% Triton X-100 for 60 min at room temperature. Detailed information regarding antibodies used for IHC can be found in Supplementary Table 3. Dilution buffer was used in lieu of primary antibody for tissue-specific negative controls. The VECTASTAIN Elite ABC kit (VECTOR Laboratories) containing biotinylated secondary antibody was then applied to the slides according to manufacturer’s instruction. For visualization, the slides were developed using SignalStain DAB Substrate Kit (Cell Signaling) according to manufacturer’s instruction, counterstained with hematoxylin and mount with permount (Fisher Scientific). Images were acquired using the EVOS FLc Imaging System (Thermo Fisher Scientific) and are presented at a magnification of 40×. Cell quantification was used using the Cell Counter plugin for ImageJ (National Institutes of Health). Five random fields including a total of 300–500 cells were captured for each antibody. Results are presented as the percentage of positive cells versus the entire counted cell population.
Exosome Isolation and Analysis
Patient plasma samples were collected at Mayo Clinic. IRB approval and informed consent were obtained. Plasma exosomes were isolated using density gradient ultracentrifugation following procedures described from Cumba-Garcia et al.22 Briefly, plasma was thawed and mixed at a ratio of 1:1 with 50% OptiPrep solution (45 ml of OptiPrep Density Gradient Medium, Sigma-Aldrich No. D1556, and 9 ml of OptiPrep diluent). 11 ml of 10% OptiPrep solution (4.4 ml of 50% OptiPrep solution, 17.6 ml of buffer A [100 ml of 2.5 M sucrose, 0.34 g (1 mM) of EDTA, 3.58 g (20 mM) of Tricine at pH 7.8, 900 ml of water]) was carefully layered onto the homogenized solution inside an ultra-clear centrifuge tube (Beckman Coulter). Samples were centrifuged at 100 000 g (Beckman Coulter Optima LE-80K ultracentrifuge) for 90 min. The top layer (10 ml) was collected and subject to another round of ultracentrifugation at 100 000 g for 16 h. The supernatant was discarded, and the exosome pellet was resuspended in a total volume of 100 μL for analysis.
Exosomes collected from cell lines were harvested from the culture media followed by centrifugation of the culture media at 3000 rpm for 10 min to remove cellular debris. The supernatant was subsequently centrifuged for 16 h at 100 000 g to concentrate exosomes. The exosome pellet was resuspended in a total volume of 100 μL–200 μL for analysis.
Results
Inhibitors of Kinase Signaling are Highly Potent Against Patient-derived H3K27M-Mutant DMG Cells
To identify potential therapeutic targets in H3K27M-mutant DMG, we performed a cell viability screen using 120 drugs either FDA-approved for clinical use or currently in clinical trial (Figure 1A and B; 70 drugs with anti-tumor cell activity shown). We screened at least three separate H3K27M-mutant (PED8, SF7761, SU-DIPG IV, SU-DIPG XIII, and SU-DIPG XVII) and three H3-wildtype high-grade glioma patient-derived cell lines including two derived from adult glioblastoma (BT114 and BT116) and one from a pediatric glioblastoma (SF9427). Consistent with prior literature on proteasome inhibition in DMG,23 bortezomib was universally efficacious against all tested cell lines but is known to have limited blood–brain barrier penetration. As a class, kinase inhibitors demonstrated good efficacy against both H3K27-mutant and H3-wildtype cell lines, including the multi-receptor tyrosine kinase inhibitors (RTKIs) ponatinib and sorafenib. While fewer JAK/STAT3 inhibitors were tested in this screen, many demonstrated reliable efficacy against all cell lines tested, indicating a previously underappreciated role for this convergent pathway in glioma.
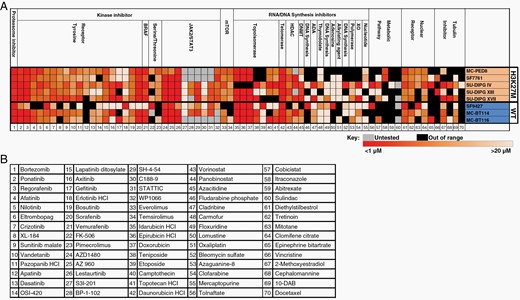
Cell viability screen for efficacious, clinically useable drugs against patient-derived H3K27M-mutant cell lines. (A) Heatmap intensity indicates the absolute IC50 with the range shown below the graph. Black indicates an IC50 outside the measurable range (ie, no efficacy), and gray indicates the drug was not tested for that cell line. Each drug (B) was tested in triplicate with 2–3 independent experiments (n = 6–9) in each cell line.
STAT3 (Y705) Activation is Prominent in H3K27M-mutant DMG
STAT3 has previously been implicated in H3K27M-DMG cell proliferation, clinical invasiveness, and therapeutic resistance.24,25 To determine the biological relevance of STAT3 to H3K27M-mutant DMG, we first leveraged previously published gene expression datasets to determine the relative expression of STAT3 in these patients and its impact on survival. In a dataset of 211 pediatric high-grade gliomas including H3K27M-mutant DMG, high relative STAT3 expression was associated with a significantly shorter overall survival (Figure 2A, log-rank P = 0.013). Moreover, in a set 27 pathology-confirmed DIPG specimens from the pre-K27M molecular era, STAT3 emerged as one of the most differentially upregulated genes in tumor specimens versus normal brain with three-fold higher expression in DIPG (Figure 2B and C).
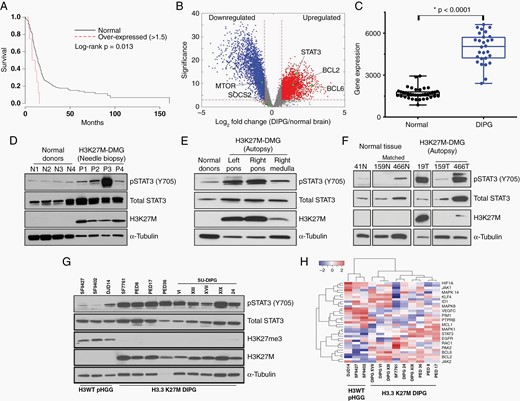
STAT3 is activated at Y705 in H3K27M-mutant DMGs. (A) Kaplan–Meier curve illustrating overall survival of patients with pediatric high-grade glioma (pHGGs) and relatively high STAT3 expression (Z ≥ 1.5× normalized mean) versus all other pHGGs (N = 211). (B and C) Differential gene expression analysis of diffuse intrinsic pontine gliomas (DIPGs); DIPG is kept as the disease designation as data were obtained in the pre-K27M era. (D–F) Western blot analysis of H3K27M-mutant DMG tumor specimens or nonneoplastic control tissue, as indicated. Two patients (159 and 466) had matched normal tissue for analysis. (G) Phospho-STAT3 (pSTAT3) expression in patient-derived H3-wildtype and H3K27M-mutant cell lines. (H) Heatmap of RNA-Seq normalized reads of known genes associated with STAT3 signaling.
We next assessed the expression of total and biologically active STAT3 (phospho-STAT3, pSTAT3) in a library of human DMG specimens and patient-derived cell lines (details in Supplementary Table 4). While total STAT3 protein expression was observed in both needle biopsy specimens of H3K27M-mutant DMG and normal cortical brain, high pSTAT3 (Y705) expression was specifically observed in tumor specimens (Figure 2D, Figure S1). We next validated this observation in autopsy specimens including three tumors with matched normal brain tissue (Figure 2E and F). In each case, activated pSTAT3 (Y705) was expressed and restricted to tumor-containing brain.
Given that STAT3 inhibitors displayed some efficacy against H3-wildtype HGG cell lines in our initial drug screen, we next set out to determine the specificity of STAT3 activation to H3K27M-mutant DMG (Figure 2G). In a panel of 12 patient-derived cell lines including three pediatric HGGs, pSTAT3 (Y705) expression was consistently higher in cells derived from H3K27M-mutant tumors. To determine relative activity of the STAT3 signaling pathway in each of these cell lines, we performed RNA-Seq and analyzed expressed transcripts for abundance of genes known to be associated with STAT3 signaling designated in the STRING database. Hierarchical clustering analysis discriminated H3-wildtype and H3K27M-mutant cell lines based on expression of protein-coding genes known to be associated with STAT3 (Figure 2H). These data indicate the pattern of STAT3 activation may be unique to glioma subtype and highlight a potential role for pSTAT3 (Y705) in H3K27M-mutant DMG.
H3F3A K27M Mutation Increases STAT3 Activation and Downstream Signaling
To further define the mechanistic role of the H3K27M mutation and STAT3 activation, we utilized CRISPR-Cas9 gene editing to create an H3K27M-knockout (H3K27M-KO) cell line (SF8628-B23), which is derived from the H3K27M-mutant DMG cell line SF8628. H3K27M knockout significantly reduced STAT3 activation as measured by baseline Y705 expression despite having no impact on total STAT3 expression (Figure 3A). H3K27M-KO cell lines remained viable in vitro but demonstrated significantly reduced proliferative capacity (Figure 3B).

H3F3A K27M mutation knockout reduces activated STAT3 levels and downstream signaling pathways. (A) Western blot of pSTAT3 (Y705) in K27M-mutant and H3K27M-KO SF8628 cells, as indicated. (B) H3K27M-KO SF8628-B23 cells remain viable in vitro but proliferate more slowly than K27M-mutant cells. (C) Volcano plot illustrating differentially expressed genes between H3K27M-KO and H3K27M-mutant cells. (D–G) Gene set enrichment analysis (GSEA) of H3K27M-mutant versus H3K27M-KO cells using gene sets including genes in the JAK-STAT signaling pathway (D), known STAT3-associated genes (E), genes with putative STAT3-responsive elements within 4kb of the transcription start site (TSS, F) and genes known to be promoter-associated with STAT3 in ChIP-Seq data in other models (G). GSEA results are presented as normalized enrichment scores (NES), P values, and false discovery rate (FDR)-corrected P values.
To determine the transcriptomic changes following knockout of the mutant H3F3A gene, we performed RNA-Seq on H3K27M and H3K27M-KO SF8628 cells. H3K27M knockout was associated with a significant transcriptomic shift with 7538 genes significantly differentially regulated between conditions (FDR < 0.05, Figure 3C). Surprisingly, compared to H3K27M-KO cells, we observed significant enrichment for the JAK/STAT signaling pathway in K27M-mutant cells, with this pathway ranked 3rd of 42 significantly enriched (P < .05) GSEA-indexed KEGG pathways (Figure 3D; normalized enrichment score [NES] 1.94, P < .001, FDR = 0.043, family-wise error rate [FWER] = 0.044; Supplementary Table 6). To investigate this further, we performed GSEA using a set of genes known to be associated with STAT3 signaling in the String functional protein association network database (string-db.org) and in previously published literature. Concordant with our previous finding, we found significant enrichment in STAT3-associated genes in K27M-mutant compared to H3K27M-KO cells (Figure 3E; NES = 2.17, P < .001, FDR = 0.113, FWER < 0.001).
Given that STAT3 acts at promoter-proximal regions as a transcription factor, we next sought to determine whether K27M-mutant cells are enriched for target genes of STAT3. Of 433 enriched GSEA-indexed gene sets associated with putative transcription factor binding sites, a STAT3-associated gene set was the most enriched gene set in K27M-mutant cells (Figure 3F, NES = 2.26, P < .001, FDR = 0.044, FWES < 0.001). To verify this finding, we used ChIPAtlas26 to leverage previously published STAT3 ChIP-Seq data across multiple cell types. In a set of 500 genes with the highest STAT3 ChIP-Seq peak average within 1kb of the transcription start site, we observed enrichment in H3K27M-mutant versus H3K27M-KO SF8628 cells (Figure 3G, NES = 1.35, P < .001, FDR = 0.113, FWES = 0.058). Taken together, these analyses indicate STAT3 signaling is upregulated in H3K27M-mutant DMG cells.
STAT3 Inhibition Reduces Cell Viability and Restores H3K27me3 In Vitro
To determine the potential therapeutic relevance of STAT3 inhibition in H3K27M-mutant DMG, we utilized CRISPR/Cas9 to genetically ablate STAT3 in SF8628 and DIPGXVII cell lines. SF8628 STAT3-knockout (KO) clones demonstrated significantly reduced proliferative capacity compared to nontargeting gRNA controls (Figure 4A and B). Of all transduced DIPGXVII cells, only two clones remained viable in vitro and did not survive passaging, underscoring the importance of STAT3 in H3K27M-DMG cell viability. Expanding on these findings, we additionally generated an inducible STAT3-knockdown model in these cell lines to address the confounding issue of cell dependency on STAT3 in clone selection. Consistent with our knockout data, inducible STAT3 knockdown demonstrated a statistically significant reduction in cellular proliferation in both cell lines (Figure S2).
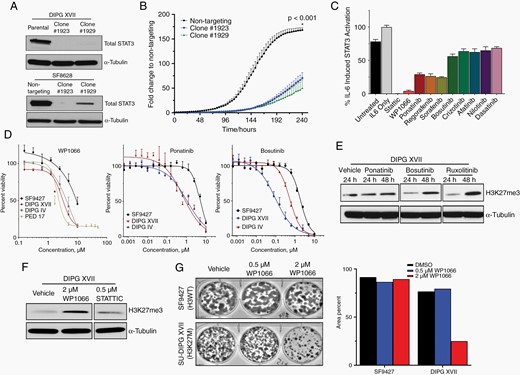
STAT3 inhibition reduces H3K27M-mutant DMG cell viability and proliferation. (A) Western blot demonstrating CRISPR/Cas9-mediated STAT3 knockout. (B) Incucyte proliferation assay of H3K27M-mutant cell line SF8628 clones following knockout of STAT3. (C) Relative anti-STAT3 activity of selected kinase/STAT3 inhibitors using an IL-6 induction assay. (D) Cell viability of indicated cell lines at escalating doses of kinase/STAT3 inhibitors. SF9427 is H3-wildtype; DIPGXVII, DIPGIV, and PED17 are H3K27M-mutant. (E, F) Relative expression of H3K27me3 by Western blot following drug treatment. (G) Clonogenic assay of H3-wildtype SF9427 and H3K27M-mutant DIPGXVII cells following treatment with WP1066.
To determine how to best translate these findings into a viable therapy for H3K27M-mutant DMG, we next returned to our drug screen results. We investigated the relative anti-STAT3 activity of a subset of kinase inhibitors, including RTKIs which demonstrated efficacy in the preliminary screen (Figure 4C). Next to the tool compound Stattic, which is known to robustly abrogate STAT3 signaling but is not suitable for clinical use, the JAK/STAT3 inhibitor WP1066 demonstrated near-complete reduction of STAT3 signaling in response to IL-6-induced stimulation. Further tightening the link between kinase signaling and cell viability in H3K27M-mutant tumors, cell viability was significantly reduced at a lower dose of multiple STAT3/kinase inhibitors in H3K27M-mutant compared to H3-wildtype cells (Figure 4D).
Given that reduced H3K27me3 is a hallmark of H3K27M-mutant DMGs, we assessed total H3K27me3 levels in cells treated with selected inhibitors of kinase signaling, including RTKIs and STAT3 pathway inhibitors. Interestingly, at 48 h posttreatment, the RTKIs bosutinib and ruxolitinib demonstrated noticeable restoration of H3K27me3 expression compared to vehicle-treated controls (Figure 4E). The STAT3 pathway inhibitor WP1066 also demonstrated a robust effect on H3K27me3 restoration (Figure 4F). As WP1066 demonstrated a combination of significant STAT3 signaling reduction, anti-tumor cell efficacy, and H3K27me3 restoration, we next determined its impact on clonogenicity of DMG cell lines. Consistent with prior observations, while WP1066 demonstrated little impact on clonogenicity of H3F3A-wildtype cells, escalating doses significantly reduced the clonogenic capacity of H3K27M-mutant cells (Figure 4G).
To determine WP1066’s epigenomic impact, we next performed ChIP-Seq to selected enhancer- and promoter-associated histone posttranslational modifications in DIPGXVII cells following a 24-hour treatment with either WP1066 or DMSO. While treatment with WP1066 resulted in few changes in H3K27Ac, H3K4me1, and H3K4me3, we observed 2087 regions differentially marked by H3K27me3 between conditions, including 1997 regions demonstrating relative H3K27me3 gain (Figure S3A). These regions were predominantly promoter-proximal, with over one-third of gained H3K27me3 peaks occurring at bivalent loci (ie, cobound by H3K4me3 and H3K27me3, Figure S3B). Gene ontology analysis of genes associated with these promoters demonstrated enrichment for developmental processes including nervous system development, indicating WP1066 impacts H3K27me3 not only globally but in a loci-specific manner (Figure S3C).
WP1066 is a multifunctional combination STAT3 pathway inhibitor that is currently in a phase-I clinical trial for adults with recurrent malignant glioma and metastatic melanoma (NCT01904123) and an additional phase-I trial for pediatric brain tumors (NCT04334863). Given a possible role for STAT3 in H3K27M-mutant DMG, the in vitro data described above and the potential for immediate clinical translation of this finding, we opted to focus on this drug for in vivo studies.
STAT3 Inhibition and H3K27M Knockout Result in Tumor Growth Arrest or Regression In Vivo
As H3K27M-KO DMG cells demonstrated reduced STAT3 activation in vitro, we implanted intracranial xenografts of K27M-mutant and H3K27M-KO SF8628 cells into athymic nude mice and performed serial bioluminescence imaging. While H3K27M-mutant xenografts demonstrated significant tumor growth over the study period, H3K27M-KO xenografts displayed minimal growth from time zero (Figure 5A).
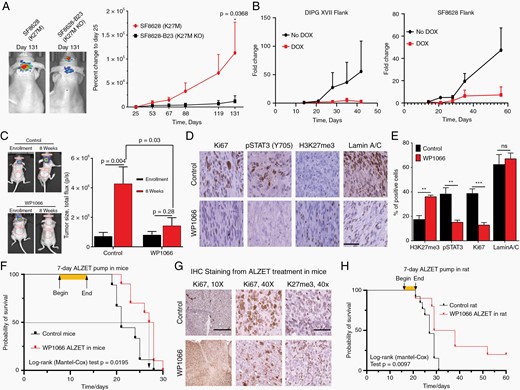
STAT3 inhibition results in tumor stasis or regression of patient-derived H3K27M-mutant DMG xenografts. (A) Bioluminescent imaging of H3K27M-KO and H3K27M-mutant SF8628 intracranial xenografts. (B) Tumor growth of patient-derived flank H3K27M-mutant xenografts as assessed by bioluminescent imaging in animals with or without doxycycline treatment. (C) Bioluminescent imaging of PED17 (H3K27M-mutant) orthotopic xenografts in mice treated with WP1066 or vehicle. Vehicle-treated mice are shown on the left, WP1066-treated mice on the right. (D, E) Relative expression of Ki67, pSTAT3 (Y705), and H3K27me3 in formalin-fixed brain sections of mice treated with WP1066 or vehicle (40×, scale bar = 100 µm). (F) Kaplan–Meier curve of ALZET-vehicle or -WP1066-treated DIPGXIII orthotopic xenografts in mice. (G) Representative immunohistochemical stains for Ki67 and H3K27me3 in formalin-fixed specimens obtained from animals in the study shown in panel F (10×, scale bar = 400 µm; 40×, scale bar = 100 µm). (H) Kaplan–Meier curve of ALZET-vehicle or -WP1066-treated DIPGXIIIp orthotopic xenografts in rats.
We next assessed the potential role of STAT3 inhibition as a therapeutic strategy using patient-derived, luciferase-tagged xenograft flank models. Using the doxycycline-inducible STAT3 knockdown cell lines described above, we observed that doxycycline-treated mice demonstrated significantly reduced tumor burden compared to untreated controls over time (Figure 5B). Following these mechanistic observations, we targeted STAT3 signaling in vivo using WP1066 delivered by oral lavage three times weekly in an intracranial xenograft model with the PED17 cell line. While vehicle-treated mice demonstrated significant tumor growth, mice treated with WP1066 were observed to have either no tumor growth or tumor regression over the study period (Figures 5C, Figure S4). Immunohistochemical analyses of WP1066-treated tumors showed both a significant decrease in pSTAT3 (Y705) and Ki67 positivity as well as a significant increase in H3K37me3 expression (Figure 5D).
To expand on these results, we next performed a validation study using a DIPGXIII orthotopic xenograft. Oral administration of WP1066 did not improve survival over the study period (Figure S5A, B). While WP1066 has demonstrated BBB permeability in other models as well as our first model, prior work indicates maximum intratumoral drug concentration of 5.8 µM 30 min following oral administration of WP1066.27 Given our observation that WP1066 has a IC50 of approximately 5 µM in DIPGXIII (Figure 1), we hypothesized that low intratumoral drug concentration contributed to these findings. To assess this possibility and maximize intratumoral drug concentration, we performed a seven-day intratumoral infusion into DIPGXIII orthotopic xenografts using an Alzet pump loaded with either WP1066 or DMSO vehicle. WP1066-Alzet-treated animals demonstrated significantly prolonged survival relative to vehicle-Alzet-treated controls (Figure 5F–G, log-rank P = 0.0195).
STAT3 is Detectable in the Plasma of H3K27M-mutant DMG Patients
Definitive molecular diagnosis is the standard of care and a prerequisite for clinical trial eligibility for patients with H3K27M-mutant DMG. While it has been demonstrated that biopsy of brainstem lesions can be done safely at experienced centers with acceptable rates of morbidity,28 there is a need to identify peripheral biomarkers that would obviate the need for biopsy. To address this need, we assessed STAT3 levels in circulating plasma extracellular vesicles (EVs) of patients with pathology-confirmed H3K27M-mutant DMG. In a limited set of clinical specimens, we found plasma EVs of pediatric patients with H3K27M-mutant DMG (N = 3), but not adult patients with glioblastoma (N = 4) or nonneoplastic CNS pathology (N = 4), had easily detectable STAT3 on Western blot (Figure 6A, Supplementary Table 5). To determine whether H3K27M-DMG cells are capable of secreting EVs containing STAT3 and thus whether circulating EVs could be tumor-derived, we isolated EVs from the media of cultured H3K27M-mutant DMG cells and assessed EV lysates for STAT3 protein expression (Figure 6B). Consistent with observations from human specimens, STAT3 was reliably present in EVs derived from H3K27M-mutant DMG cells. To determine whether exosomes containing STAT3 may also function as a response to STAT3 inhibitor therapy, we performed Western blot to whole cell lysates and EVs derived from either vehicle- or WP1066-treated DIPGXVII cells (Figure 6C). WP1066 treatment significantly abrogated STAT3-containing EV production, implicating this as a potential biomarker for response to STAT3 inhibitor therapy. Intriguingly, while H3K27me3 increase in whole cell lysates treated with WP1066, expression of this histone posttranslational modification decreased in EVs.
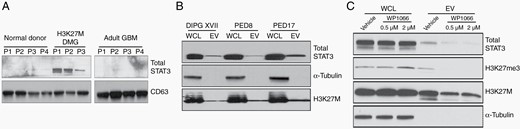
STAT3 is present in plasma extracellular vesicles (EVs) of patients with H3K27M-mutant DMG. Western blots demonstrating total STAT3 expression in (A) H3K27M-mutant DMG patients compared to normal controls and adult glioblastoma, (B) patient-derived cell line EVs obtained from culture media, and (C) in culture media of DIPGXVII cells treated with the STAT3 inhibitor WP1066 versus vehicle.
Discussion
Using a cell viability-based drug screen, we identified kinase signaling broadly and STAT3 signaling specifically as a potentially biologically relevant, targetable pathway in H3K27M-mutant DMG. Activated STAT3 is over-expressed H2K27M-mutant tumor tissue, and STAT3 is detectable in circulating plasma of patients harboring these tumors. Moreover, we observed STAT3 inhibition using either inducible shRNA or small molecule inhibition significantly ameliorated growth of H3K27M-mutant DMG xenografts in vivo, indicating STAT3 inhibition may be a viable therapeutic strategy for this deadly disease.
Activation of the JAK/STAT signaling pathway has been long associated with cancer.29–31 The STAT protein family is comprised of transcription factors that, when activated, have wide-ranging downstream effects that promote tumorigenesis.29,32–37 STAT3, in particular, has been shown to promote tumorigenesis, immune escape, and therapy resistance in cancer cells.29,38 In adult glioblastoma, STAT3 has been shown to be constitutively activated and significantly influences the molecular progression of this tumor in adults.39,40 Previous efforts have demonstrated that high-grade gliomas are therapeutically vulnerable to STAT3 inhibition.41 To date, limited data are available on the role of STAT3 in pediatric glioma, but recent evidence indicates that ACVR1 mutations cooperate with H3F3A mutations to activate STAT3 signaling in H3K27M-mutant DMG.42 Taken together with our observation that STAT3 is a peripherally detectable, therapeutically responsive biomarker in this tumor, these findings indicate STAT3 should be considered as a therapeutic target in H3K27M-mutant DMG.
Numerous STAT3 inhibitors have been developed to date, but there are no FDA-approved STAT3 inhibitors at this time. Currently, WP1066, a brain-penetrant STAT3 pathway inhibitor, is in phase-I clinical trials for both adult recurrent glioma and metastatic melanoma (NCT01904123) and high-grade pediatric brain tumors refractory to standard therapy (NCT04334863). Our data demonstrate that WP1066 has excellent anti-STAT3 activity in vitro and displays potent anti-tumor activity and on-target effects in vivo using intracranial xenograft models. One potential criticism of this study is the use of direct intratumoral drug infusion as a means to achieve therapeutic efficacy with WP1066 in an orthotopic xenograft model. There are two key aspects to consider in this regard. First, the DIPGXIII cell line, while forming rapidly lethal orthotopic xenografts, displays an IC50 of approximately 5 uM to WP1066, which approximates the highest observed intracranial concentration of this drug in intracranial xenograft models.27 Second, direct intracranial drug injection in the form of convection-enhanced delivery (CED) has been demonstrated safe in a phase-I clinical trial for DMG,43 and this approach has become an increasingly popular tool to maximize drug concentration in brain tumor models.44–46 With these considerations in mind, we consider direct intracranial drug infusion a reasonable, translatable approach to assessing drug efficacy in xenograft models of DMG.
While the data shown here demonstrate STAT3 is a biologically relevant and druggable target in H3K27M-mutant DMG, our study has several additional limitations. First, while our results clearly implicate kinase and STAT3 signaling in glioma cell survival, our study is limited in the scope of drugs tested in vitro and in vivo. Ongoing efforts in our laboratory are aimed at high-throughput drug screens to test additional experimental compounds with potential anti-tumor efficacy. Second, observations of activated STAT3 expression in clinical specimens are limited by tissue and plasma availability owing to the low incidence of the disease under study. To address this need, we are actively recruiting and enrolling additional patients in observational and interventional studies to facilitate the accrual of valuable clinical specimens. Finally, additional in vivo studies using genetically engineered mouse models would be ideal for studying the role of STAT3 inhibition in an immune-competent setting. Given the known role of immune modulation in DMG and the role of JAK/STAT signaling in immune cell activation, these studies are of paramount importance.
Acknowledgments
We gratefully thank the generous support of Dr. Michelle Monje (Stanford University), the International DIPG/DMG Registry (Cincinnati Children’s Hospital Medical Center), Dr. Fang Dong (Zhejiang University) and Dr. Zhiguo Zhang (Columbia University).
Funding
National Institute of Neurological Disorders and Stroke (K08 NS092891 to DJD); Brains Together For a Cure Foundation (award #10 NCE to DJD); National Center for Advancing Translational Sciences (TR002380 to E.A.P.); National Institute of General Medical Sciences (T32GM 65841 to J.B.A.).
Conflict of interest statement. The authors have no conflicts of interest to declare.
Authorship statement. Conception and design: L.Z., E.H.H., D.J.D. Acquisition of funding for the study: D.J.D. Data acquisition: L.Z., C.L.N., C.A.D., J.C., V.M.L., T.P., E.A.P., J.B.A., F.H.H., R.S., J.G., T.K., R. Simons, J.W., R. Siada. Data analysis and interpretation of results: L.Z., C.L.N., E.A.P., J.B.A., P.D., S.A.J., D.J.D. Drafting of manuscript: L.Z., C.L.N., V.M.L., D.J.D. Critical review of manuscript prior to submission: all authors.
References



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}