





Abstract
Cancer immunotherapy has made remarkable advances with over 50 separate Food and Drug Administration (FDA) approvals as first- or second-line indications since 2015. These include immune checkpoint blocking antibodies, chimeric antigen receptor-transduced T cells, and bispecific T-cell–engaging antibodies. While multiple cancer types now benefit from these immunotherapies, notable exceptions thus far include brain tumors, such as glioblastoma. As such, it seems critical to gain a better understanding of unique mechanistic challenges underlying the resistance of malignant gliomas to immunotherapy, as well as to acquire insights into the development of future strategies. An Immuno-Oncology Think Tank Meeting was held during the 2019 Annual Society for Neuro-Oncology Scientific Conference. Discussants in the fields of neuro-oncology, neurosurgery, neuro-imaging, medical oncology, and cancer immunology participated in the meeting. Sessions focused on topics such as the tumor microenvironment, myeloid cells, T-cell dysfunction, cellular engineering, and translational aspects that are critical and unique challenges inherent with primary brain tumors. In this review, we summarize the discussions and the key messages from the meeting, which may potentially serve as a basis for advancing the field of immune neuro-oncology in a collaborative manner.
Immunotherapy of cancer has advanced from an experimental stage to an established therapeutic pillar for many first- and second-line indications. In particular, humanized antibodies blocking the negative costimulatory molecules, cytotoxic T-lymphocyte–associated protein (CTLA)-4, programmed cell death (PD)-1, or its ligands, such as PD-L1, have revolutionized the treatment of many metastatic solid tumors. Current FDA approvals of immune checkpoint inhibitors, with companion biomarkers,1 for the treatment of malignancies include melanoma, lung cancer, triple-negative breast cancer, bladder cancer, liver cancer, renal cell carcinoma, microsatellite instable colon cancer, and others. FDA approvals have also included chimeric antigen receptor (CAR)-transduced T cells targeting CD19 for refractory B-cell lymphoma and acute lymphoblastic leukemia.1 Similarly, the use of bispecific T-cell–engaging antibodies targeting CD19 or antibody-drug conjugates targeting CD30 was recently granted FDA approval, and the FDA has granted more than 50 separate approvals to immune-based therapies since 2015 (https://www.cancerresearch.org/scientists/immuno-oncology-landscape/fda-approval-timeline-of-active-immunotherapies).
Although multiple cancer types have been successfully treated with immunotherapies, a notable exception includes primary malignant gliomas. Recent randomized clinical trials with PD-1 blockade in unselected O6-methylguanine-DNA methyltransferase (MGMT)-unmethylated newly diagnosed and recurrent glioblastoma (GBM),2 vaccines targeting tumor-associated and tumor antigens,3,4 and replication-competent retrovirus administration5 have not demonstrated a clinical benefit in this patient population. To more effectively understand the resistance of GBM to immunotherapy and to foster inter-cancer collaborations, the authors hosted an Immuno-Oncology Think Tank Meeting at the 2019 Annual Society for Neuro-Oncology Scientific Conference. Experts in the field of brain cancer were paired with other scientific experts whose expertise was extracranial cancer and/or fundamental immunology. In consultation with the organizing committee, the interdisciplinary sessions focused on the tumor microenvironment (TME), myeloid cells, T-cell dysfunction, other immune cell populations, immunomodulatory factors, targetable antigens, metastatic brain tumor differences, viral immunotherapies, imaging technologies, CAR-T adoptive transfer strategies, and clinical trial design. An overview from these sessions, as well as key points, is presented below. We also propose a roadmap for future immunotherapy research based on insights gained from this opportunity.
Immune Microenvironment of GBM
Studies of the GBM TME and tumor-infiltrating leukocytes (TILs) highlight the complex and immunosuppressive environment within the tumor that underlies the resistance to immunotherapy. Among these mechanisms are the release of immunosuppressive factors, recruitment of immunosuppressive cells, exclusion of anti-tumor immune effector cells, and expression of cell-surface inhibitory ligands.6 Relative to other cancers that have responded to immunotherapy, GBM has a lower frequency of CD3+ T cells, a higher frequency of myeloid cells (monocytes, macrophages, microglia), and a lower expression of PD-L1 and lower prevalence of PD-1-expressing TILs.7–9 The lower T-cell infiltration and lower expression of inhibitory markers make it more challenging for immune checkpoint blockade in GBM to exert a strong therapeutic effect. In addition, macrophages found within GBM have a higher expression of phenotypic markers of immunosuppression and are associated with poor survival, suggesting that improving immunotherapeutic interventions should include altering macrophage characteristics or abundance.7,10,11
Poor Status of T Cells
Tumor-infiltrating leukocytes (TILs) constitute a small population of immune cells within the TME and represent a highly heterogeneous population. In GBM and other cancers, TILs include both cancer-specific cells and nonspecific bystanders.12 To improve the efficacy of immunotherapy, it will be essential to distinguish bystander CD8+ T cells from cancer-specific CD8+ T cells.13 In lung and colorectal cancer, many of the CD8+ TILs are specific for unrelated antigens, such as Epstein-Barr virus (EBV), cytomegalovirus (CMV), or influenza virus (flu).12 Although these cancer-unrelated tumor-infiltrating T cells can express various markers associated with their active participation in an inflammatory response, such as PD-1, TIGIT, HLA-DR, and CD69, they consistently lack the expression of CD39,12 which is an ectonucleotidase implicated as having immunosuppressive effects14 and expressed by chronically stimulated and terminally exhausted T cells.15 Similarly, GBM has virus-specific cells in the TME, with a notable enrichment of Influenza-specific T cells as compared with other tumor types and with other viral antigen specificities16 (Figure 1). As the presence of viral antigens in the GBM TME has not been supported by the Pan-Cancer Analysis of Whole Genomes (PCAWG),17 the role of virus-specific T cells in the TME remains unknown. Yet, their presence offers a potential source of tumor-targeting T cells.18
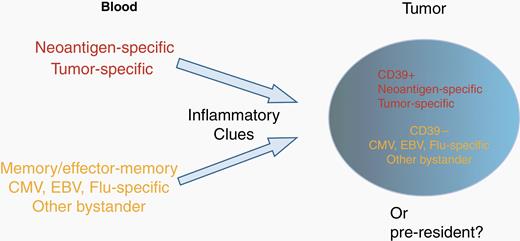
Bystander T cells in the tumor microenvironment. Schematic representation of the source and nature of T cells present in tumors. Tumors contain neoantigen-specific, tumor-specific, and viral-specific T cells that may be distinguished by CD39 expression. Concepts based on results are presented in Simoni et al.12
Among the challenges to effective immunotherapy include significant T-cell dysfunction, which can be attributed to poor T-cell infiltration into the tumor and the highly immunosuppressive nature of the TME.6,19 T-cell defects can be the result of exhaustion (hypofunctional state due to sustained expression of inhibitory receptors), ignorance (the absence of response due to either anatomical barriers or insufficient antigen presentation), anergy (an inactive state following an antigen encounter), tolerance (programmed induction of unresponsiveness or deletion of T-cell clones to prevent autoimmunity), and senescence (a hypofunctional state resulting from shortened telomeres) (Figure 2).19 In addition, there are several metabolic factors that hinder T-cell function, such as tumor-induced nutrient competition and hypoxia.19
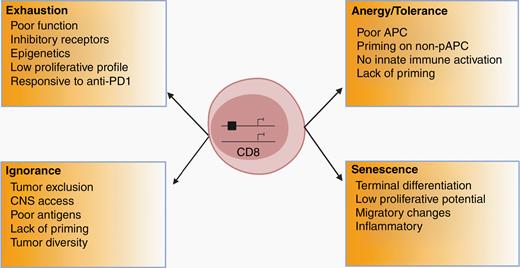
Potential types of T-cell dysfunction relevant for neuro-oncology. Summary of the major factors contributing to T-cell dysfunction.
Tumor- or myeloid-mediated T-cell exclusion, poor access to the CNS, and poor T-cell priming due to high tumor diversity and few suitable antigens all likely contribute to the decreased infiltration of functional T cells.19,20 Each of these factors should be further investigated in order to elucidate the mechanisms and components contributing to this dysfunction. For example, further interrogation into the mechanisms of T-cell exclusion has revealed a role for remote organs. Naïve T cells have been shown to accumulate in the bone marrow of GBM patients and mice, which appears to be the result of tumor-mediated internalization of sphingosine-1-phosphate receptor 1 (S1P1) on T cells.21 In mouse models, blocking S1P1 internalization releases T cells and restores anti-tumor cytotoxicity, providing a possible avenue for therapeutic intervention. Likewise, the identification of common pathways in tumor-infiltrating T cells, which are excluded or suppressed by tumor-derived metabolites, such as kynurenine or 2-hydroxyglutarate,22,23 may result in therapeutic strategies to increase T-cell resilience and function.
In the context of persistent antigen stimulation, as seen in chronic infection or cancer, T cells are driven into a state of exhaustion, which is characterized by the loss of cytokine production and cytotoxicity.24 This exhausted state is often mediated by transcriptional changes and epigenetic programming.25 Tox, an HMG-box transcription factor, has been identified as a key regulator of CD8+ T-cell exhaustion in mice.25 Exhausted CD8 T cells also exhibit increased expression of inhibitory ligands, such as PD-1, which can be targeted via immune checkpoint blockade to reinvigorate T-cell activity and stimulate an anti-tumor immune response.19 Currently, it is not known what the antigenic targets of chronic T-cell reactivity are, but several groups are currently studying this concept.
To leverage the benefits of immunotherapy, the mechanisms contributing to T-cell dysfunction in GBM need to be further explored. Questions yet to be answered should address the following: What are the key immunosuppressive pathways or cell types? How does priming to CNS antigens occur? What regulates immune cell homing to the brain? Addressing these questions requires a highly combinatorial and multifaceted approach which should include high dimensional immune profiling platforms, better preclinical models, and creative clinical trial design, such as neoadjuvant immunotherapies involving cell-based methods, checkpoint inhibitors, and immune enhancers.
| Key Points |
|---|
| • GBM has poor T-cell infiltration, high myeloid infiltration, and low PD-L1/PD-1 expression |
| • A large subset of TILs are cancer nonspecific and are characterized by the lack of CD39 expression. TILs in GBM appear to be enriched for Influenza-specific T cells |
| • GBM patients have decreased T-cell numbers and functions both locally in the TME and systemically in the circulation |
| • T-cell dysfunction is attributed to exhaustion, ignorance, anergy, tolerance, senescence, and metabolic pressures |
| Key Points |
|---|
| • GBM has poor T-cell infiltration, high myeloid infiltration, and low PD-L1/PD-1 expression |
| • A large subset of TILs are cancer nonspecific and are characterized by the lack of CD39 expression. TILs in GBM appear to be enriched for Influenza-specific T cells |
| • GBM patients have decreased T-cell numbers and functions both locally in the TME and systemically in the circulation |
| • T-cell dysfunction is attributed to exhaustion, ignorance, anergy, tolerance, senescence, and metabolic pressures |
| Key Points |
|---|
| • GBM has poor T-cell infiltration, high myeloid infiltration, and low PD-L1/PD-1 expression |
| • A large subset of TILs are cancer nonspecific and are characterized by the lack of CD39 expression. TILs in GBM appear to be enriched for Influenza-specific T cells |
| • GBM patients have decreased T-cell numbers and functions both locally in the TME and systemically in the circulation |
| • T-cell dysfunction is attributed to exhaustion, ignorance, anergy, tolerance, senescence, and metabolic pressures |
| Key Points |
|---|
| • GBM has poor T-cell infiltration, high myeloid infiltration, and low PD-L1/PD-1 expression |
| • A large subset of TILs are cancer nonspecific and are characterized by the lack of CD39 expression. TILs in GBM appear to be enriched for Influenza-specific T cells |
| • GBM patients have decreased T-cell numbers and functions both locally in the TME and systemically in the circulation |
| • T-cell dysfunction is attributed to exhaustion, ignorance, anergy, tolerance, senescence, and metabolic pressures |
Myeloid Cells as Mediators of Immunosuppression and Tumor Progression
Myeloid cells represent the most abundant immune cell type infiltrating GBMs, where their content can range between 40% and 70% of the total cell population.26,27 These myeloid cells are a heterogeneous population with primarily tumor-promoting functions.11,28 Macrophage activation status has been shown to span a broad spectrum of phenotypes that are highly dependent on the stimuli present in a specific tissue microenvironment.11 The paradigm defining macrophages as either classically activated anti-tumor M1 macrophages or immunosuppressive M2 macrophages provides points of reference; however, recent data indicate the differentiation of GBM-infiltrating macrophages is a continuous process, characterized by the large numbers of relatively immature myeloid cells that can express both canonical M1 and M2 markers at the same time.29,30 These cells are likely to contribute to the outcomes of GBM patients treated with immune therapies given their high abundance and immunosuppressive functions.
Recent studies have underscored the importance of cell ontogeny for the overall myeloid cell distribution and activation. Single-cell–based analyses by the Diaz lab demonstrated that, in contrast to microglia, blood-derived myeloid cells aggregate in distinct compartments within GBM tumors and associate with distinct GBM cell types.29,30 These findings indicate that despite being exposed to similar tumor environmental stimuli, myeloid cells retain functional differences based on programming defined by their progenitors. Differences also exist in the activation profiles of different myeloid populations.31,32 These findings suggest that myeloid cell responses in the context of targeted therapies are varied and their contributions to the tumor’s adaptive resistance to treatment need to be considered.
A deeper understanding of the source and biological function of GBM-associated myeloid cells would also improve efforts to target them with greater specificity than achieved previously. Vascular endothelial growth factor (VEGF) produced by tumors has been found to induce the development of myeloid-derived suppressor cells (MDSCs) through mechanisms dependent on signal transducer and activator of transcription 3 (STAT3).33 STAT-3 is a transcription factor known to promote tumorigenesis and facilitate T-cell dysfunction.11 Activation of STAT3 in brain tumor-infiltrating macrophages has been demonstrated in both preclinical models and patient-derived samples.34 Inhibiting STAT-3 in mice and in immune cells from GBM patients leads to a restoration of T-cell function and reversal of immune suppression mediated by macrophages and microglia.34 In addition to STAT-3 inhibition, blockade of VEGF or its receptor could overcome this immunosuppressive effect. These findings suggest that agents that reprogram the transcriptional signature and cellular interactions within the TME can increase the effectiveness of immunotherapies, such as vaccines and CAR-T adoptive transfer therapy.
As the largest number of infiltrating myeloid cells has been shown to be blood-derived, an opportunity is available to impact the polarization and homing of blood-circulating monocytes prior to their arrival at the tumor site, such as targeting chemokines.35,36 Immune-targeting therapies are often administered and exert their primary effect systemically. For example, the target of the intravenously injected anti-CD40 antibody has been shown to be predominantly circulating monocytes.37 These monocytes were then able to traffic to nonlymphoid organs and induce differentiation of tissue-resident CD8+ T cells, which is a critical step in the development of long-lasting anti-tumor immunity. Elucidating more precisely the biological functions of tumor-associated myeloid cells provides further understanding of how these populations interact with additional lines of treatment, such as oncolytic viruses and adoptively transferred cytotoxic lymphocytes. Finally, such in-depth functional studies are likely to yield novel strategies for the therapeutic repolarization of GBM-associated myeloid cells, which might provide greater benefits than pan-myeloid depletion.
It is also important to recognize how the heterotypic relationship between tumor and myeloid cells impacts the evolution of tumors. Genetic and epigenetic variations within tumor cells affect not only the growth of the tumor cells themselves, but also how they interact with the stroma and immune components of TME. These broader effects can be surmised through empirical and unbiased systematic cross-species analyses of different tumor subtypes and their corresponding TME phenotypes. Given the reciprocal nature of these heterotypic interactions, applying pressure on one cell type through therapeutic interventions may affect the phenotype and function of the other. Recently, in models of PTEN-deficient GBM,38 Chen et al. demonstrated that PTEN deficiency in GBM cells leads to upregulation of the potent macrophage chemoattractant lysyl oxidase (LOX) in a YAP-1-dependent manner. The LOX-dependent upregulation in β1 integrin receptor signaling in blood-circulating monocytes drives their infiltration into GBM tissue where they acquire a tumor-associated macrophage (TAM) phenotype and promote GBM survival and angiogenesis via secretion of secreted phosphoprotein 1 (SPP1). Perturbation of these interactions through the inhibition of LOX signaling reduces TAM infiltration and inhibits tumor growth. This synthetic lethality approach offers a novel therapeutic target but is specific to a particular GBM genotype, underscoring the vast heterogeneity that exists within the TME. In addition, using a gain-of-function screen of epigenetic regulators, Chen et al. identified the circadian regulator CLOCK as a top hit that not only enhances stem-cell self-renewal and anabolic metabolism, but also recruits tumor-promoting microglia into the TME through CLOCK-mediated transcriptional upregulation of the novel chemokine OLFML3. In GBM models, depletion of CLOCK or OLFML3 extends overall survival and reduces intratumoral microglia density.39 Thus, a detailed and systemic evaluation of the tumor cellular and molecular composition is warranted, both at diagnosis (and ideally, through the biopsy of multiple tumor locations) and in a longitudinal format if the mechanisms of adaptive resistance are to be revealed. Once such mechanisms are discovered through the use of ever-evolving model systems, careful consideration of the benefits and drawbacks of these models must be applied in order to generate successful and informative combination clinical trials.
The immunological TME of GBM warrants further study and should involve the definition of the distinct immune cell populations and their interactions. Questions to be addressed include: what are the key cell-cell interactions in the TME? What role can macrophages play in immunotherapy? What are the frequencies of tumor-specific T cells? What is attracting virus and particularly Influenza-specific T cells to the TME and can they be repurposed for immunotherapy? To address this, several methods should be employed to visualize and define the immune topographical landscape of GBM.
| Key Points |
|---|
| • Macrophages are overrepresented in the glioma TME and are highly immunosuppressive |
| • It is necessary to determine how TAM activation and tumor distribution are guided by the ontogeny of the different myeloid cell types and by the underlying genetics of the tumor |
| • Perform empirical and unbiased systematic validation of TAM-targeting therapies (alone or in combination with other therapies) on both genetic and pharmacological levels to yield positive outcomes without high levels of systemic toxicity |
| Key Points |
|---|
| • Macrophages are overrepresented in the glioma TME and are highly immunosuppressive |
| • It is necessary to determine how TAM activation and tumor distribution are guided by the ontogeny of the different myeloid cell types and by the underlying genetics of the tumor |
| • Perform empirical and unbiased systematic validation of TAM-targeting therapies (alone or in combination with other therapies) on both genetic and pharmacological levels to yield positive outcomes without high levels of systemic toxicity |
| Key Points |
|---|
| • Macrophages are overrepresented in the glioma TME and are highly immunosuppressive |
| • It is necessary to determine how TAM activation and tumor distribution are guided by the ontogeny of the different myeloid cell types and by the underlying genetics of the tumor |
| • Perform empirical and unbiased systematic validation of TAM-targeting therapies (alone or in combination with other therapies) on both genetic and pharmacological levels to yield positive outcomes without high levels of systemic toxicity |
| Key Points |
|---|
| • Macrophages are overrepresented in the glioma TME and are highly immunosuppressive |
| • It is necessary to determine how TAM activation and tumor distribution are guided by the ontogeny of the different myeloid cell types and by the underlying genetics of the tumor |
| • Perform empirical and unbiased systematic validation of TAM-targeting therapies (alone or in combination with other therapies) on both genetic and pharmacological levels to yield positive outcomes without high levels of systemic toxicity |
Dendritic Cells
A critical step in the generation of a robust antigen-specific immune response is the interaction between professional antigen-presenting cells (APCs), which capture and present protein and lipid antigens, and CD8+ or CD4+ T cells. Dendritic cells (DCs) are well characterized for their ability to capture antigen, migrate to secondary lymphoid organs, and interact with cells of the adaptive arm of the immune system. Their role in the TME, however, is not fully understood. Studies from the Gajewski lab have demonstrated that the expression of the transcription factor, Batf3, in DCs is required for the recruitment of CD8+ effector T cells to the TME in a murine model of melanoma.40 The role of DCs, and their ability to cross-present tumor antigens, may be even more crucial in the microenvironment of GBM given the absence of the afferent arm of the immune response in the brain parenchyma.41 Decades of studies in the fields of infection and CNS autoimmune disorders have shown that APCs are able to infiltrate the parenchyma and initiate antigen-specific immune responses.42,43 Given sufficient priming and recruiting chemokine gradients, APCs can traffic through the meninges, choroid plexus, and the blood-brain barrier (BBB), particularly when perturbations of the endothelial cell tight junctions of the BBB have occurred due to tumor growth.44,45 However, elucidating the key stages of tumor growth, when APCs exert their most important anti-tumor immune function, remains a large gap in the community’s understanding. Furthermore, developing strategies for the differentiation of myeloid cells, already present at the tumor site, into DCs, or alternatively, for the recruitment of DCs from the circulation, could significantly benefit the induction of a productive anti-tumor immune response. One such strategy might be to induce the production of Flt3L in the TME, using adenoviral-mediated gene delivery, resulting in the influx of DCs and brain tumor-specific immune response, as described by King and colleagues.46 Alternatively, recent studies by the Mitchell lab demonstrated the possibility of using Lin−CCR2+ hematopoietic stem cells, which traffic to the intracranial tumor site and differentiate into mature DCs, to overcome resistance to immune checkpoint therapy.47,48 These results indicate that multiple routes are available for the generation of functional APCs at the tumor site, and their contributions to a productive anti-GBM immune response are likely important and significant.
A different aspect of APC function in tumor growth and development also needs to be considered. As professional APCs, DCs are known for their potent ability to prime naïve T cells and initiate an anti-tumor response in the periphery. However, as chronic antigen stimulation may result in T-cell exhaustion, the precise contribution of APCs to the development of T-cell dysfunction needs to be carefully examined. Within the glioma TME, multiple cell types can present antigens, including conventional DCs, TAMs, astrocytes, as well as the cancer cells themselves. In order to present tumor-derived antigens, cells need to employ either the more frequent mechanism of cross-presentation, where extracellular peptides/proteins are phagocytosed, processed endogenously, and cross-presented, or the lesser-known pathway of cross-dressing, where preformed antigen-class I major histocompatibility (MHC) molecules are transferred from one cell to another.49 How these mechanisms operate within TAMs, which are the most abundant APC within tumors, is not well known, and elucidating their contribution to antigen presentation past the initial states of priming may reveal novel therapeutic opportunities. In order to study these pathways most efficiently, accurate model selection is critical as certain models better represent the mechanisms of antigen presentation than do others. Furthermore, cell-type–specific perturbations in those models would be necessary in order to more precisely define differences in the activity of the many different APCs. Those can be accomplished through the use of cell-specific Cre models or the temporal/spatial control of cell depletion through the use of diphtheria toxin receptor-engineered mouse strains. One such recent study, by the Li lab, employed the transgenic breast cancer model MMTV-PyMT (mouse mammary tumor virus-polyoma middle tumor T-antigen) crossed with several mouse lines carrying APC-specific perturbations. Through detailed characterization of the phenotypic changes in both APCs and T cells, the authors demonstrate that while IRF8-expressing DCs are critical for the initial priming of cytotoxic T cells, IRF8 expression in TAMs leads to CTL exhaustion. This effect was reversed, and tumor growth was suppressed following macrophage-specific IRF8 ablation (https://www.biorxiv.org/content/10.1101/2020.03.12.989731v1). These findings were extended in human renal carcinoma samples, where a high score for IRF8 signature-expressing TAMs was highly associated with T-cell exhaustion markers and was a negative predictor of patients’ long-term survival. These observations highlight the important contributions different APCs have and how heterogeneous these effects can be given the large spectrum of cells capable of performing antigen presentation.
| Key Points |
|---|
| • The appropriate migration and differentiation of DCs within the TME is critical for the priming of antigen-specific T-cell responses. There is a need for developing strategies to this end |
| • The adoptive transfer of hematopoietic stem cells may enhance the migration and differentiation of DCs within the TME |
| • There is a need to understand the contributions of microglia and astrocytes to the process of antigen presentation in the context of GBM tumors |
| • Gene delivery of Flt3L and/or GM-CSF to the TME may be employed to enhance differentiation of monocytes into APCs or recruit additional APCs |
| • Whether and how different APC populations drive T-cell exhaustion is unknown |
| • Development of models that accurately recapitulate and allow for robust study of antigen presentation in glioma is needed |
| Key Points |
|---|
| • The appropriate migration and differentiation of DCs within the TME is critical for the priming of antigen-specific T-cell responses. There is a need for developing strategies to this end |
| • The adoptive transfer of hematopoietic stem cells may enhance the migration and differentiation of DCs within the TME |
| • There is a need to understand the contributions of microglia and astrocytes to the process of antigen presentation in the context of GBM tumors |
| • Gene delivery of Flt3L and/or GM-CSF to the TME may be employed to enhance differentiation of monocytes into APCs or recruit additional APCs |
| • Whether and how different APC populations drive T-cell exhaustion is unknown |
| • Development of models that accurately recapitulate and allow for robust study of antigen presentation in glioma is needed |
| Key Points |
|---|
| • The appropriate migration and differentiation of DCs within the TME is critical for the priming of antigen-specific T-cell responses. There is a need for developing strategies to this end |
| • The adoptive transfer of hematopoietic stem cells may enhance the migration and differentiation of DCs within the TME |
| • There is a need to understand the contributions of microglia and astrocytes to the process of antigen presentation in the context of GBM tumors |
| • Gene delivery of Flt3L and/or GM-CSF to the TME may be employed to enhance differentiation of monocytes into APCs or recruit additional APCs |
| • Whether and how different APC populations drive T-cell exhaustion is unknown |
| • Development of models that accurately recapitulate and allow for robust study of antigen presentation in glioma is needed |
| Key Points |
|---|
| • The appropriate migration and differentiation of DCs within the TME is critical for the priming of antigen-specific T-cell responses. There is a need for developing strategies to this end |
| • The adoptive transfer of hematopoietic stem cells may enhance the migration and differentiation of DCs within the TME |
| • There is a need to understand the contributions of microglia and astrocytes to the process of antigen presentation in the context of GBM tumors |
| • Gene delivery of Flt3L and/or GM-CSF to the TME may be employed to enhance differentiation of monocytes into APCs or recruit additional APCs |
| • Whether and how different APC populations drive T-cell exhaustion is unknown |
| • Development of models that accurately recapitulate and allow for robust study of antigen presentation in glioma is needed |
CD1d-Restricted Natural Killer T (NKT) Cells
NKT cells represent a diverse population of T-lineage lymphocytes which, unlike conventional T cells, recognize antigen presented by CD1d, an MHC class I-like molecule.50 NKT-cell receptors recognize primarily glycolipids, such as α-galactosylceramide (α-GalCer) and sulfatide, and play significant roles in the regulation of tumor immunity in a highly context-dependent fashion. Different NKT subsets may recognize different antigens and mediate productive or suppressive immune responses against tumors. Tumor patients often present with a suppressed number of NKT cells, although the precise mechanisms leading to this are unknown. Studies by numerous research groups have reported that glycolipids are enriched in brain tumor samples and likely contribute significantly to the maintenance of GBM stem cells.51 In addition, expression of glucosylceramide synthase in GBM samples was shown to reduce the efficacy of temozolomide (TMZ) treatment by Giussani et al., thereby implicating glycolipids in mechanisms of resistance.52 Sulfatides present in myelin sheaths in the CNS and in some tumor cell membranes can activate immunosuppressive type II NKT cells that can promote tumor growth.53 Despite the increasing volume of evidence indicating the important role of glycolipids in the tumorigenesis of GBM, very little is known about the precise quantitative and qualitative differences of the glycolipid composition of healthy and malignant glial tissues. Given this lack of in-depth understanding of the quantity and distribution of NKT-cell antigens within brain tumors, NKT-cell activity in the TME remains unclear and presents a vast opportunity for therapeutic exploration.
| Key Points |
|---|
| • A better understanding of glycolipid composition between healthy and malignant glial tissues as well as glycolipid synthesis pathways could be important for effective anti-tumor immune responses |
| • NKT cells are an understudied lymphocyte population that recognize lipid antigens and could play a role in the anti-tumor immune response. There is a need to understand the antigenic properties of these glycolipids for NKT-mediated recognition by the immune system |
| • Above approaches will likely need to be combined with other treatment modalities such as immune checkpoint blockade, vaccination, radiation therapy, and local chemotherapy |
| Key Points |
|---|
| • A better understanding of glycolipid composition between healthy and malignant glial tissues as well as glycolipid synthesis pathways could be important for effective anti-tumor immune responses |
| • NKT cells are an understudied lymphocyte population that recognize lipid antigens and could play a role in the anti-tumor immune response. There is a need to understand the antigenic properties of these glycolipids for NKT-mediated recognition by the immune system |
| • Above approaches will likely need to be combined with other treatment modalities such as immune checkpoint blockade, vaccination, radiation therapy, and local chemotherapy |
| Key Points |
|---|
| • A better understanding of glycolipid composition between healthy and malignant glial tissues as well as glycolipid synthesis pathways could be important for effective anti-tumor immune responses |
| • NKT cells are an understudied lymphocyte population that recognize lipid antigens and could play a role in the anti-tumor immune response. There is a need to understand the antigenic properties of these glycolipids for NKT-mediated recognition by the immune system |
| • Above approaches will likely need to be combined with other treatment modalities such as immune checkpoint blockade, vaccination, radiation therapy, and local chemotherapy |
| Key Points |
|---|
| • A better understanding of glycolipid composition between healthy and malignant glial tissues as well as glycolipid synthesis pathways could be important for effective anti-tumor immune responses |
| • NKT cells are an understudied lymphocyte population that recognize lipid antigens and could play a role in the anti-tumor immune response. There is a need to understand the antigenic properties of these glycolipids for NKT-mediated recognition by the immune system |
| • Above approaches will likely need to be combined with other treatment modalities such as immune checkpoint blockade, vaccination, radiation therapy, and local chemotherapy |
Prioritizing Immunosuppressive Molecular Factors to Target Therapeutically
Soluble factors should also be explored in the context of immunotherapy for GBM. Of interest are mediators of immune suppression, such as indoleamine and tryptophan 2,3-dioxygenase (IDO, TDO) and transforming growth factor-beta (TGF-β).
IDO and TDO are tryptophan metabolizing enzymes with additional nonenzymatic functions54 that are elaborated by mononuclear cells in the TME. In patients and mouse models, IDO and TDO are associated with decreased overall survival.55,56 In both humans and mice, IDO1 increases in the brain with advanced aging.57–59 This coincides with GBM occurrence, suggesting that aging-increased IDO1 effects should be studied in the context of patient treatment strategies. Patients might benefit from treatments combining IDO and/or TDO inhibition with immunotherapy, although recent studies in melanoma were negative.60
TGF-β in the TME plays a role in several immune regulatory pathways. It has 3 isoforms that act on the same receptor but in different locations, and with varying effects on cancer.61 TGF-β3 may be beneficial in some cancers, whereas isoforms 1 and 2 are immunosuppressive and support the tumor growth.61 TGF-β suppresses CD8+ T cells and helps activate regulatory T cells (Tregs), thereby promoting tumor immune escape.62 In a series of mouse studies, it was found that TGF-β blockade can markedly reduce tumor size, can synergize with vaccines, and that TGF-β1/2 blockade, when combined with vaccine and anti-PD-1 therapy, can significantly increase survival.61,63,64 However, in recent studies of galunisertib, an oral TGF-β-inhibitor alone or in combination with radiochemotherapy65 or lomustine66 failed to show improved overall survival in patients with newly diagnosed or recurrent GBM. These negative results emphasize the need for thorough biomarker-driven combination therapy studies exploring the capability of TGF-β blockade to synergize with other immunotherapies (Figure 3).
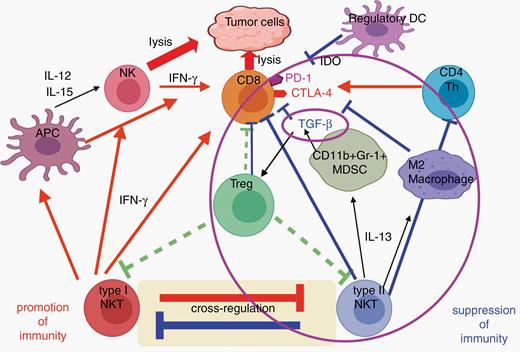
Interaction of cells and molecules as potential targets for immunotherapy. A large number of immunoregulatory cells, cell-surface molecules, and secreted cytokines and chemokines regulate the immune response to tumors and help define the tumor microenvironment. Effective immunotherapy with vaccines or adoptively transferred T cells will likely require overcoming many of these immunosuppressive agents. To block each one individually is a Herculean task, so we need to identify nodes at which these suppressive pathways intersect, to allow simultaneous inhibition of many of them. Transforming growth factor (TGF)-β may be one such node, as it plays a critical role in immune escape as it is able to suppress CD8+ T cells and activate regulatory T cells. TGF-β is made by many cells, including tumor cells, as well as myeloid-derived suppressor cells (MDSCs) when stimulated by interleukin (IL)-13 from type II natural killer T (NKT) cells. Type II NKT cells also inhibit protective type I NKT cells and make IL-13 that can induce M2 macrophages. All of these and the Tregs induced by TGF-β can inhibit CD8 anti-tumor T cells. Thus, TGF-β and the cells it acts upon are central to a complex circuity involving several different cell types and cytokines, positioning TGF-β at the intersection of multiple immunosuppressive mechanisms that could be targeted simultaneously by blocking this one cytokine.
Several soluble immune regulatory factors exist in the TME that can limit the effectiveness of immunotherapies. By characterizing these factors more extensively, the therapeutic benefits of treatments may be enhanced. Questions to be explored include: How does aging-increased IDO1 affect survival in elderly GBM patients? How do age-mediated changes contribute to the decreased immunotherapeutic efficacy in GBM patients? What are the key immune regulatory points in the TME? What is the role of TGF-β in responses to PD-1 blockade and other immunotherapies?
| Key Points |
|---|
| • Immunosuppressive factors in the TME can be targeted in combination with immunotherapy to increase treatment effectiveness |
| • IDO and TDO are immunosuppressive and IDO1 increases with advanced aging. IDO1 effects should be studied in the context of patient treatment strategies |
| • TGF-β is immunosuppressive and its blockade synergizes with other immune treatments |
| Key Points |
|---|
| • Immunosuppressive factors in the TME can be targeted in combination with immunotherapy to increase treatment effectiveness |
| • IDO and TDO are immunosuppressive and IDO1 increases with advanced aging. IDO1 effects should be studied in the context of patient treatment strategies |
| • TGF-β is immunosuppressive and its blockade synergizes with other immune treatments |
| Key Points |
|---|
| • Immunosuppressive factors in the TME can be targeted in combination with immunotherapy to increase treatment effectiveness |
| • IDO and TDO are immunosuppressive and IDO1 increases with advanced aging. IDO1 effects should be studied in the context of patient treatment strategies |
| • TGF-β is immunosuppressive and its blockade synergizes with other immune treatments |
| Key Points |
|---|
| • Immunosuppressive factors in the TME can be targeted in combination with immunotherapy to increase treatment effectiveness |
| • IDO and TDO are immunosuppressive and IDO1 increases with advanced aging. IDO1 effects should be studied in the context of patient treatment strategies |
| • TGF-β is immunosuppressive and its blockade synergizes with other immune treatments |
Targetable Antigens and Molecular Signatures
Can Neoantigens Be Replaced by Tumor-Associated Antigens?
Most GBM tumors initially present with a low mutational burden, and the majority of neoantigens are the products of private mutations and are not consistently expressed between patients, resulting in an extremely low number of bona fide GBM-specific and widely targetable neoantigens.67,68 One possible solution is to use RNA-sequencing data and analyze further for potentially immunogenic, differentially expressed nonmutant genes. However, this approach is extremely laborious and is perhaps not the most optimal one. Newer methods, such as those presented by Dutoit et al. rely on mining and exploiting already published data, and generating additional information on the immunogenicity and HLA (human leukocyte antigen)-binding preferences of different peptides in GBM samples.69–71 Alternatively, unbiased searches for immunogenic peptides can be performed through a proteomics-based method in tumor protein fractions that have previously been shown to induce activation of T cells in patients.72,73 These approaches require several key investigative steps to ensure the efficacy and safety of the proposed targets. To validate the immunogenicity of new GBM targets, new tumor-associated antigens (TAAs) need in-depth characterization. This characterization should validate the expression and the level of immunogenicity by gauging the level of response to the same TAA in T cells derived from a large series of patients, and finally characterizing the reactivity of T cells derived from healthy donors against the proposed TAA. In cases where diagnoses and circumstances allow for the development of a personalized list of targets, single-cell RNA-sequencing data has been shown to be suitable for the identification of multiple TAAs in a single patient. Finally, clinical efforts evaluating newly discovered TAAs might benefit from more robustly selected patient populations. In particular, elucidating the preexisting levels of T-cell infiltration in patients’ samples, as well as their clinical parameters (age, extent of tumor resection, etc.), might provide valuable information on whether patients are able to mount a significant immune response and potentially benefit from immunotherapy.
| Key Points |
|---|
| • Exploit already generated and available data to combine with novel proteomics-based searches in order to expand the data set of GBM-relevant TAAs, thereby expanding the repertoire and understanding of the antigenic properties of TAAs in GBM |
| • Characterize the heterogeneity, frequency of TAA recognition, and safety profile of immunogenic TAAs |
| • Re-evaluate immunotherapy clinical trials for the expression, presentation, and recognition of well-characterized TAAs in order to develop more effective combination therapies |
| • Develop more robust selection criteria for patients entering immunotherapy clinical trials to assure patients are “immune-responsive” |
| Key Points |
|---|
| • Exploit already generated and available data to combine with novel proteomics-based searches in order to expand the data set of GBM-relevant TAAs, thereby expanding the repertoire and understanding of the antigenic properties of TAAs in GBM |
| • Characterize the heterogeneity, frequency of TAA recognition, and safety profile of immunogenic TAAs |
| • Re-evaluate immunotherapy clinical trials for the expression, presentation, and recognition of well-characterized TAAs in order to develop more effective combination therapies |
| • Develop more robust selection criteria for patients entering immunotherapy clinical trials to assure patients are “immune-responsive” |
| Key Points |
|---|
| • Exploit already generated and available data to combine with novel proteomics-based searches in order to expand the data set of GBM-relevant TAAs, thereby expanding the repertoire and understanding of the antigenic properties of TAAs in GBM |
| • Characterize the heterogeneity, frequency of TAA recognition, and safety profile of immunogenic TAAs |
| • Re-evaluate immunotherapy clinical trials for the expression, presentation, and recognition of well-characterized TAAs in order to develop more effective combination therapies |
| • Develop more robust selection criteria for patients entering immunotherapy clinical trials to assure patients are “immune-responsive” |
| Key Points |
|---|
| • Exploit already generated and available data to combine with novel proteomics-based searches in order to expand the data set of GBM-relevant TAAs, thereby expanding the repertoire and understanding of the antigenic properties of TAAs in GBM |
| • Characterize the heterogeneity, frequency of TAA recognition, and safety profile of immunogenic TAAs |
| • Re-evaluate immunotherapy clinical trials for the expression, presentation, and recognition of well-characterized TAAs in order to develop more effective combination therapies |
| • Develop more robust selection criteria for patients entering immunotherapy clinical trials to assure patients are “immune-responsive” |
Unraveling Molecular Signatures from Tumor Resections
Given the inability of current immunotherapeutic treatments to generate long-term stable responses, evaluation of the immune response to therapy on the molecular level will require subtler and more discerning approaches in situ. One example of an emerging new technology that is able to provide detailed proteomic and transcriptomic information is the Digital Spatial Profiler (DSP) system developed by Nanostring Technologies. This platform currently allows the quantitation of up to 50 proteins or thousands of genes in a small, user-defined region of interest, using formalin-fixed, paraffin-embedded (FFPE) tissue slices, while preserving the architecture of a tumor sample and without the need for additional sample collection. Previous bulk transcriptomic approaches have had very limited success in distinguishing responders from nonresponders in GBM trials, and one reason is the high level of inter- and intratumoral heterogeneity within the immune component in these tumors.74 Through the use of in situ proteomic or transcriptomic technologies, the community can begin addressing the subtler differences in protein expression in a highly localized region, thus allowing the effect of the immediate microenvironment to be observed.75 Novel approaches, such as this, are needed in order to more fully understand the behavior of the TME in GBM patients after recurrence or in response to therapy, particularly immunotherapy.
| Key Points |
|---|
| • Develop and exploit novel, more powerful technologies that can provide high-resolution single-cell type analyses without disrupting the architectural context |
| • Define molecular changes that can distinguish responders from nonresponders in GBM clinical trials |
| Key Points |
|---|
| • Develop and exploit novel, more powerful technologies that can provide high-resolution single-cell type analyses without disrupting the architectural context |
| • Define molecular changes that can distinguish responders from nonresponders in GBM clinical trials |
| Key Points |
|---|
| • Develop and exploit novel, more powerful technologies that can provide high-resolution single-cell type analyses without disrupting the architectural context |
| • Define molecular changes that can distinguish responders from nonresponders in GBM clinical trials |
| Key Points |
|---|
| • Develop and exploit novel, more powerful technologies that can provide high-resolution single-cell type analyses without disrupting the architectural context |
| • Define molecular changes that can distinguish responders from nonresponders in GBM clinical trials |
Understanding GBM Immune Resistance by Learning From Brain Metastases
Metastatic tumors to the brain confer a dire prognosis with significantly worse outcomes than metastases to other organs. In melanoma, CNS metastases have an overall survival in months (mOS) of 2.5 months, compared with metastases to the liver and digestive tract (mOS 5.5 months), lung (mOS 13 months), or subcutaneous tissue and lymph nodes (mOS 20.8 months).76 Several recent clinical trials have demonstrated that checkpoint blockade immunotherapy can induce objective clinical responses, and even complete responses, in selected patients with small, asymptomatic, and nonsteroid-dependent brain metastases.77,78 Thus, brain metastases of melanoma appear similarly responsive to immunotherapy as extracranial disease, possibly attributable to diminished integrity of the BBB compared to GBM and thus permissive to the entry of cytotoxic T cells. In small numbers of patients with brain metastases from non–small cell lung cancer, objective responses were also achieved with single-agent PD-1 blockade, and further studies are ongoing in both groups of patients using novel combination therapies based on PD-1 with or without CTLA-4 blockade.79 Despite these benefits with immunotherapy in tumors from immuno-responsive malignancies metastatic to the brain, we have not seen similar success with immunotherapy in primary brain tumors such as GBM, with failures of anti-PD1 therapy in Checkmate 143,2 Checkmate 498, and Checkmate 548.80 Such findings suggest that the intrinsic immune response directed to the tumor may dictate the efficacy of immunotherapy, and not necessarily the microenvironment. Thus, an understanding of the immunological mechanisms underlying the “seed and soil” hypothesis of metastasis may provide an explanation for the discrepancy in response to immunotherapy.81
Recent two reports compared the immune landscape of primary vs metastatic brain cancers using high throughput analyses.82,83 Concordantly with previous studies, they found TAMs dominated in gliomas (up to 80% of leukocytes) whereas lymphocytes were abundant in metastatic brain tumors (up to 50%). These studies consistently demonstrated that immune cell composition and their respective phenotypic and functional features are dependent on the disease type in the CNS.
The anti-tumor immune response directed toward brain metastases may critically involve the systemic priming of T-cell responses before the development of metastatic disease in the brain,84 which may account for how extracranial metastases and brain metastases are rejected by the immune system following successful checkpoint blockade immunotherapy. Such findings suggest that inadequate antigen presentation by tumor-infiltrating DCs may be one of the limiting factors for immunotherapy in primary brain tumors. A similar sensitivity and resistance to immunotherapy may also be intrinsic to the timing of treatment, as a survival benefit was demonstrated when PD-1 blockade was administered neoadjuvantly (before surgery) both in GBM75 and extracranial solid tumors.85–89 A similar benefit was not shown in adjuvant-only administration of the same immunotherapy in mouse models.90 Regardless, based on what we know about the anti-tumor immune response directed at brain metastases, a potential solution may be to facilitate extracranial priming with vaccines,91 CD40 agonists,92 toll-like receptor (TLR) agonists,93 cytoreductive surgery,75 or radiation94 to maximize the efficacy of immunotherapy for GBM.
| Key Points |
|---|
| • GBM does not respond to immunotherapy with the same success as metastatic brain tumors |
| • The limited response of GBM to immunotherapy may be dependent upon the seed (tumor subtype, tumor-intrinsic characteristics) and the soil (brain microenvironment) |
| • Priming of the immune system through surgery, radiation, extracranial CD40 agonists, extracranial TLR agonists, or vaccines may improve responses to checkpoint inhibition |
| Key Points |
|---|
| • GBM does not respond to immunotherapy with the same success as metastatic brain tumors |
| • The limited response of GBM to immunotherapy may be dependent upon the seed (tumor subtype, tumor-intrinsic characteristics) and the soil (brain microenvironment) |
| • Priming of the immune system through surgery, radiation, extracranial CD40 agonists, extracranial TLR agonists, or vaccines may improve responses to checkpoint inhibition |
| Key Points |
|---|
| • GBM does not respond to immunotherapy with the same success as metastatic brain tumors |
| • The limited response of GBM to immunotherapy may be dependent upon the seed (tumor subtype, tumor-intrinsic characteristics) and the soil (brain microenvironment) |
| • Priming of the immune system through surgery, radiation, extracranial CD40 agonists, extracranial TLR agonists, or vaccines may improve responses to checkpoint inhibition |
| Key Points |
|---|
| • GBM does not respond to immunotherapy with the same success as metastatic brain tumors |
| • The limited response of GBM to immunotherapy may be dependent upon the seed (tumor subtype, tumor-intrinsic characteristics) and the soil (brain microenvironment) |
| • Priming of the immune system through surgery, radiation, extracranial CD40 agonists, extracranial TLR agonists, or vaccines may improve responses to checkpoint inhibition |
Viral Therapy and Immunotherapy
Oncolytic viruses are genetically engineered or natural strains of viruses that can infect tumor cells, replicate, and “biodistribute through the tumor.” This creates an inflammatory environment in the tumor causing influx of neutrophils, natural killer cells, macrophages, B cells, and T cells. This may be the fastest way to convert an “immunologically naive” tumor-like GBM into an inflammatory hot tumor (Figure 4). Oncolytic viruses that kill tumor cells and release tumor antigens in this inflammatory environment can convert the tumor into an endogenous vaccine. Multiple clinical trials in GBM have been conducted with herpes, polio, measles, adenovirus, reovirus, and parvoviruses. In addition, oncolytic viruses can be combined with immunotherapies, such as checkpoint inhibitors.95
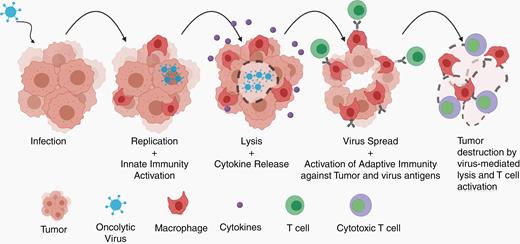
Replication-competent viruses infect tumor cells and activate immune responses. As they lead to tumor cell cytotoxicity and spread an infection to surrounding tumor cells, innate immune cells, such as macrophages, are activated and release cytokines, inflaming the tumor microenvironment. Lysis of tumor cells is thought to improve antigen presentation and infiltration of activated effector T cells against tumor and viral antigens. This leads to durable antitumor responses (adapted from Chiocca et al.96).
Emerging data from preclinical and clinical studies suggest that viral replication and persistence do not always correlate with anti-tumor effect and patient outcome. In the mouse GBM model (C2TA) treated with an oncolytic herpes simplex virus (HSV) rQNestin-34.5, tumor volume reduction directly correlated with increased viral replication/infection and expansion of functional T cells against tumor and viral antigens.97 On the other hand, the data from a Phase Ib clinical trial in patients with recurrent GBM treated with oncolytic HSV G207 virus in the neoadjuvant setting indicated poor viral replication but the presence of an inflammatory infiltrate (CD3+ T cells, CD8+ T cells, and macrophages) in the posttreatment resected tumor.98 Tumor biopsies from the longest surviving patient from this trial showed high amounts of HSV antigen, viral DNA, and RNA transcripts, suggesting active viral replication and biodistribution within the GBM tissue of this patient. In contrast, viral antigen was detected in other patients but without transcripts, indicating the absence of viral replication. It is not very clear if viral antigen persistence is beneficial to the anti-tumor response or would result in chronic antigen exposure.
Another line of evidence supporting viral replication, persistence, and immunostimulatory activity is the data from syngeneic GBM model with retroviral replicating vector (RRV) Toca511-mediated prodrug activator gene therapy in a syngeneic GBM model.99,100 These studies reported an increase in CD4+ T helper cells, CD8+ CTL, and a bystander effect of virus killing of MDSCs, TAMs, and monocytes. In early-phase clinical trials of Toca 511 in recurrent high-grade glioma patients, the responder population exhibited a direct correlation between viral replication, persistence, and inflammatory immune profile in the resected tumors. The resected tumors from responders had a higher frequency of activated memory CD4+ T cells and reduced accumulation of macrophages when compared to the nonresponders.101 These studies suggest that, in responders, viral therapy may synergize with immunotherapy.
The 3-year patient survival rates across different Phase I and II oncolytic viral therapy (replicating retrovirus,102,103 parvovirus,104 poliovirus,105 and adenovirus106) are around 20%.96 Does this survival statistic suggest that all responders are immunologically equal, and they will respond irrespective of the kind of oncolytic viral therapy used? Distinct virus-specific gene expression patterns were induced in infected tumor cells, suggesting a differential effect by different viruses on different tumors. This implies that it is possible to combine two or more different viruses in the same patient to achieve an additive or synergistic effect. A recent study107 interrogated the impact of oncolytic measles virus on a panel of patient-derived xenograft lines by RNA sequencing and gene set enrichment analysis. A unique gene signature was identified that was predictive of response to measles virus, which suggests that it may be possible to select patients who would benefit from this therapy based on pretreatment gene signature analysis.
The standard of care treatment for GBM with radiation and TMZ induces myelotoxicity and lymphopenia, while corticosteroid use causes T-cell apoptosis and cytokine depletion. In fact, recent vaccine studies have shown deleterious effects of corticosteroids for induction of vaccine-specific T-cell responses.108,109 Likewise, corticosteroids may negatively impact outcomes in cancer patients treated with checkpoint inhibitors,110 and no usage of corticosteroids at baseline was associated with longer overall survival (OS) in recurrent GBM patients treated with nivolumab (CheckMate 143 trial).2 However, radiation therapy and TMZ, if used judiciously, may theoretically synergize with viral therapy. Both these agents cause tumor lysis, cross-priming, an abscopal effect, depletion of Tregs, and rebound expansion of effector CD8+ T cells. The use of oncolytic parvovirus or measles virus together with radiation therapy has been demonstrated to have synergistic activity.111,112 Similarly, the antitumor effects of TMZ were enhanced when used in combination with Newcastle disease virus.113
There appears to be a need to keep patients on immunotherapy trials for longer time in order for effective anti-tumor immune responses to fully develop. Overall, the timeline for achieving partial response has been 1-2 years, and complete response may take about 3 years in the GBM patient population. For example, subgroup analysis from the Phase III trial with Toca 511 appears to support the need for patients to be on the trial for a longer period of time. There is also a need for better evaluation criteria to assess and monitor the efficacy of viral therapy. Pseudoprogression, necrosis, influx of T cells, and staining for residual virus (to indicate virus persistence) are some of the parameters to be considered in evaluating the outcome of virotherapy. Ideally, tissue biopsies should be attempted pre- and post-viral therapy to assess the biological impacts of the viral therapy. It is also important to perform preclinical mouse studies to systematically address questions arising from clinical trials, such as use of oncolytic virus in combination with steroids or bevacizumab, and to optimize the timing of checkpoint inhibitors when used in conjunction with viral therapy.
| Key Points |
|---|
| • More in-depth understanding is needed in terms of viral replication, persistence, immunogenicity for each of the virus platforms |
| • Multiplexing oncolytic viral and vector-mediated gene therapy—by arming the virus or viral vector with cytokines, chemokines, and checkpoint inhibitors |
| • Preclinical studies should be performed to optimize the timing of checkpoint inhibitors when used in conjunction with viral therapy |
| Key Points |
|---|
| • More in-depth understanding is needed in terms of viral replication, persistence, immunogenicity for each of the virus platforms |
| • Multiplexing oncolytic viral and vector-mediated gene therapy—by arming the virus or viral vector with cytokines, chemokines, and checkpoint inhibitors |
| • Preclinical studies should be performed to optimize the timing of checkpoint inhibitors when used in conjunction with viral therapy |
| Key Points |
|---|
| • More in-depth understanding is needed in terms of viral replication, persistence, immunogenicity for each of the virus platforms |
| • Multiplexing oncolytic viral and vector-mediated gene therapy—by arming the virus or viral vector with cytokines, chemokines, and checkpoint inhibitors |
| • Preclinical studies should be performed to optimize the timing of checkpoint inhibitors when used in conjunction with viral therapy |
| Key Points |
|---|
| • More in-depth understanding is needed in terms of viral replication, persistence, immunogenicity for each of the virus platforms |
| • Multiplexing oncolytic viral and vector-mediated gene therapy—by arming the virus or viral vector with cytokines, chemokines, and checkpoint inhibitors |
| • Preclinical studies should be performed to optimize the timing of checkpoint inhibitors when used in conjunction with viral therapy |
Challenges for CARs and TCR (T-Cell Receptor) Therapy: Antigen Heterogeneity, Tumor Escape, and Trafficking
The key challenges for effective chimeric antigen receptor-engineered T cells (CAR-T cells) for GBM therapy include many of the topics that have been discussed above, including overcoming pathways of immunosuppression, addressing tumor heterogeneity, and promoting trafficking and infiltration into these highly invasive tumors. Investigators are exploring various strategies to boost the trafficking efficiency of CAR-T cells to GBM and other brain tumors. Given that GBM almost never metastasizes to extra-CNS organs, one approach has been to evaluate locoregional modes of CAR-T cell delivery, primarily intratumoral/intracavitary (ICT) and intracerebroventricular (ICV) administration. Such regional delivery modalities can help in bypassing some of the brain-specific anatomical barriers. Preclinical studies comparing routes of administration in orthotopic brain tumor models114–116 demonstrated that regional delivery of CAR-T cells (ICT and ICV) mediate superior antitumor activity and long-term survival benefit as compared to intravenous (IV) delivery.114 In early-phase clinical trials, locoregional delivery of CAR-T cells via a reservoir catheter device has been shown to be well tolerated in patients with evidence of antitumor activity, as well as providing the opportunity of liquid biopsy to follow local therapy-dependent changes in the CNS. Locoregional delivery of CAR-T cells may improve trafficking and tumor infiltration, but additional studies addressing the expansion and persistence of T cells are needed to improve treatment efficacy. To date, clinical response to CAR-T cell therapy has been limited to rare individual cases,117 and several obstacles contribute to this therapeutic challenge.
First, GBM’s remarkable heterogeneity in antigen expression provides a strong tumor defense against CAR-T cells designed to target one specific antigen. Indeed, antigen escape—or the escape of tumor cells through the lack or loss of the targeted antigen—has been a major cause of CAR-T cell treatments in GBM.118 In response to this challenge, CARs have been designed to perform multi-antigen recognition through either hardwired approaches such as tandem-CARs and dual-CAR designs, or configurable antigen recognition designs, such as convertible CARs and zipCARs.119–122 The latter designs increase antigen recognition capability by either sequential or simultaneous addition of scFv-adaptor molecules. The drawback of this approach is the requirement for multimolecular interaction, in which adaptor molecules must find T cells as well as tumor cells at the right place and time in order to have efficacy. Whether such multimolecular interactions will work in the clinical setting remains to be tested.
An alternative strategy to tackle heterogeneous antigenic landscape of the tumor is to engage endogenous immune responses and induce antigen spreading. This can be done by overexpressing molecules, such as CD40L, FLt3L, and CXCR4 antagonists, in therapeutic T cells. In this strategy, therapeutic T cells are designed to not only provide front-line attack but also activate endogenous immune cells, such as APCs, to amplify the anti-tumor response.
In addition to antigen heterogeneity leading to tumor escape, CAR-T cell therapy for GBM also faces challenges posed by a highly immunosuppressive TME that can either exclude T cells entirely, or render T-cell dysfunction despite tumor infiltration. To promote recruitment to and infiltration of tumor cells, T cells have been engineered with chemokine receptors that complement the chemokine profile of the tumor. CXCR1- or CXCR2-modified CAR-T cells have shown improved migration, persistence, and antitumor response in IL-8–producing orthotopic GBM models.123 Additionally, the targeting of integrins and adhesion molecules on endothelial cells within the tumor ecosystem can potentially improve the intratumoral trafficking of therapeutic T cells.124,125 Altering the chemokine composition of the tumor using an armed oncolytic virus, such as RANTES-expressing adenovirus, is another approach for enhancing T-cell recruitment.126
Upon entering the TME, T cells must contend with potent immunosuppressive factors such as TGF-β, which has been shown to promote T-cell exclusion and dysfunction in a variety of solid tumors.127,128 The blockade of immunosuppressive mediators, such as TGF-β, can promote a T-cell–inclusive TME that facilitates adoptive T-cell therapy for GBM. A novel approach for combating immunosuppressive mediators in the TME is to convert them into activating signals for CAR-T therapy. Recent work by Chen and colleagues demonstrated the ability to engineer CARs responsive to soluble antigens, including immunosuppressive cytokines, such as TGF-β.129 In particular, a CAR that recognizes the mature form of TGF-β and not the latent form has been shown to convert TGF-β into a potent activator of T-cell effector function.129 In addition, the TGF-β CAR outcompetes endogenous receptor and blocks endogenous TGF-β signaling, and it inhibits the conversion of naïve CD4+ T cells into Tregs in the presence of TGF-β.130 The TGF-β CAR itself does not trigger cytotoxicity, thus, it must be combined with a receptor that recognizes a surface-bound tumor antigen for direct tumor killing, potentially in the tandem-CAR or dual-CAR formats previously developed for the purpose of overcoming antigen escape.122,131
| Key Points |
|---|
| • Locoregional delivery of CAR-T cells can help in bypassing anatomical barriers involved in trafficking from circulation |
| • Strategies are needed to engineer T cells such that they are resistant to suppressive TME or target multiple antigens to overcome tumor heterogeneity, as well as rational combination with other agents that promote CAR-T-cell function and/or endogenous immune response, are likely needed to unleash the full potential of CAR therapy for GBM |
| Key Points |
|---|
| • Locoregional delivery of CAR-T cells can help in bypassing anatomical barriers involved in trafficking from circulation |
| • Strategies are needed to engineer T cells such that they are resistant to suppressive TME or target multiple antigens to overcome tumor heterogeneity, as well as rational combination with other agents that promote CAR-T-cell function and/or endogenous immune response, are likely needed to unleash the full potential of CAR therapy for GBM |
| Key Points |
|---|
| • Locoregional delivery of CAR-T cells can help in bypassing anatomical barriers involved in trafficking from circulation |
| • Strategies are needed to engineer T cells such that they are resistant to suppressive TME or target multiple antigens to overcome tumor heterogeneity, as well as rational combination with other agents that promote CAR-T-cell function and/or endogenous immune response, are likely needed to unleash the full potential of CAR therapy for GBM |
| Key Points |
|---|
| • Locoregional delivery of CAR-T cells can help in bypassing anatomical barriers involved in trafficking from circulation |
| • Strategies are needed to engineer T cells such that they are resistant to suppressive TME or target multiple antigens to overcome tumor heterogeneity, as well as rational combination with other agents that promote CAR-T-cell function and/or endogenous immune response, are likely needed to unleash the full potential of CAR therapy for GBM |
Lack of Adequate Imaging Technologies
Imaging is the standard noninvasive technique to measure the response to therapy in the absence of a biomarker to assess tumor progression or regression. However, early discrimination of pseudoprogression (inflammatory enlargement mimicking tumor growth) from disease progression remains a clinical challenge.132,133
Attempts to address the limitations of conventional MRI and the RANO (Response Assessment in Neuro-Oncology) criteria in the setting of immunotherapy have been made through the mRANO134 and iRANO criteria,135 The mRANO criteria take into account pseudoresponse from bevacizumab/steroids and pseudoprogression from treatment-related changes through standardization of imaging acquisition with the international brain tumor imaging protocol (BTIP), consideration of lesion volume rather than bi-dimensional measurements, and the use of the postsurgical MRI as baseline to assist in clinical trial stratification during randomization. The iRANO criteria propose that within the first 6 months of initiation of an immunotherapy, therapy may be continued with repeat surveillance MRIs in 3 months or less, if the patient remains clinically stable or improved.
Several advanced MRI techniques show promise in delineating responses to therapy.132 Perfusion MRI through dynamic susceptibility contrast imaging, dynamic contrast-enhanced imaging, and atrial spin labeling add value in determining progression vs pseudoprogression. However, perfusion may be confounded by factors, such as tumor vascularity (transfer coefficient ktrans) or fever which may increase cerebral blood volume to an area.
Diffusion-weighted sequencing characterizes the high cell density of tumor. However, immune infiltration into a tumor may likewise increase density. Additionally, postoperative cellular injury in the setting of surgical trauma, vascular injury, or devascularization of tumor may contribute to diffusion restriction.136
The labeling of DCs with iron oxide or indium may noninvasively evaluate the fate of adoptively transferred cells.137,138 However, detrimental effects on cell viability, migration, differentiation, and immune function have limited its use in cellular imaging.139 Further investigation in advanced MRI techniques may explore changes in diffusion characteristics of other lymphoid organs, such as the spleen or lymph nodes to infer systemic immune responses to immunotherapy.140,141
The limitations of anatomic imaging have spurred the exploration and development of novel imaging techniques, including magnetic resonance spectroscopy (MRS), metabolic pH with chemical exchange saturation transfer (CEST), and novel positron emission tomography (PET) probes.142,143
MRS allows for the noninvasive measurement of metabolites to help improve the diagnostic value of conventional MRI. By measuring isotopes of 1H, 13C, or 31P, it is possible to correlate derangements in concentrations of metabolites with tumor grade.144 Identification of metabolites of immunotherapy or inflammation may provide a marker to identify pseudoprogression.
CEST is a noninvasive pH-weighted modality that characterizes tissue acidosis via amine protons on glutamine from abnormal tumor perfusion and metabolism. Acidic signatures in malignant GBM correlate with histology of active and pseudopalisading tumor, perfusion abnormalities, lactate on MRS, and elevated FDOPA PET uptake.145 Even in the setting of bevacizumab use, residual or emerging regions of acidity predicted areas of tumor progression.146
Amino acid PET has been evaluated in brain tumors, with preliminary positive results with 11C-methyl-l-methionine (MET), O-(2-[18F]fluoroethyl)-l-tyrosine (FET), and 3,4-dihydroxy-6-6[18F]-fluoro-l-phenylalanine (FDOPA).147,148 The mechanism appears to be mediated by increased transport of large neutral amino acids through transport systems LAT1 and LAT2.
| Key Points |
|---|
| • The challenge of distinguishing pseudoprogression from progression leads to the early removal of patients from studies, curtailing our ability to evaluate the long-term benefits of immunotherapy |
| • Advanced MRI techniques including perfusion MRI, diffusion-weighted MRI, and iron-oxide or indium-labeled MRI may help to improve diagnostic decision-making |
| • The limitations of anatomic imaging have spurred exploration and development of novel imaging techniques, including MRS, metabolic pH with CEST, and novel PET probes |
| Key Points |
|---|
| • The challenge of distinguishing pseudoprogression from progression leads to the early removal of patients from studies, curtailing our ability to evaluate the long-term benefits of immunotherapy |
| • Advanced MRI techniques including perfusion MRI, diffusion-weighted MRI, and iron-oxide or indium-labeled MRI may help to improve diagnostic decision-making |
| • The limitations of anatomic imaging have spurred exploration and development of novel imaging techniques, including MRS, metabolic pH with CEST, and novel PET probes |
| Key Points |
|---|
| • The challenge of distinguishing pseudoprogression from progression leads to the early removal of patients from studies, curtailing our ability to evaluate the long-term benefits of immunotherapy |
| • Advanced MRI techniques including perfusion MRI, diffusion-weighted MRI, and iron-oxide or indium-labeled MRI may help to improve diagnostic decision-making |
| • The limitations of anatomic imaging have spurred exploration and development of novel imaging techniques, including MRS, metabolic pH with CEST, and novel PET probes |
| Key Points |
|---|
| • The challenge of distinguishing pseudoprogression from progression leads to the early removal of patients from studies, curtailing our ability to evaluate the long-term benefits of immunotherapy |
| • Advanced MRI techniques including perfusion MRI, diffusion-weighted MRI, and iron-oxide or indium-labeled MRI may help to improve diagnostic decision-making |
| • The limitations of anatomic imaging have spurred exploration and development of novel imaging techniques, including MRS, metabolic pH with CEST, and novel PET probes |
PET may also be used to evaluate tumor markers such as PD-(L)1. In a first-in-human evaluation of non–small cell lung cancer, Zirconium-labeled nivolumab, and Fluor-labeled anti-PD-L1 Adnectin were successfully used to image PD-(L)1 expression of tumors and could be correlated with response to therapy.149 However, there exist limitations in intracranial PET-tracer uptake secondary to the BBB.150
The efficient access to and sharing of imaging remains a considerable challenge. By streamlining the ability to share data, it is possible to harness the power of advanced imaging in large trials to address questions, such as the impact of immune-related inflammation on survival. By leveraging radiomics and artificial intelligence to ground image analysis in pathology and immunological profiles, it may be possible to finally use imaging to identify pseudoprogression and predict response.151–153
Preclinical Models
While adoptive T-cell therapies, such as TCR-transgenic and CAR-T cells, can be studied in xenograft models using established human cell lines or patient-derived xenografts in immunocompromised mice, most immunotherapies modulating the endogenous immune system require novel syngeneic mouse glioma cell lines, such as SB28.154,155 SB28 cells present clinically relevant characteristics, such as the low level of mutation load similar to human GBM, invasive growth and resistance to PD-1 and CTLA-4 double blockade therapy.154 While de novo genetically engineered mouse models more thoroughly recapitulate genetic lesions found in human gliomas, to study therapies directed against human glioma neoepitopes, mice with a human MHC system have been established.22,156 There is an increasing availability of patient avatar models, where mice are reconstituted with a human immune system. Although these models in principle can more faithfully predict response to immunotherapies for individual patients, they are complex and require more extensive efforts and time to derive information for individual treatment decisions. While these models should be utilized more extensively in a variety of preclinical evaluations, the limitations emphasize the need for thoroughly designed, biomarker-driven clinical trials.
Clinical Trial Design
At least 50 clinical trials have evaluated immune-mediated therapies for GBM since 1997.157 However, nearly all have failed in a randomized Phase III context. Immunological rejection of tumor cells requires a complete cascade of immune mechanisms, including antigen presentation, activation, and tumor-homing of effector cells as well as mitigation of immunosuppression. As such, the field recognizes the strong need of rational combination regimens. We would like to stress that, when we design combination regimens, it is critical to validate that each of the therapeutic agents has adequate levels of intended biological activities even if the agent did not show therapeutic efficacy as monotherapy. For example, if a vaccine is combined with a checkpoint blockade agent, the vaccine itself should be able to induce robust expansion of antigen-specific effector cells. It is therefore important to critically review biological data from relevant early-phase studies.
To effectively measure immunologic outcomes in small cohorts of patients, it is necessary to develop a complex and coordinated process in the form of smartly designed clinical trials.
Critical issues in the clinical evaluation of immunotherapies include: (1) appropriate clinical trial design, (2) establishment of dosing, timing, and sequencing of therapy, (3) identification of immunological baselines, (4) determination of time points for evaluation, and (5) development of relevant biomarkers for future interventions.
The factorial approach in clinical trial design allows for a randomized, multiple-arm study evaluating a combination of several therapies simultaneously and efficiently.158 Given the urgent need for effective therapies, the factorial approach lends to efficiency—but potentially at the cost of loss of statistical power, inability to account for unexpected synergistic interactions of therapies, and variability due to intra-patient or inter-patient changes in dosing.159,160 The first Phase II factorial study in newly diagnosed GBM involved 8 arms with 20 participants per arm. The primary objective compared the efficacy of adjuvant TMZ to adjuvant TMZ plus up to three additional agents (isotretinoin, celecoxib, and/or thalidomide). The secondary objective compared individual arms and doublet vs triplet therapy.161 Perhaps more importantly, this seminal investigation established the feasibility of factorial clinical trials in neuro-oncology.
Neoadjuvant studies provide a systematic method of evaluating hypotheses regarding mechanisms and biology (Figure 5). By applying a therapy and then undergoing resection to obtain tissue, neoadjuvant studies make possible the ability to isolate and distinguish immune effects from treatment and develop specific biomarkers to evaluate the effects in future interventions.75 Furthermore, neoadjuvant perturbations may isolate immune pharmacodynamic effects, and evaluations of combinations of therapies may provide regulatory implications by attributing clinical effects to combinations. The optimal time points for evaluation remain incompletely characterized. For initial anti-tumor effects, the immune response peaks between 1-3 weeks. For adaptive resistance, longitudinal re-sampling will be necessary.162 It is also paramount to keep in mind that early biological effects may not necessarily parallel late clinical effects. The baseline immune landscape and precise dosing, timing, and sequencing of therapies warrant further investigation.
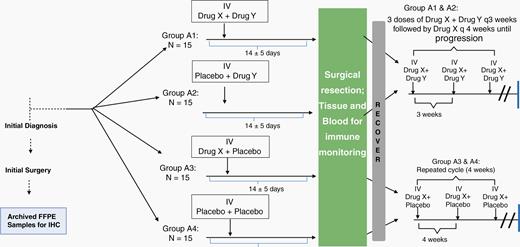
Neoadjuvant trial design schema. A factorial approach facilitates the simultaneous evaluation of several combinations of agents (Drug X + placebo, Drug Y + placebo, placebo + placebo, Drug X + Drug Y). By applying a therapy and then undergoing resection to obtain tissue, neoadjuvant studies provide a systematic method of isolation of pharmacodynamics, immune, and clinical effects of treatment.
Through a globally coordinated effort, we are able to ask and answer questions faster in patients.163 The GBM Adaptive Global Innovative Learning Environment (GBM AGILE),163 Individualized Screening Trial of Innovative Glioblastoma Therapy (INSIGHT),164 and the NCT Neuro Master Match165,166 are novel, multi-arm, platform trials using Bayesian adaptively randomized screening to identify the treatment arm with the most promising effect on OS compared to a common control, while balancing clinical factors and biomarkers. This design makes it possible to seamlessly add or drop experimental arms as patient accrual continues. As we design clinical trials and discover biomarkers, it will be important to biobank cerebrospinal fluid, serum, plasma, and tissue for comparisons across multiple treatments and over time in a longitudinal evaluation.
| Key Points |
|---|
| • Clinical trials involving a factorial design allow for the simultaneous evaluation of multiple therapies at once |
| • The GBM AGILE, INSIGHT, and the NCT Neuro Master Match represent the next wave of smartly designed clinical trials |
| • Window-of-opportunity, neoadjuvant Phase 0 studies provide a systematic method of evaluating therapeutic mechanisms |
| Key Points |
|---|
| • Clinical trials involving a factorial design allow for the simultaneous evaluation of multiple therapies at once |
| • The GBM AGILE, INSIGHT, and the NCT Neuro Master Match represent the next wave of smartly designed clinical trials |
| • Window-of-opportunity, neoadjuvant Phase 0 studies provide a systematic method of evaluating therapeutic mechanisms |
| Key Points |
|---|
| • Clinical trials involving a factorial design allow for the simultaneous evaluation of multiple therapies at once |
| • The GBM AGILE, INSIGHT, and the NCT Neuro Master Match represent the next wave of smartly designed clinical trials |
| • Window-of-opportunity, neoadjuvant Phase 0 studies provide a systematic method of evaluating therapeutic mechanisms |
| Key Points |
|---|
| • Clinical trials involving a factorial design allow for the simultaneous evaluation of multiple therapies at once |
| • The GBM AGILE, INSIGHT, and the NCT Neuro Master Match represent the next wave of smartly designed clinical trials |
| • Window-of-opportunity, neoadjuvant Phase 0 studies provide a systematic method of evaluating therapeutic mechanisms |
Conclusions and Moving Forward
The Think Tank meeting provided unique and privileged opportunities in which investigators in CNS and non-CNS tumor and immunology fields had problem-oriented discussions and shared novel insights that can be evaluated in CNS tumors. Moving forward, novel technologies in the assessment of TME and cell engineering, such as CAR-T cells, should be integrated with other important aspects that we discussed in this meeting, such as innovative clinical trial design and imaging. To further develop our discussions and accelerate clinical translation, future meetings of this series should include representatives of other areas, including but not limited to industry and regulatory agencies.
The Roadmap
Most importantly, taking valuable insights from this roundtable, we propose a roadmap to assist future studies to accelerate progress and collaborations. In particular, this revolves around six issues that need to be addressed concurrently (Figure 6):
The roadmap.
(1) A comparison between the anti-tumor immune response in successfully treated extracranial solid tumors should help the brain tumor field better understand what is lacking or missing in GBM patients treated with immunotherapy.
(2) New high-dimensional technologies, both using single-cell data and in situ, should provide our field a more complete understanding of the immune cell types present within GBM and can be compared with new data from extracranial malignancies.
(3) While we should continue extending our understanding on these issues, we already know some of the unique challenges of CNS cancers, such as BBB and blood-tumor barrier. We must enhance our engineering efforts by collaborating with relevant experts to develop more effective T-cell and antibody-based therapies, for example. These efforts should be integrated with considerate trial designs with the window of opportunity.
(4) Properly designed early-phase “window of opportunity” clinical trials in patients can yield an enormous amount of new information regarding how discrete immunotherapies influence the microenvironment of GBM.
(5) Collaborative ventures and the sharing of primary data between investigators can help to speed the understanding of how different immunotherapeutic interventions alter the anti-tumor immune response.
(6) Finally, the development of a regulatory group to help offer many of the new immunotherapeutic agents, frequently restricted to patients with extracranial tumors, could dramatically enhance the scientific understanding of the anti-tumor immune response in brain tumor patients.
Funding
National Institute of Neurological Disorders and Stroke R35 NS105068-01(H.O.), National Cancer Institute P50CA211015 (R.M.P.).
Acknowledgments
The authors thank the Society for Neuro-Oncology and their Staff for their tremendous contributions to the arrangements and coordination for the meeting.
Authorship statement. Writing and editing of the original manuscript by all authors. Revision by P.C., F.C., P.B.W., M.G., M.P., R.M.P., and H.O. We declare no parts of the manuscript have been previously published or submitted concurrently to any other journal and all co-authors have read and approved of its submission to this journal.
Conflicts of interest statement. Y.C.: an inventor on a patent related to the TGF-beta CAR technology discussed in the publication. E.A.C.: currently an advisor to Advantagene Inc., Alcyone Biosciences, Insightec, Inc., DNAtrix Inc., Immunomic Therapeutics, Sangamo Therapeutics, Seneca Therapeutics and has equity interest in DNAtrix, Immunomic Therapeutics, Seneca Therapeutics; he has also advised Oncorus, Merck, Tocagen, Ziopharm, Stemgen, NanoTx., Ziopharm Oncology, Cerebral Therapeutics, Genenta. Merck, Janssen, Karcinolysis, Shanghai Biotech. He has received research support from NIH, US Department of Defense, American Brain Tumor Association, National Brain Tumor Society, Alliance for Cancer Gene Therapy, Neurosurgical Research Education Foundation, Advantagene, NewLink Genetics, and Amgen. He also is a named inventor on patents related to oncolytic HSV1 and noncoding RNAs. R.A.D.: founder, director, and/or advisor of Tvardi Therapeutics, Nirogy Therapeutics, Stellanova Therapeutics, Asylia Therapeutics, and Sporos Bioventures. B.M.E.: For Profits—Medicenna—Paid Consultant—Significant (>$20k), MedQIA, LLC—Paid Consultant, Ad Board, Neosoma—Paid Consultant, Agios Pharmaceuticals—Paid Consultant, Ad Board, Siemens—Paid Consultant, Research Grant, Ad Board, Janssen Pharmaceuticals—Research Grant, Imaging Endpoints—Paid Consultant, Kazia/Novogen—Paid Consultant, Ad Board, VBL—Research Grant, NW Biopharmaceuticals—Paid Consultant, Ad Board, Oncoceutics Inc.—Consultant, BeiGene—Consultant, BBI—Consultant, Tocagen—Consultant; Non-Profits: Global Coalition for Adaptive Research (GCAR)—Consultant, NIH/NCI Cancer Imaging Steering Committee—Paid Consultant, National Brain Tumor Society—Research Grant, American Cancer Society—Research Grant. J.R.H.: the founder and board member of the following companies: Indi Molecular, PACT Pharma, Isoplexis, and Sofie Biosciences. A.B.H.: serves on the scientific advisory board of Caris Life Science and WCG Oncology Board, he received licensing fees from Celldex Therapeutics and DNAtrix, received clinical trial funding from Merck and Celularity, and received study drug from Moleculin. N.K.: consultant for Abintus Bio, GenVivo, and SillaJen Biotherapeutics. L.M.L.: Consultant fees—JW Creagene, InSightec, Arbor Pharmaceuticals; Research grants to institution—Northwest Biotherapeutics, Merck. M.L.: Research Support—Arbor, BMS, Accuray, DNAtrix, Tocagen, Biohaven, Kyrin-Kyowa; Consultant—Tocagen, SQZ Technologies, VBI; CMO—Egret Therapeutics, Patent, Focused radiation + checkpoint inhibitors, Non-research Consultant – Stryker. E.W.N.: a co-founder, advisor, and shareholder of ImmunoScape Pte. Ltd. and an advisor for Neogene Therapeutics and Nanostring Technologies. H.O.: Consultant fees: Bristol-Myers Squibb, Agios Pharmaceuticals, Grant/Research support from: Agios Pharmaceuticals. Royalties as Inventor of: the H3.3K27M TCR, IL-13Ra2 (345-353:1A9V) peptide, and EGFRvIII-CAR for which an exclusive licensing agreement has been executed with Tmunity, Inc., Stemline, Inc., and Novartis Pharma, respectively. M.P.: reports non-financial support from Roche, personal fees and non-financial support from Bayer, personal fees from Novartis, personal fees from Apogenix, non-financial support from Pfizer, personal fees from Affiris outside the submitted work. In addition, M.P. has a patent EP2753315B1 licensed to Bayer, a patent EP2800580B1 issued, a patent US20180155403A1 pending, a patent US20180246118A1 pending, a patent US20170254803A1 pending, a patent WO2018146010A1 licensed to Bayer, a patent WO2019101643A1 licensed to Bayer, a patent WO2019101647A1 licensed to Bayer, a patent WO2019101641A1 licensed to Bayer, and a patent WO2019101642A1 licensed to Bayer. P.Y.W.: Research Support—Agios, Astra Zeneca/Medimmune, Beigene, Celgene, Eli Lily, Genentech/Roche, Kazia, MediciNova, Merck, Novartis, Oncoceutics, Vascular Biogenics, VBI Vaccines; Advisory Board—Agios, Astra Zeneca, Bayer, Boston Pharmaceuticals, Immunomic Therapeutics, Karyopharm, Kiyatec, Puma, Taiho, Vascular Biogenics, Deciphera, VBI Vaccines, Tocagen, Voyager, QED, Imvax, Elevate Bio; Speaker—Merck, Prime Oncology. All other authors declared none.
References
Author notes
These authors contributed equally to this work.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}