





Abstract
Cellular heterogeneity is a hallmark of advanced cancers and has been ascribed in part to a population of self-renewing, therapeutically resistant cancer stem cells (CSCs). Glioblastoma (GBM), the most common primary malignant brain tumor, has served as a platform for the study of CSCs. In addition to illustrating the complexities of CSC biology, these investigations have led to a deeper understanding of GBM pathogenesis, revealed novel therapeutic targets, and driven innovation towards the development of next-generation therapies. While there continues to be an expansion in our knowledge of how CSCs contribute to GBM progression, opportunities have emerged to revisit this conceptual framework. In this review, we will summarize the current state of CSCs in GBM using key concepts of evolution as a paradigm (variation, inheritance, selection, and time) to describe how the CSC state is subject to alterations of cell intrinsic and extrinsic interactions that shape their evolutionarily trajectory. We identify emerging areas for future consideration, including appreciating CSCs as a cell state that is subject to plasticity, as opposed to a discrete population. These future considerations will not only have an impact on our understanding of this ever-expanding field but will also provide an opportunity to inform future therapies to effectively treat this complex and devastating disease.
Diversity within the CSC compartment is shaped by a series of cell intrinsic and extrinsic interactions.
CSCs are not a discrete population but rather a state that is subject to evolutionary principles.
Introduction—the Evolutionary Ground State
Cancer and developmental biology remain closely linked and continue to provide unique insights into one another. The concept of tumors representing aberrantly developing organs has been extended to posit that the cellular heterogeneity present in advanced cancers is driven by a self-renewing cancer stem cell (CSC) population. In the past few decades, the presence of cancer cells in stemlike states has been demonstrated across multiple advanced cancers including glioblastoma (GBM). The past decade in the GBM field has shaped our understanding of CSCs as multiple heterogeneous and fluid cellular phenotypes rather than a singular, discrete cell type. As a result, we have come to appreciate that CSCs are a product of their interactome and have the ability to adapt to metabolic, immune and microenvironmental changes, as well as therapeutic stimuli, and to simultaneously modify their microenvironment to support their own persistence. In this review, we provide a historical background on CSCs and discuss recent developments on CSCs in established aspects of cancer including genetic alterations, self-renewal signaling, tumor initiation, and therapeutic resistance. In addition, we discuss the evolving role of epigenetic and metabolic signatures in CSCs and evidence for functional interactions of CSCs with non-tumor cell types in GBM. As our understanding of GBM continues to evolve, these advances call for a more holistic view of GBM that incorporates the CSC state as a driver that is subject to the evolutionary principles of variance, inheritance, selection, and time.
A History of CSCs
CSCs are functionally defined as tumor cells with 2 main properties: self-renewal and capacity for initiation of tumors that maintain the cellular heterogeneity of the parental tumor. Self-renewal refers to the ability to produce at least one daughter cell that maintains stem cell identify upon division1 and can be assessed through in vitro sphere formation capacity, while a more precise determination of stem cell frequency can be estimated through in vitro limiting dilution analysis and in vivo tumor formation.1 More stringently, long-term self-renewal is measured through serial colony formation assays and serial transplantation in vivo.2 The existence of CSCs in GBM was initially shown in clinical samples from patients with recurrent GBM, where specimens contained clones capable of anchorage-dependent growth, generation of tumor cells containing multiple lineage markers, and formation of tumors with cellular heterogeneity in orthotopic xenografts.2–4 In addition, self-renewal, as read out by neurosphere formation in culture, was a positive predictor of poorer overall survival in GBM patients.5 Consistently, primary tumors contained single GBM cells that could also give rise to clonal cell lines exhibiting multipotency as well as long-term proliferation and self-renewal. These initial studies showed differential function between CSCs and non-stem tumor cells based on enrichment for the cell surface glycoprotein CD133.3 The proportion of CD133+ cells in primary tumors is quite variable. Early studies estimated the proportion of CD133+ cells in the tumor to be between 19.5% and 28%,3 while later studies with expanded patient numbers found this frequency to range 0.5‒10%6 and 1‒50%.7 Meanwhile, sphere formation rate (required but not sufficient criteria to demonstrate stemness) from unsorted primary tumors has been estimated to be 0.5–31%2 and 20–24%.3 Importantly, increased proportion of CD133+ cells correlates with shorter overall survival for GBM patients, supporting the functional relevance of this population.6,7 However, this single marker was not sufficient to identify the CSC population across all tumors, and there are several examples of CD133− CSC populations.8 Based on these caveats, a series of additional cell surface markers that show differences in self-renewal and tumor initiation have been validated (Table 1). Despite the utility of these markers for enrichment and allowing the study of stemlike cells in primary GBM specimens, it has become clear that tumor-initiating potential is not restricted to cells expressing these surface markers. By cell surface marker expression and functional profiling, CSC isolation is relatively high in GBM compared with other tumors.9 This may relate to factors such as the ability of CSCs to persist, and therefore expand, within the hostile tumor environment of GBM.10 Altogether, these findings support the notion of therapeutic targeting of these population(s).
| Putative CSC marker |
|---|
| CD133 |
| CD15 |
| A2B5 |
| L1CAM |
| Integrin α6 |
| CD44 |
| ALDH |
| ABCG2 |
| Putative CSC marker |
|---|
| CD133 |
| CD15 |
| A2B5 |
| L1CAM |
| Integrin α6 |
| CD44 |
| ALDH |
| ABCG2 |
| Putative CSC marker |
|---|
| CD133 |
| CD15 |
| A2B5 |
| L1CAM |
| Integrin α6 |
| CD44 |
| ALDH |
| ABCG2 |
| Putative CSC marker |
|---|
| CD133 |
| CD15 |
| A2B5 |
| L1CAM |
| Integrin α6 |
| CD44 |
| ALDH |
| ABCG2 |
In addition to sphere formation as an assessment of self-renewal, single-cell assessment of cell division in CSCs can be informative. When primary human GBM-derived CD133+ cells were subjected to ex vivo growth, symmetric stem cell-expanding divisions were the primary mode of cell division observed, followed by asymmetric divisions (production of one stem cell and one differentiated daughter cell), which maintain the stem cell pool while also increasing tumor heterogeneity.11 This is in contrast to normal neural stem cells (NSCs), which primarily undergo asymmetric self-renewing divisions during neurogenesis.12 One described mechanism underlying this disparity is enrichment of the transcription factor HMGA1 in the CD133+ population of CSCs, leading to downregulation of the endocytic, asymmetric division-promoting protein Numb.13 Examination of the segregation of CSC markers in mitosis revealed that in some mitotic events in CSCs, CD133, but not other CSC markers, was asymmetrically segregated.11 A likely explanation is that CD133 and other CSC markers represent different subgroups of CSC populations.
Genetically engineered mouse models have also been widely used in GBM to model tumor development and dynamics in an immunocompetent setting. These models hold potential for studying CSC populations due to the ability to label CSC populations with NSC-specific promoters. Although these models do not recapitulate all human CSC findings, some markers including CD133,14,15 ABCG1,15 ABCG2,16 and Sox2,17 have been found to be expressed in populations with CSC properties in mouse models of glioma and GBM. In both mouse and human, the number of CD133+ cells needed for tumor initiation was found to be 100-fold less than their CD133− counterparts.14,18 Additional studies on genetic mouse models hold promise in that they provide opportunity for the prospective isolation, study and monitoring of CSCs from tumor initiation through disease progression, treatment, and recurrence.
Mosaicism of Discrete CSC Populations
The conceptual framework for CSCs and cellular heterogeneity has been adapted from adult neurogenesis. Specifically, the application and interpretation of the neurosphere assay has been widely used for the assessment of CSCs in GBM.19 When applied to a mixed population of neural cell types, emergence of multipotent neurospheres retroactively indicated the presence of NSCs. Similarly, sphere generation from GBM biopsies or tumor tissue harvested from animal models suggested the presence of CSCs. The original model for adult neurogenesis held that NSCs residing within the subventricular zone (SVZ) represented a single homogenous population responsible for generating all of the new interneurons that eventually integrate into the olfactory bulb. Initial experimental designs supported this notion by revealing NSC activity from the bulk population of SVZ astrocytes. When applied to GBM, the assertion was therefore made that a single population of CSCs would be sufficient to generate the full spectrum of heterogeneous cell types within the tumor. Over the course of time, continued research has challenged this framework in both cases. For adult neurogenesis, the current view embraces the idea of an NSC mosaic. Rather than one parental NSC population, multiple, non-overlapping and non-interconvertible NSCs occupy anatomically precise locations with the SVZ. These distinct NSCs give rise to specific subsets of olfactory interneurons.20
Similarly, recent work suggests that CSCs in GBM may also resemble a mosaic. For example, the coexistence of multiple, morphologically distinct, non-interconvertible CSC populations within single biopsies from a series of patients has been described.21 These CSC populations each satisfied the in vitro requirements of long-term self-renewal via sphere formation. Interestingly, when transplanted into the brains of immune compromised mice, the individual CSC populations each generated a limited repertoire of the cells present in the original patient biopsy. Even more importantly, the individual CSC populations demonstrated unique profiles of therapy resistance. Administration of any single agent or combination of 2 agents effectively eliminated a small number of treatment-sensitive CSC populations. However, the remaining treatment-resistant populations quickly filled the vacated space. Thus, just as adult neurogenesis is driven by a collection of distinct NSC populations, the development and progression of GBM represents the concerted effort of multiple CSC populations.
The concept of CSC mosaicism is also supported by recent single cell RNA sequencing efforts. For example, contrasting transcriptional signatures were generated from non-stem tumor cells cultured in 10% serum, and CSCs cultured in defined, serum-free conditions. When the RNA from hundreds of individual tumor cells isolated from 5 separate patients was compared with these signatures, the authors observed that the primary, patient-derived tumor cells occupy positions continuously across a spectrum of transcriptional states between non-stem tumor cells and CSCs.22 The majority of these tumor cells would be expected to satisfy the functional definition of CSCs, as observed by other studies21; however, single-cell RNA sequencing confirmed that individual phenotypic and potentially functional differences are likely difficult to appreciate when the relatively low-resolution neurosphere assay is utilized. Moreover, it remains unclear if the multitude of CSC populations revealed by single cell RNA sequencing are a reflection of independent clones or a result of plasticity that shapes a limited number of original CSC clones. In addition, it is unclear how this mosaicism informs the cell of origin of CSCs, as this remains a key unanswered question.
The concept of mosaicism is further supported by studies of tumor evolution. As early as 1940, neuropathologists described GBM as a diffuse and disconnected disease spread across the brain. They observed no evidence suggesting that the tumor emerged or was sustained by a central or singular aggressive focus.23 These early inferences have been supported by recent studies in humans and mouse models using distinct methodology that concluded that GBM actually comprises multiple, non-overlapping malignant gliomas, which evolve over time and anatomical space.24,25 These separate but commingled tumors work in concert with one another and present holistically as GBM. Again, each of these separate tumors are populated by numerous, distinct tumor cells that satisfy the functional definition of CSCs. However, it is important to note that, while the CSC population shares some functional characteristics, individual CSC lineages are likely not the same, may not share the same cell of origin, and are sensitive to different drug combinations. Thus, a singular CSC targeting scheme will likely be ineffective.
CSCs and Therapeutic Resistance
Standard therapy in GBM includes surgical resection, radiation and chemotherapy (specifically the alkylating/methylating agent temozolomide [TMZ]). Poor prognosis in GBM has been attributed to several factors including resistance to conventional therapies, which is enhanced in CSCs and ultimately drives evolutionary selection. In both human patients and mouse models, therapeutic resistance occurs due to a variety of mechanisms, including enhanced DNA damage response and drug efflux pumps.4,26–32 In addition, several signaling pathways, cellular processes, and cell states have been linked to resistance, including autophagy,33 gap junction proteins,34,35 activation of the Wnt and Notch pathways directly or via prosurvival proteins,36–38 and activation of the mesenchymal program.39–42 CSC mosaicism adds an additional layer to the numerous underlying mechanisms of therapy resistance in GBM (Fig. 1).
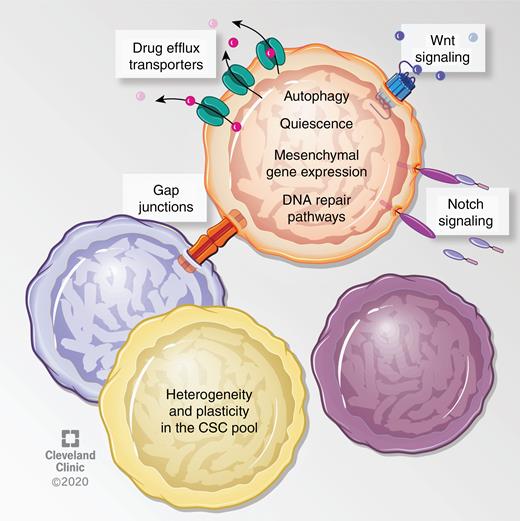
Described mechanisms of therapeutic resistance in GBM CSCs. Several studies have ascribed therapy resistance to features of GBM CSCs that prevent or antagonize the effects of these therapies. For instance, slowly dividing/quiescent GBM cells, which exhibit higher tumor initiating frequency, were resistant to radiation and chemotherapy.28,30,32 In addition, CD133+ GBM cells express higher levels of drug efflux transporters such as ABCG2, ABCC1, and ABCC3, and also highly express DNA repair enzymes such as MGMT.27,28,31,133 In addition, several pathways, processes and cellular states have been linked to radiation resistance. For instance, CD133+ GBM cells express higher levels of autophagy related proteins LC3, ATG5, and ATG12, radiation specifically prompted LC3-II expression in CD133+ GBM cells, and inhibition of autophagy sensitized cells to radiation.33 Inhibition of the gap junction protein connexin-46 or -43 sensitized TMZ resistant cells to TMZ-mediated apoptosis.34,35 Activation of the Wnt and Notch pathways have been described in radioresistant GBM CSCs, and radiosensitization could be achieved by inhibiting Wnt signaling or with gamma secretase inhibitors, which target the Notch pathway.36,37 In a similar manner to Notch, genetic targeting of receptor tyrosine kinase c-Met or its ligand sensitized cells to TMZ.38 Furthermore, primary patient-derived GBM CSCs with mesenchymal-like gene expression profiles were radioresistant, and radiation induced a proneural to mesenchymal gene expression profile in GBM cells,40,42 suggesting an enrichment or expansion of the resistant cells upon treatment. Knockdown of the master regulator of the epithelial to mesenchymal transition, ZEB1, sensitized GBM CSCs to chemotherapy through reducing expression of MGMT and c-MYB.41
Efficacious treatment strategies in preclinical GBM models have often failed to yield therapeutically relevant results in the clinic. While this phenomenon is not specific to GBM, researchers are beginning to understand the underlying reasons and overcome these limitations. Coexisting CSC populations, a variety of niches, and multiple expression-based subtypes existing in the same tumor43 logically result in a tumor containing subclones of differing drug sensitivities, as recently demonstrated.21 Preserving tumor heterogeneity when performing laboratory research on primary GBM specimens derived from resected tumors is a major challenge. The existence of multiple CSC niches indicates individual models may be missing vital subclones that exist in non-resectable portions of the tumor. In fact, differences have been observed between resected GBM cells and residual tumor cells remaining after surgery, as well as between core and marginal tumor regions, including differences in proliferation, invasion, self-renewal, cell surface antigen profile, mutational spectrum, and chemotherapy/radiation sensitivity.41,44,45 This heterogeneity highlights the importance of identifying the genetic and phenotypic characteristics of subclones and correlating these characteristics with drug sensitivity to predict treatment responses and inform second line treatments.21 One way in which these limitations have been addressed is through improved ex vivo modeling of GBM, such as tumor organoids (discussed further in a later section). These approaches are becoming standard and are likely to provide additional insight into therapeutic response.46 Collaborative studies such as the Glioma Longitudinal Analysis (GLASS) Consortium have documented cellular and molecular evolution in gliomas throughout treatment and recurrence in order to better understand treatment resistance.47 As CSCs are implicated in therapy resistance, undertaking such studies with isolated CSCs are essential to characterizing how they evolve after chemo-radiation regimens.
Cell Intrinsic Factors Driving CSCs—the Root of Evolutionary Inheritance and Variation
CSC Signaling Networks
The molecular features underlying self-renewal phenotypes in CSCs include dependence on pluripotency transcription factors and co-opting of core developmental programs. For example, Olig2, a transcriptional repressor necessary for maintaining replication competence in oligodendrocyte precursors, is necessary for glioma development.48 The expression of OLIG2 together with 3 other neurodevelopmental transcription factors, POU3F2, SOX2, and SALL2, was sufficient to induce a CSC state in differentiated glioma cells.49 Similarly, co-expression of OLIG2, SOX2, and ZEB2 has been implicated in maintaining GBM CSCs.50 Several other developmentally important factors are highly expressed and functionally important for GBM CSC growth, including Nestin, sonic hedgehog (SHH), Notch, SOX2, ZEB1, and ZFX.1 These developmental factors, including SHH and Notch, have been the basis for CSC targeting strategies that are at various stages of clinical evaluation (extensively reviewed by Saygin et al9). Receptor tyrosine kinases expressed on neural stem and progenitor cells and CSCs—including FGFR2, EGFR, and PDGFRα—activate these transcription factors to maintain the CSC state.51–53 In fact, expression of these receptors was exploited in early studies to identify and expand tumorigenic, NSC-like GBM cells from primary tumors.2,19 Given the functional importance of the above-mentioned master transcription factors that maintain the undifferentiated state and promote proliferation programs, disrupting these programs and forcing differentiation has been one objective of GBM therapy. One example is the use of bone morphogenic proteins (BMPs) such as BMP4, which drastically reduced tumor initiation and disease penetrance in mouse models of GBM54. This was ascribed to reduction in self-renewal through the induction of asymmetric cell division, diminishing the CSC pool, and pushing cells into mature post-mitotic states.55 However, extensive heterogeneity has been observed in BMP4 responsiveness in other studies,56,57 highlighting the importance of assessing therapeutic responses across multiple CSC populations.
Genetic Alterations
The phenotypic heterogeneity of GBM can be attributed, in part, to genetic changes present in these tumors. A number of genetic alterations frequently found in GBM, such as EGFR and PDGFRA amplifications, as well as mutations in PTEN, NF1, PDGFRA, and TP53, are common features of many tumor types.58 In addition, FGFR-TACC (transforming acidic coiled-coil) fusions are found in gliomas, resulting in constitutive activation of FGFR signaling as well as chromosomal instability and aneuploidy.59 In contrast, isocitrate dehydrogenase (IDH1) mutations, which lead to a CpG island methylator phenotype (G-CIMP), are preferentially enriched in GBM compared with other cancers and have prognostic value in clinical settings.60 It is important to note that, while IDH1 mutations act as drivers in the setting of low grade gliomas, GBM CSCs from IDH1 mutant tumors often do not exhibit the G-CIMP phenotype. This may be partially explained by the observation that mutant IDH1 plays an important role in tumor initiation but is dispensable for tumor maintenance.61,62 Further investigation is necessary to understand why some mutations, such as IDH1, are not commonly found in CSCs. While uncovering the genetic landscape of GBM has provided a more comprehensive understanding of this disease, there has been limited clinical success in therapeutically targeting commonly mutated genes. Molecular subtypes have been identified within GBM tumors based on integration of gene expression and mutational profiles,63,64 but their link to the cell of origin for an individual tumor or CSC population remains unclear. The clinical significance of these continually evolving subtypes has begun to emerge.63–65 For example, the mesenchymal subtype is associated with more aggressive, recurrent tumors63,66 and a transition to a mesenchymal state has been observed in GBM CSCs after exposure to radiation therapy, indicating a role for cellular subtypes in therapeutic resistance.42 Importantly, these assessments have been made from heterogeneous GBM tumors and the extent to which genetic alterations are present CSCs, as well as the heterogeneity of these alterations at the single cell level, remains unclear. However, both of these questions may be directly addressed with emerging single cell whole genome sequencing platforms.
Although CSCs from primary and recurrent tumors share common mutations, recurrent CSCs can accumulate lesions and exhibit distinct mutation patterns relative to primary CSCs.67 These findings also highlight the functional significance of genetic mutations in GBM CSCs and their contribution to tumor recurrence and therapeutic resistance, which remain a major challenge in the treatment of this disease.67 A recent study utilized CRISPR-Cas9 dropout screens in GBM CSCs to help uncover genetic vulnerabilities that may be used to target proliferation and the stem cell state.68 Importantly, this work also addressed underlying genetic factors that contribute to TMZ resistance in GBM CSCs, opening up potential avenues for the use of combination therapies to improve efficacy.68 There are also well-established differences in microRNAs and emerging contributions of long noncoding RNAs and amplification of oncogenes in extrachromosomal DNA69 that underlie the evolution of tumor heterogeneity in GBM (however, these topics are not covered in this review). As the ability to interrogate molecular genetics improves, additional insights are likely to emerge that can inform the CSC state.
Epigenetic Alterations
Epigenetic alterations contribute to the maintenance of heterogeneous CSC states in GBM and may serve as potential targets for drug development. Several key processes are involved in the epigenetic regulation of GBM, including DNA methylation and chromatin remodeling.
Aberrant DNA methylation commonly occurs in cancer states and is a well-characterized feature of GBM. In the context of CSCs, it was found that GBM CSCs displayed unique patterns of hyper- and hypomethylation that are not observed in the bulk primary tumor or NSCs and may contribute to disease pathogenesis.70 These findings are further supported by observations of differential regulation of methylation and demethylation in GBM CSCs compared with NSCs.71 Additional studies characterizing methylation patterns and correlating them to gene expression and therapeutic responses will broaden our understanding of the contribution of DNA methylation to tumorigenesis.
Characterization of the chromatin landscape in GBM is an evolving field. GBM CSCs exhibited several genomic regions with unique enhancer profiles compared with NSCs; for example, HOX gene clusters gained H3K4me1 and H3K27ac peaks in CSCs.71 Through profiling of histone modifications, 2 distinct groups of GBM CSCs were defined by unique enhancer profiles and transcription factor programs.72,73 Single-cell ATAC (assay for transposase-accessible chromatin) sequencing of GBM CSCs revealed heterogeneity in the stem cell pool, with cells falling into 3 main states characterized by promoter accessibility of genes involved in immune processes, neural development, or extracellular matrix and angiogenesis.74 These studies underscore the importance of transcriptional regulation in the heterogeneity of GBM CSCs.
Metabolic Alterations
CSCs within GBM tumors have robust flexibility to alter their metabolic activity in response to ever-changing nutrient availability and various tumor microenvironments (Fig. 2). Perhaps the most well-studied of these metabolic perturbations in cancer cells is the shift in glucose utilization from oxidative phosphorylation to aerobic glycolysis known as the Warburg effect. In the context of GBM, this process contributes to CSC maintenance,75 and recent data demonstrate that an “anti-Warburg effect,” or the switch to an oxidative phenotype from a glycolytic one, may drive the differentiation of GBM cells and inhibit tumor growth.76 Conversely, some studies reveal an important role for oxidative phosphorylation in driving the CSC phenotype.77 Furthermore, inhibition of oxidative metabolism, but not glycolysis, reduced the clonogenic potential of these CSCs.77,78 These seemingly incompatible findings further underscore the heterogeneity and metabolic plasticity of CSCs in GBM.
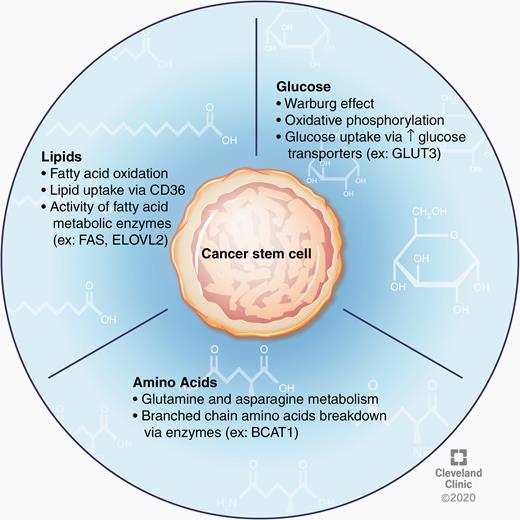
CSC nutrient metabolism. CSC plasticity is further demonstrated by the ability of these cells to alter substrate utilization as an adaptive advantage in states of limited nutrient availability. In the context of glucose metabolism, the Warburg effect, as well as oxidative phosphorylation, have been shown to contribute to the CSC phenotype. CSCs can also scavenge glucose from nutrient-poor environments by upregulating surface expression of glucose transporters. Additionally, investigation into lipid metabolism in these cells has revealed an important role for fatty acid oxidation and lipid uptake in CSC proliferation and maintenance. Amino acid metabolism in GBM CSCs is an emerging area of study but existing data provide evidence for the importance of glutamine, asparagine, and branched chain amino acid metabolism in regulating the stem cell state.
One of the most potent regulators of tumor glucose metabolism is oxygen availability. Regions of hypoxia are a characteristic feature of GBM tumors and are known to augment the Warburg effect in CSCs.79 Hypoxia has also been broadly implicated as a driver of the CSC state through upregulation of stem cell markers and enhanced clonogenicity.80 Importantly, oxygen deprivation enhances CSC tumorigenicity in GBM via hypoxia inducible factors (HIFs); for example HIF-2α signaling promoted proliferation and self-renewal.82 CSC self-renewal has also been shown to rely on HIF-1α and phosphorylation of signal transducer and activator of transcription 3 (STAT3),81 and under hypoxic conditions CSCs secrete factors promoting STAT3 phosphorylation.83 HIF-1α has also been implicated in dedifferentiation of glioma cells, which promotes the stem cell state and contributes to chemoresistance.84 Additionally, GBM CSCs can influence endothelial cells in the tumor microenvironment; it has been observed that GBM CSCs secrete factors, including vascular endothelial growth factor (VEGF), which stimulate the proliferation of endothelial cells and promote vessel formation.85 HIFs also promote upregulation of glucose transporters on tumor cells, which allows them to survive in nutrient-poor conditions. Specifically, upregulation of the neuronal glucose transporter, GLUT3, has been shown to correlate with poor patient prognosis and was more recently suggested to contribute to therapeutic resistance to bevacizumab, a VEGF inhibitor.86,87
While examination of glucose utilization has undoubtedly been the main focus of tumor metabolism studies in GBM, alterations in other pathways have also been explored (Fig. 2). Investigations into the fate of glucose uptake in GBM tumors revealed that less than 50% of the acetyl-coenzyme A was derived from blood glucose, suggesting that alternative metabolic substrates significantly contribute to tumor energetics.88 Upon further investigation, it was observed that fatty acid oxidation enzymes are active in GBM cells and that inhibition of this process reduces their proliferative capacity.89 Indeed, several enzymes involved in fatty acid metabolism, including FABP7, FAS, ACSVL3, and ELOVL2, have been independently investigated and shown to aid in tumorigenicity and maintenance of the stem cell state.90–93 Furthermore, inhibition or knockdown of FAS, ACSVL3, and ELOVL2 resulted in reduced CSC proliferation and loss of stemness.91–93 Uptake of lipids may also be crucial for survival of GBM CSCs, as evidenced by work showing that CD36 enrichment in GBM CSCs enhances the uptake of oxidized phospholipids and drives proliferation.94 Although understudied, amino acid metabolism may also play an important role in CSC biology. It was recently shown that glutamine metabolism enables the stem cell state, revealing that proliferation is inhibited in the absence of glutamine while tumor growth is enhanced by increasing the activity of the glutamate synthetase enzyme during glutamine deprivation.95 Additionally, depletion of asparagine by L-asparaginase inhibited proliferation of both GBM CSC lines and patient-derived tumor models,96 and the enzyme BCAT1, which catalyzes the breakdown of branched-chain amino acids, increased cell proliferation in IDH1 wildtype GBM tumors.97 Overall, the metabolic profile and substrate utilization of GBM CSCs critically regulate the stem cell state and may provide druggable targets for treating GBM. However, further study is warranted to further elucidate the respective contributions of each of these metabolic processes, their interactions, and pathways that can be targeted for treatment.
Cell Extrinsic Interactions—Shaping CSCs Through Evolutionary Selection
CSC-Immune Interactions
Nonneoplastic cells play a prominent role in promoting GBM progression; for example, immune cells have been reported to account for up to 30% of the GBM tumor mass. These cell populations generally adopt an immunosuppressive phenotype, protecting the tumor from immune rejection and subsequently promoting tumor growth.98 Several studies have characterized the GBM immune landscape and revealed the predominant hematopoietic lineages involved in glioma biology to include T cells, microglia, and infiltrating myeloid cells.99–102
Brain-resident microglia first exhibit antitumor properties but are quickly altered by tumor cells to support malignant growth (reviewed by Sevenich103). Meanwhile, bone marrow–derived monocytes are recruited early to the tumor and accelerate disease.102,103 Within human GBMs, the primary myeloid populations (tumor associated myeloid cells [TAMs]) have been found to be myeloid-derived suppressor cells (MDSCs) and microglia, but macrophages are also present.99,104 In human gliomas, infiltration of myeloid cells correlated with grade of histological malignancy,105 a feature that has been recapitulated in genetic mouse models of GBM.106 Of note, the composition of immune infiltrates likely depends on the mutational and gene expression profile(s) of the tumor, and there are marked differences in these populations in the context of IDH1 and NF1 mutations.98
CSCs use several mechanisms to evade immune recognition.107 For instance, CD133+ cells isolated from human GBM expressed low levels of receptors necessary for T-cell activation such as MHC II, CD40, CD80, and CD86 and expressed moderate levels of the co-inhibitory molecule B7-H1 (PD-L1). In addition, CSC conditioned media inhibited T-cell proliferation, led to apoptosis of activated T cells, and expanded regulatory T cell (Treg) populations. While co-culture with CSCs or incubation with CSC conditioned media inhibited allogeneic T-cell proliferation, this was not observed for differentiated GBM cells.107,108 Interestingly, a similar phenomenon has been observed in NSCs, where NSC-secreted factors inhibited T cell proliferation.109 CSCs also increased Treg induction and decreased interferon production from T cells, effects that were amplified under hypoxic conditions.108 GBM CSC medium inhibited macrophage phagocytosis108,110 and resulted in macrophage-mediated inhibition of T-cell proliferation.110 CSCs have been specifically shown to promote increased pSTAT3, decreased pSTAT1, and increased migration in macrophages—all characteristics of a tumor-promoting macrophage phenotype.110,111 GBM CSC-derived exosomes also exhibit immunomodulatory properties. Specifically, these exosomes were shown to be internalized by monocytes, leading to expression of immunosuppressive myeloid markers such as CD80, CD163, and CD206 and phosphorylation of STAT3, as well as expression of the T-cell checkpoint ligand PD-L1.112,113 Several cytokines produced by CSCs have been shown to mediate immunosuppression through various mechanisms (Fig. 3, Table 2). Many other secreted factors are produced by GBM cells and can lead to recruitment of macrophages into the tumor and promotion of immunosuppression—such as GM-CSF, CX3CL1, and CXCL12—although the specific contribution from CSCs has not been studied. There are numerous mechanisms by which recruited TAMs and other immune populations foster glioma progression and invasion, including promotion of angiogenesis, CSC maintenance and further immunosuppression, which have recently been reviewed elsewhere.98
Immune modifying factors secreted by GBM CSCs and their proposed mechanisms of immune suppression. References include only those studies showing CSC-specific secretion and/or effects
| Factors Secreted by GBM CSCs | Effects on Immune System | References |
|---|---|---|
| Fibrinogen-like protein 2 (FGL2) | CD103+ dendritic cell differentiation, PD1 expression, immunosuppressive macrophage and MDSC expansion, Treg expansion | 135,136 |
| Galectin-3 | Apoptosis of T cells | 137 |
| Macrophage colony-stimulating factor (CSF1) | Immunosuppressive polarization of macrophages | 138 |
| Macrophage inhibitory cytokine 1 (MIC1) | Inhibition of phagocytosis in macrophages and microglia | 110 |
| Macrophage migration inhibitory factor (MIF) | Recruitment of MDSCs and mast cells, downregulation of NKG2D on natural killer and CD8+ T cells | 139–141 |
| Monocyte chemoattractant protein-1 (MCP1/CCL2) | Microglia recruitment, Treg migration | 102,110,111 |
| Osteopontin | Migration of tumor-supportive macrophages | 142 |
| Periostin | Recruitment of immunosuppressive GAMs | 143 |
| Prostaglandin E2 (PGE2) | Putative regulation of Th17 cell differentiation | 111 |
| Transforming growth factor beta 1 (TGFβ1) | Downregulation of MHCII on myeloid cells, inhibition of mature T cell proliferation and cytotoxic activity, proliferation of Tregs | 144,145 |
| Vascular endothelial growth factor (VEGF) | Inhibition of dendritic cell maturation and function | 146 |
| Versican | Induction of MT1-MMP expression in microglia | 147 |
| WISP1 | Survival of tumor-supportive GAMs | 148 |
| Factors Secreted by GBM CSCs | Effects on Immune System | References |
|---|---|---|
| Fibrinogen-like protein 2 (FGL2) | CD103+ dendritic cell differentiation, PD1 expression, immunosuppressive macrophage and MDSC expansion, Treg expansion | 135,136 |
| Galectin-3 | Apoptosis of T cells | 137 |
| Macrophage colony-stimulating factor (CSF1) | Immunosuppressive polarization of macrophages | 138 |
| Macrophage inhibitory cytokine 1 (MIC1) | Inhibition of phagocytosis in macrophages and microglia | 110 |
| Macrophage migration inhibitory factor (MIF) | Recruitment of MDSCs and mast cells, downregulation of NKG2D on natural killer and CD8+ T cells | 139–141 |
| Monocyte chemoattractant protein-1 (MCP1/CCL2) | Microglia recruitment, Treg migration | 102,110,111 |
| Osteopontin | Migration of tumor-supportive macrophages | 142 |
| Periostin | Recruitment of immunosuppressive GAMs | 143 |
| Prostaglandin E2 (PGE2) | Putative regulation of Th17 cell differentiation | 111 |
| Transforming growth factor beta 1 (TGFβ1) | Downregulation of MHCII on myeloid cells, inhibition of mature T cell proliferation and cytotoxic activity, proliferation of Tregs | 144,145 |
| Vascular endothelial growth factor (VEGF) | Inhibition of dendritic cell maturation and function | 146 |
| Versican | Induction of MT1-MMP expression in microglia | 147 |
| WISP1 | Survival of tumor-supportive GAMs | 148 |
Immune modifying factors secreted by GBM CSCs and their proposed mechanisms of immune suppression. References include only those studies showing CSC-specific secretion and/or effects
| Factors Secreted by GBM CSCs | Effects on Immune System | References |
|---|---|---|
| Fibrinogen-like protein 2 (FGL2) | CD103+ dendritic cell differentiation, PD1 expression, immunosuppressive macrophage and MDSC expansion, Treg expansion | 135,136 |
| Galectin-3 | Apoptosis of T cells | 137 |
| Macrophage colony-stimulating factor (CSF1) | Immunosuppressive polarization of macrophages | 138 |
| Macrophage inhibitory cytokine 1 (MIC1) | Inhibition of phagocytosis in macrophages and microglia | 110 |
| Macrophage migration inhibitory factor (MIF) | Recruitment of MDSCs and mast cells, downregulation of NKG2D on natural killer and CD8+ T cells | 139–141 |
| Monocyte chemoattractant protein-1 (MCP1/CCL2) | Microglia recruitment, Treg migration | 102,110,111 |
| Osteopontin | Migration of tumor-supportive macrophages | 142 |
| Periostin | Recruitment of immunosuppressive GAMs | 143 |
| Prostaglandin E2 (PGE2) | Putative regulation of Th17 cell differentiation | 111 |
| Transforming growth factor beta 1 (TGFβ1) | Downregulation of MHCII on myeloid cells, inhibition of mature T cell proliferation and cytotoxic activity, proliferation of Tregs | 144,145 |
| Vascular endothelial growth factor (VEGF) | Inhibition of dendritic cell maturation and function | 146 |
| Versican | Induction of MT1-MMP expression in microglia | 147 |
| WISP1 | Survival of tumor-supportive GAMs | 148 |
| Factors Secreted by GBM CSCs | Effects on Immune System | References |
|---|---|---|
| Fibrinogen-like protein 2 (FGL2) | CD103+ dendritic cell differentiation, PD1 expression, immunosuppressive macrophage and MDSC expansion, Treg expansion | 135,136 |
| Galectin-3 | Apoptosis of T cells | 137 |
| Macrophage colony-stimulating factor (CSF1) | Immunosuppressive polarization of macrophages | 138 |
| Macrophage inhibitory cytokine 1 (MIC1) | Inhibition of phagocytosis in macrophages and microglia | 110 |
| Macrophage migration inhibitory factor (MIF) | Recruitment of MDSCs and mast cells, downregulation of NKG2D on natural killer and CD8+ T cells | 139–141 |
| Monocyte chemoattractant protein-1 (MCP1/CCL2) | Microglia recruitment, Treg migration | 102,110,111 |
| Osteopontin | Migration of tumor-supportive macrophages | 142 |
| Periostin | Recruitment of immunosuppressive GAMs | 143 |
| Prostaglandin E2 (PGE2) | Putative regulation of Th17 cell differentiation | 111 |
| Transforming growth factor beta 1 (TGFβ1) | Downregulation of MHCII on myeloid cells, inhibition of mature T cell proliferation and cytotoxic activity, proliferation of Tregs | 144,145 |
| Vascular endothelial growth factor (VEGF) | Inhibition of dendritic cell maturation and function | 146 |
| Versican | Induction of MT1-MMP expression in microglia | 147 |
| WISP1 | Survival of tumor-supportive GAMs | 148 |
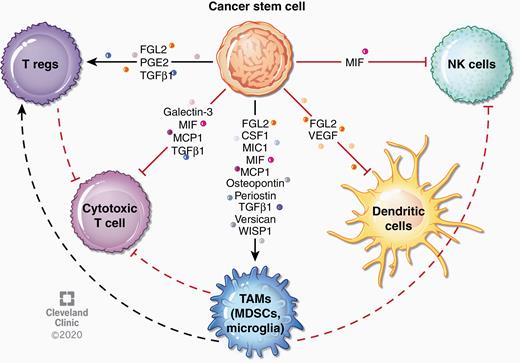
Immune modulation by CSCs. CSCs can alter tumor-associated immunity by secretion of factors that directly and indirectly suppress immune responses. For instance, several factors secreted by CSCs recruit TAMs, which subsequently suppress adaptive immune responses. Some factors have been found to inhibit natural killer cell and dendritic cell responses. CSC-derived factors can also directly affect T cell responses, leading to expansion of regulatory T cells or inhibition of cytotoxic T cells. Effects of specific cytokines are described in more detail in Table 2.
While these mechanisms provide paradigms through which CSCs communicate with and alter the immune activation status of the tumor microenvironment, further study is required across several areas, including the origin and plasticity of GAMs, response of these populations to standard of care and steroids, and validation of preclinical findings in human tumors.
Neural Interactions
GBMs grow adjacent to normal neural tissue and directly interact with cells of the neural lineage, including neurons, astrocytes, and oligodendrocytes. Moreover, there is well-documented interaction between GBM cells and neural structures, including the myelinated fiber tracts that make up the primary anatomy of the brain. Tumor cells residing here present as either collective tendrils or as a diffuse array of individual cells loosely patterned on established white matter pathways. Classic work suggested that glioma cells with the ability to invade, a property hypothesized to be a CSC phenotype, may be uniquely suited to the invasive niche because of an intrinsic capacity for myelin proteolysis.114 It has been observed that the transcription factor ZEB1, which is critical for self-renewal, also functions within this putative niche.41 While early histological studies revealed the preservation of neuronal fibers within the GBM tumor microenvironment,23 there has been little information available on neuronal contributions to gliomagenesis. It was recently demonstrated that spastic glutamatergic neurons form a synapse-like structure115 on a subpopulation of GBM tumor cells and release soluble neuroligin-3, which is sufficient to drive GBM proliferation.116 Further, while neuronal innervation has not been directly visualized, receptors for a number of other neurotransmitter systems have been identified on patient-derived GBM models. Moreover, intact gamma aminobutyric acid‒evoked signaling was recently demonstrated in cultured human GBM cells.117 Other groups have identified the dopamine receptors DRD2118 and DRD4119 on GBM cells and GBM CSCs, with DRD2 receptor activation resulting in hyperproliferation of cultured human GBM cells, while proliferation and survival of GBM cells was compromised through DRD4 inhibition. There are also examples though which GBM cells directly interact with astrocytes, including via gap junction-mediated communication120 and secreted factors to drive invasion.121 While there is far less information available that details how GBM tumor cells interact with neural populations, these relationships are clearly present and contribute to the overall progression of the disease.
Emerging Regulatory Mechanisms that Amplify Evolutionary Processes and Models that Recapitulate This Complexity
Organoid Culture of CSCs
Three-dimensional organoid culture systems complement CSC sphere culture methods and allow the study of interacting cell populations. Such models recapitulate the cellular and genetic diversity of the parental tumors,122 and may allow for superior modeling of tumor progression. Organoids grown from primary GBM specimens were shown to exhibit regional heterogeneity with a proliferative outer rim and less proliferative hypoxic core harboring quiescent CSCs and non-CSC tumor cells. These regions were differentially sensitive to standard treatments (as in primary tumors123) and better recapitulated diffuse and infiltrative properties of human GBM when implanted into mice.124 In addition, co-culture models of embryonic stem cell–derived cerebral organoids with GBM CSCs have been generated in order to better recapitulate the tumor microenvironment. GBM CSCs in these models invade and proliferate in the cerebral organoid, develop connexin-43+ gap junctions connecting various tumor subpopulations, and model other features of primary tumors such as necrosis. CSCs within these organoids are also less sensitive to standard therapies.125 Furthermore, organoids originating from larger tumor specimens (1 mm in diameter) that are never dissociated to single cells were able to model the cell-cell interactions, tumor heterogeneity, and tumor microenvironmental features such as hypoxic gradients and vasculature of primary human tumors.126 Single cell RNA sequencing of organoid models demonstrates their recapitulation of the diversity of cellular states found within the primary tumor.126,127 Organoids may be useful for testing therapeutic interference with niches hosting various CSC populations. Overall, 3D models that preserve multiple cellular states existent in the primary tumor may serve as improved tools for therapeutic screening of combination therapies that simultaneously target different tumor states and interactions.
Sex Differences
Sexual dimorphism in GBM has emerged as an important consideration in the study and treatment of the disease. Recent work has revealed that male and female GBM tumors differ, not only in their incidence, presentation, and prognosis but also in their molecular characteristics and response to therapy.128 The molecular mechanisms underlying these observations are gradually being explored and provide intriguing insight into potential therapeutic targets. Sex-specific genetic alterations have been observed in multiple studies. Recently, investigators discovered that female astrocytes demonstrate a more robust protection against malignant transformation than male astrocytes. Specifically, female, but not male astrocytes respond to cytotoxic stress with increased p16 and p21 activity and cell cycle arrest. In contrast, male astrocytes continue to proliferate in spite of the cytotoxic damage. This obstinate mitotic activity results in the steady accumulation of chromosomal abnormality, neoplastic transformation, and ultimate in vivo tumorigenesis.129 In a separate study, it was observed that transformed male astrocytes of the mesenchymal GBM subtype underwent more inactivation of the retinoblastoma tumor suppressor and were more proliferative and stem-like than their female counterparts.130 Finally, a transcriptional profiling study led to the identification of distinct molecular subtypes in male and female GBM tumors and demonstrated that alterations in cell cycle signaling correlated with increased survival in male patients while alterations in cell invasion correlated with increased survival in female patients.128 Furthermore, these differences in signaling pathways manifested as differences in drug response between males and females, with observed increases in the sensitivity of female mouse and human GBM cells to TMZ.128 Metabolic differences in male and female glioma tumors were characterized using a dataset from glioma patients to demonstrate that survival outcomes in males, but not females, may be stratified by measuring glycolysis.131 In addition, recent evidence has suggested that MDSC heterogeneity may also have a sexual dimorphism, with implications on sex-specific immune suppression mechanisms and opportunities for sex-specific immunotherapies.132 These studies suggest that a variety of genetic, metabolic, and immune alterations may contribute to sex differences in the context of GBM. Investigation of the stem cell properties of male and female GBM tumors as well as the individual contribution of sex hormones and sex chromosomes are important areas of future study.
Conclusion
Since the initial description of CSCs in GBM, progress has been made with respect to the phenotypes and underlying molecular mechanisms that define and drive these cells. Additionally, several areas have emerged that potentially alter CSC regulation including metabolic alterations, immune system and neural interactions, and sex differences, which likely contribute to differences in GBM prevalence, therapeutic response, and survival (Fig. 4). There has also been a concerted effort to leverage this ever-expanding knowledge to develop more effective therapies for GBM, which remains a top priority for the field. As technology continues to develop and allow for an increase in biological insight using fewer cells, recognition of CSCs as a state, as opposed to a discrete population, may provide a conceptual framework of evaluation that can benefit from evolution concepts such as variation, inheritance, selection and time. Outstanding questions for variation include the mosaicism of CSC populations, the ability to transition in and out of the CSC state, and how these states are shaped by distinct niches. Outstanding questions regarding inheritance include core genetic and epigenetic aspects of the CSC state and the fidelity to which they are inherited upon subsequent generations, as well as the balance between clonal expansion and plasticity within the CSC state. Outstanding questions for selection include how therapies, including next-generation strategies, alter the CSC state, potentially through promotion of self-renewal, and how cell extrinsic interactions serve as a selective pressure to modify, enhance, and amplify the CSC state. Finally, outstanding questions for time include how the CSC state evolves over generations and at what time scale changes occur relative to therapeutic response and recurrence. Answering these questions is the next step in our evolving understanding of CSCs and provides additional insight into therapeutic development efforts.
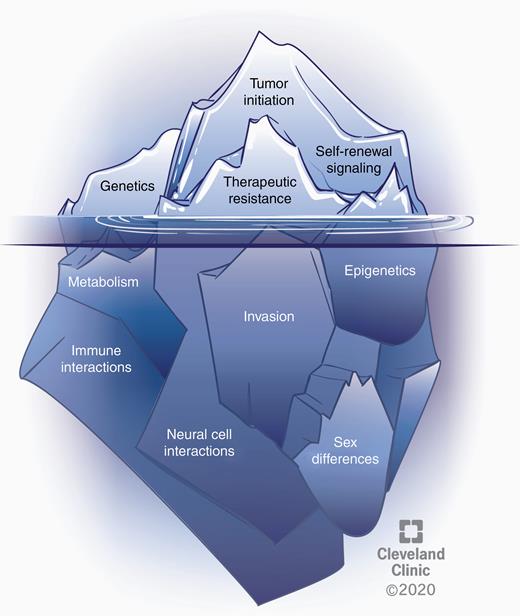
Several phenomena underlie the CSC state and warrant further exploration. The CSC state is shaped by cell intrinsic alterations and cell extrinsic interactions that contribute to clonal evolution and heterogeneity in GBM. Well-studied, established aspects of CSCs in GBM include genetic alterations, self-renewal signaling, tumor initiation potential, and therapeutic resistance (represented above the water). Several emerging areas likely affecting the CSC state include vast epigenetic changes, metabolic alterations, drivers of invasion, the role of immune and neural cell interactions, and sex differences (below the water). CSCs must be considered in the context of all of these features and interactions.
Funding
Work in the Lathia laboratory relevant to this review is currently or has been funded by the National Institute of Health (R01 NS117104, NS109742, NS089641, NS083629 (JDL), 1F32CA213727 (DJS)), Case Comprehensive Cancer Center National Institute of Health T32 CA059366 (KM, DJS), the Sontag Foundation (JDL), Cleveland Clinic Brain Tumor Center of Excellence (JDL), Cleveland Clinic VeloSano Bike Race (JDL), Case Comprehensive Cancer Center (JDL), American Brain Tumor Association Research Collaboration Grant (JDL).
Acknowledgments
We thank the members of the Lathia laboratory and Cleveland Clinic Brain Tumor Research & Therapeutic Development Center of Excellence for thoughtful discussions. We thank Dr Erin Mulkearns-Hubert for editorial assistance and Ms Amanda Mendelsohn for illustration assistance. We regret not being able to include a comprehensive reference list due to space limitations.
Conflict of interest statement. The authors declare no conflicts of interests.
Authorship statement. All authors contributed to the writing of this review and approved of the final version of the submitted manuscript.
References
Author notes
These authors contributed equally.



{kind=link}
{kind=link}
{kind=link}
{kind=link}