





Abstract
Diffuse midline glioma, formerly DIPG (diffuse intrinsic pontine glioma), is the deadliest pediatric brainstem tumor with median survival of less than one year. Here, we investigated (i) whether direct delivery of adenovirus-expressing cluster of differentiation (CD)40 ligand (Ad-CD40L) to brainstem tumors would induce immune-mediated tumor clearance and (ii) if so, whether therapy would be associated with a manageable toxicity due to immune-mediated inflammation in the brainstem.
Syngeneic gliomas in the brainstems of immunocompetent mice were treated with Ad-CD40L and survival, toxicity, and immune profiles determined. A clinically translatable vector, whose replication would be tightly restricted to tumor cells, rAd-Δ24-CD40L, was tested in human patient–derived diffuse midline gliomas and immunocompetent models.
Expression of Ad-CD40L restricted to brainstem gliomas by pre-infection induced complete rejection, associated with immune cell infiltration, of which CD4+ T cells were critical for therapy. Direct intratumoral injection of Ad-CD40L into established brainstem tumors improved survival and induced some complete cures but with some acute toxicity. RNA-sequencing analysis showed that Ad-CD40L therapy induced neuroinflammatory immune responses associated with interleukin (IL)-6, IL-1β, and tumor necrosis factor α. Therefore, to generate a vector whose replication, and transgene expression, would be tightly restricted to tumor cells, we constructed rAd-Δ24-CD40L, the backbone of which has already entered clinical trials for diffuse midline gliomas. Direct intratumoral injection of rAd-Δ24-CD40L, with systemic blockade of IL-6 and IL-1β, generated significant numbers of cures with readily manageable toxicity.
Virus-mediated delivery of CD40L has the potential to be effective in treating diffuse midline gliomas without obligatory neuroinflammation-associated toxicity.
1. Ad-CD40L therapy improves survival through induction of immune cell infiltration.
2. CD4 T cells are important in Ad-CD40L therapy.
New, aggressive therapies for pediatric brainstem tumors including diffuse midline gliomas are urgently required. CD40/CD40L engagement has generated potent antitumor responses in anatomic locations where high levels of inflammation are acceptable. Our study here is the first to investigate whether such CD40L-induced inflammation in brainstem tumors can be tolerated. We demonstrate here that when CD40L expression was tightly restricted to tumor cells, Ad-CD40L significantly prolonged survival of brainstem tumor–bearing mice. Depletion of CD4 T cells abolished Ad-CD40L therapy and decreased levels of antigen-presenting cells. A replication competent adenovirus engineered to replicate only in tumor cells (rAd-Δ24-CD40L) had cytopathic effects on patient-derived diffuse midline glioma cells and improved survival of mice bearing brainstem tumors in vivo. Our results show that it is possible to develop a novel, potentially highly effective immunotherapy for diffuse midline glioma, without unacceptable toxicity, which can be translated into clinical trials.
Brainstem gliomas represent about 10–15% of pediatric central nervous system tumors.1 Of these, diffuse midline glioma (formerly diffuse intrinsic pontine glioma [DIPG]) is the major cause of death, with survival rates of fewer than 9 months from diagnosis.1 Focal radiation to the pons remains the main therapy for newly diagnosed disease and the addition of chemotherapy, targeted therapies, differentiation agents, and radiation sensitizers has not significantly impacted outcomes. Moreover, surgery is not an option due to the critical location within the brain.2 Therefore, new therapeutic modalities are urgently needed.
Cancer immunotherapy has dramatically improved treatment of several tumors with immune checkpoint inhibitors and chimeric antigen receptor T-cell therapies, although not appreciably so far in children with brain tumors.3 Oncolytic virotherapy utilizes engineered viruses which preferentially replicate in tumor cells, leading to cell death and stimulation of the immune system and has been clinically investigated in brain tumors.4 Immunotherapies for tumors in the brainstem pose unique potential dangers associated with inflammation in this location. However, we have shown that diverse immunotherapies can extend survival in mice bearing brainstem tumors without catastrophic neurological toxicity,5 opening the path for testing novel immunotherapies for this disease.
Cluster of differentiation (CD)40 ligand (CD40L:CD154) is expressed both as a membrane-bound form on activated helper T cells and as a soluble form by platelets.6 CD40/CD40L engagement develops T helper (Th)–dependent immune responses, provides proliferation/differentiation signals to B cells, and triggers maturation of antigen-presenting cells (APCs) for induction of cytotoxic T lymphocytes.7 Therefore, agonistic antibodies against CD40, and viral vectors expressing CD40L, have been successfully tested against several malignancies, including glioblastoma,8 and are associated with priming cytotoxic CD8+ T cells, reducing regulatory T cells, and activating tumor-associated macrophages.9
In models of subcutaneous melanoma, where local inflammation was well tolerated, we demonstrated that CD40L expressed from a replication-defective adenoviral vector (Ad-CD40L) significantly enhanced survival and primed antitumor CD8+ T cells.10,11 Others have shown that Ad-CD40L activates dendritic cells, increases the intratumoral Teffector/Tregulatory cell ratio,12 and combines with checkpoint inhibitors to eradicate tumors.13 Therefore, we investigated whether Ad-CD40L could be an effective treatment for brainstem tumors, while simultaneously monitoring whether the inflammation associated with antitumor therapy could be controlled/tolerated. Using immunocompetent models to assess the efficacy/toxicity balance of adenovirus expressing CD40L, we show here that tumor-restricted expression of CD40L represents a novel, potentially highly effective immunotherapy for diffuse midline glioma with an acceptable toxicity profile.
Materials and Methods
Additional detailed methods are provided in the Supplementary Material.
Cell Lines
GL261, CT2A, and 293A cell lines were grown in DMEM (HyClone) supplemented with 10% fetal bovine serum (Life Technologies). Mixed astrocytes were cultured in DMEM (HyClone) supplemented with 10% fetal bovine serum (Life Technologies), 1% penicillin/streptomycin (Corning), and 1% sodium pyruvate (Sigma). SJPDGF1 and SF7761 diffuse midline glioma neurospheres were cultured in trichoderma selective media.
Viruses
Ad-GFP or Ad-CD40L serotype 5 replication-defective adenoviruses express the green fluorescent protein (GFP) or murine CD40L, respectively, under the cytomegalovirus promoter in E1.11 Two replication-competent adenoviruses—rAd-Δ24-luc and rAd-Δ24-CD40L—are expressing luciferase or murine CD40L in which 24 bp of E1A was deleted.14–16
In Vivo Studies
All studies were approved by the Mayo Foundation Institutional Animal Care and Use Committee. Female C57BL/6 mice (6–8 mice/group) were challenged (2 µL) with tumor cells or virus via stereotactic implantation using established coordinates in the frontal lobe or brainstem.17
Flow Cytometry
Brain-infiltrating leukocytes (BILs) were harvested in Roswell Park Memorial Institute medium from perfused brains. BILs were prepared using dounce homogenization and Percoll gradient.18 Immune cells were analyzed using FlowJo 10.
Histology and Immunofluorescence
Brains were fixed in 10% formalin and stained with hematoxylin and eosin by Mayo Clinic Histology Core Facility. Immunofluorescence staining was as described.19
RNA-Sequencing Analysis of Gene Expression
Ten days established GL261 tumor-bearing mice were intratumorally injected (2 µL) with phosphate buffered saline (PBS) or 5 × 106 plaque-forming units (pfu) Ad-GFP or Ad-CD40L. On day 8, brains were dissected for RNA extraction (RNeasy, Qiagen). RNA samples were then subjected to RNA-seq analysis at the Genome Analysis Core, Mayo Clinic.
Tumor-Specific Immunoglobulin G Detection
Sera were harvested on day 8 post-virus injection and used as primary antibody. Stained were 1 × 105 GL261 cells, or mixed astrocytes (1:10 dilution) at 4°C for 30 min, and 1:100 of Alexa Fluor 488 goat anti-mouse immunoglobulin (Ig)G (heavy + light chain) antibody (Invitrogen) was added for flow analysis.
Viability Assays
GL261 cells were plated at 1 × 104 cells for overnight; 24 hours later, GL261, SJPDGF1, or SF7761 cells (1 × 105) were infected with viruses at different multiplicity of infections (MOIs). At indicated time points, cell viability was assessed by alamarBlue Cell Viability Reagent (ThermoFisher Scientific).
For CD4+ T cell co-culture assays, GL261 cells were seeded in 96-well plates in triplicate and treated with CD4+ T cells at an effector:target ratio of 10:1. Then, 48 h later, wells were washed gently 3x in PBS to remove non-adherent cells. Surviving cells were trypsinized and resuspended in PBS and viable cells were counted (viability was assessed by alamarBlue Cell Viability Reagent [ThermoFisher Scientific]).
Luciferase Assay
SJPDGF1 cells (1 × 105) were infected with rAd-∆24-luc at different MOIs. At indicated time points, luciferase was assessed by Steady-Glo Luciferase Assay System (Promega).
Statistics
The Mantel–Cox log-rank test was used to analyze Kaplan–Meier survival curves. Student’s t-tests were used for in vitro and ex vivo analysis where appropriate. Statistical significance was determined at P < 0.05. All analyses were performed using GraphPad Prism 8 software.
Results
Restricting Ad-CD40L to Tumor Cells Induces Rejection Without Toxicity
To test the hypothesis that ligation of the CD40/CD40L axis would promote brainstem tumor clearance with manageable toxicity, we used the GL261/C57BL/6 immunocompetent models of tumor growth in the brainstem. Although not derived from pediatric diffuse midline gliomas, we have shown that GL261 can be reproducibly grown as transplantable tumors in the pons in the anatomic location that mirrors diffuse midline glioma growth in patients.5
Infection of GL261 cells with replication-defective Ad-GFP or Ad-CD40L at various MOIs (0.1–10) did not induce direct cytotoxicity, but transgene expression was detected by 48 h post-infection (Figure 1A, B). To determine the efficacy of CD40L as a mediator of immune rejection in the brainstem, GL261 cells were pre-infected with Ad-GFP or Ad-CD40L immediately prior to implantation into the brainstem using established coordinates.17 Implantation of tumor cells pre-infected with Ad-GFP (MOI 10) did not prolong survival of mice relative to mice treated with PBS (Figure 1C). However, mice implanted with GL261 cells pre-infected with Ad-CD40L at MOI of 1 or 10 (but not 0.1) rejected tumors completely with no overt toxicity (Figure 1C). To determine whether surviving mice developed long-term immunologic memory, they were rechallenged with the same tumor cells in the frontal lobe. While naïve mice succumbed to tumor by day 40, all mice which survived the brainstem challenge rejected the frontal lobe rechallenge (Figure 1D). These results suggest that highly targeted delivery of Ad-CD40L to tumors in the brainstem might improve survival with manageable toxicity and represents a valuable model to explore further as a novel immunotherapy of diffuse midline glioma.
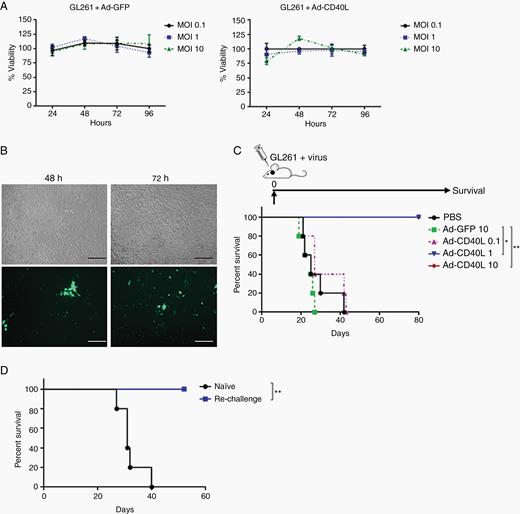
Restricted expression of CD40L in tumor cells exerts antitumor response. (A) GL261 cells were infected with different MOIs of Ad-GFP or Ad-CD40L, and cell viability was measured using MTT assay. Error bars ± SD. (B) GL261 cells were infected with Ad-GFP (MOI 1) and GFP expression was detected using florescence microscopy (20x). Scale bar, 100 µm. (C) Survival curves of mice treated with GL261 cells pre-infected with viruses and implanted into the brainstem (day 0). (D) Survival curves of mice which survived in (C) and were rechallenged with parental GL261 in the frontal lobe. *P ≤ 0.05, **P ≤ 0.01.
Ad-CD40L Induces Recruitment of Immune Cells into the Tumor Microenvironment
To characterize the mechanisms by which Ad-CD40L induced antitumor responses in the brainstem, we analyzed the induction of BILs 8 days following implantation of tumor cells pre-infected with PBS, Ad-GFP, or Ad-CD40L (MOI 1). Treatment with Ad-CD40L induced a profound infiltration of CD45+ cells (Figure 2A), largely composed of CD4+ and CD8+ T cells, CD19+ (B) cells, and natural killer cells (Figure 2B). Ad-CD40L induced CD11b+Gr1+ cell infiltration (Figure 2C) and importantly activated class II and CD86 expression in CD11c+ and CD45midCD11b+ cells (Figure 2D, E). As shown by bromodeoxyuridine labeling, Ad-CD40L significantly promoted local proliferation of activated (CD44+) CD4+ T, CD8+ T, and CD19+ (B) cells (Figure 2F). In separate studies, we have also observed significant proliferation of adoptively transferred tumor-specific T cells in the draining lymph nodes following intratumoral expression of CD40L.
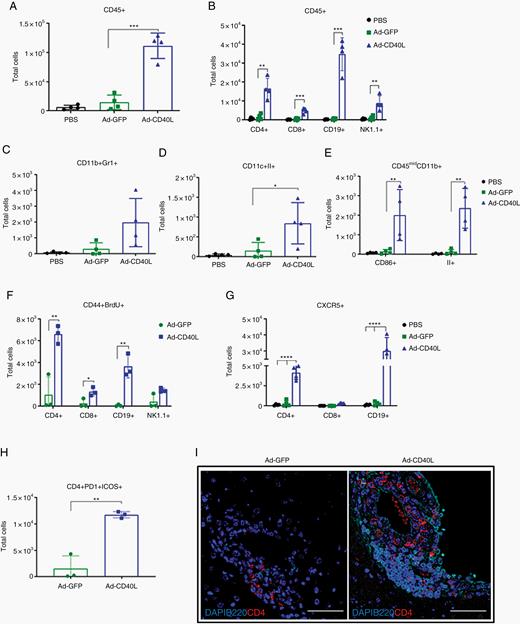
Ad-CD40L induces immune cell infiltration in brain tumors. Mice were implanted with GL261 tumors pre-infected with PBS, Ad-GFP, or Ad-CD40L (MOI 1) on day 0. Mice were given bromodeoxyuridine (BrdU) on day 7, and brains were perfused and harvested on day 8 for flow analysis. Numbers of total (A) CD45+, (B) CD4+ T, CD8+ T, CD19+, NK1.1+, (C) CD11b+Gr1+, (D) CD11c+II+, (E) CD45midCD11b+, (F) CD44+BrdU+, (G) CXCR5+, (H) CD4+PD1+ICOS+. Error bars ± SD (n = 4). Student’s t-test: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. (I) Representative images (40x) of brains day 8 post-implantation stained with DAPI, B220, and CD4. Scale bar, 50 µm.
In addition, Ad-CD40L induced recruitment of CXCR5+CD4+ T and B cells (Figure 2G). Consistent with C-X-C chemokine receptor 5 binding to C-X-C chemokine ligand 13, a chemokine important in the generation of secondary/tertiary lymphoid structures (TLS), these CD4+ T cells expressed programmed cell death 1 and inducible co-stimulator (ICOS), which are markers of follicular CD4+ T cells (Figure 2H). Similarly, CD4+ T cells and B220+ B cells clustered around the meninges following Ad-CD40L treatment, reminiscent of TLS formation (Figure 2I).
CD4+ T Cells Are Critical for Ad-CD40L Therapy
To identify critical cellular types for therapy, we performed multiple antibody-mediated depletions of CD4+ T, CD8+ T, or B cells. Of these, only depletion of CD4+ T cell significantly diminished the total number of BILs (Figure 3A), under conditions which we confirmed led to depletions of CD4+ T, CD8+ T, or B cells (Figure 3B). Depletion of B cells did not alter the number of CD4+ or CD8+ T cells. However, CD4 depletion reduced the number of CD19+CD40+ and CD19+II+ cells (Figure 3C) and CD11c+CD40+, CD11c+II+, CD11b+F4/80+, or CD11b+Gr1+ cell numbers were all significantly decreased (Figure 3D, E). CD4 depletion also suppressed microglia activation as evidenced by levels of CD45midCD11b+CD86+ and CD45midCD11b+II+ cells (Figure 3F).

CD4 depletion abrogates Ad-CD40L therapy. Immune cells were depleted 3 days before implanting pre-infected cells. Brains were harvested on day 8. Number of total (A) BILs, (B) CD45+, (C) CD19+, (D) CD11c+, (E) CD11b+, (F) CD45midCD11b+ cells. Error bars ± SD (n = 3). Student’s t-test: *P ≤ 0.05, **P ≤ 0.01. (G) Survival curves of immune cell-depleted mice implanted with pre-infected GL261 cells. **P ≤ 0.01. (H) Survival curves of rechallenge of surviving mice from (G). All treatments **P ≤ 0.01 over naïve control. (I, J) Mice with 10 day established GL261 tumors were injected with PBS or with 5 × 106 pfu of Ad-GFP or Ad-CD40L. Five days later, CD4+ T cells were purified from splenocytes by magnetic bead separation. CD4+ T cells pooled from 3 mice per treatment group were co-cultured with 104 GL261 target cells pretreated for 24 h with murine IFN-γ (20 U/mL) at an effector:target ratio of 10:1 (triplicate wells). The number of surviving GL261 cells 48 h later is shown (I) for GL261 cells co-cultured with: 1: no CD4+ T cells; 2: CD4+ T cells from naïve mice (no tumor, no treatment); 3: CD4+ T cells from tumor bearing mice treated with PBS; 4: CD4+ T cells from tumor bearing mice treated with Ad-GFP; 5: CD4+ T cells from tumor bearing mice treated with Ad-CD40L. J. Supernatants harvested from these cultures were assayed for levels of IFN-γ by enzyme-linked immunosorbent assay.
Despite the fact that Ad-CD40L therapy recruited both CD8+ T cells and B cells, depletion of neither CD8+ T nor B cells significantly altered survival compared with the IgG control. In contrast, CD4+ T cell depletion drastically reduced Ad-CD40L therapy, showing CD4+ T cells were absolutely and obligatorily required for therapy (Figure 3G). Regardless of whether mice survived following depletion of either CD8+ T or B cells, all of these surviving mice developed long-term antitumor memory responses against a rechallenge with GL261 cells in the frontal lobe (Figure 3H). In addition to providing helper function to generate intrinsic antitumor immune responses, CD4+ T cells have also been reported to have direct cytotoxicity against tumor cells.20,21 In this respect, CD4+ T cells isolated from spleens of mice treated with Ad-CD40L significantly inhibited the proliferation of target GL261 cells in vitro compared with CD4+ T cells from untreated mice, or from mice treated with Ad-GFP (Figure 3I). This inhibition of tumor cell proliferation may be contributed by some direct cytotoxicity and/or through a directly cytostatic effect exerted by the CD4+ T cells. This anti-proliferative activity on GL261 targets was also associated with low, but detectable levels of interferon-gamma (IFN-γ) secretion by the CD4+ T cells from Ad-CD40L-treated mice. In contrast, neither inhibition of proliferation against GL261 cells nor IFN-γ secretion was observed from CD4+ T cells from mice treated with PBS or Ad-GFP (Figure 3J).
CD4+ T cell mediated anti-proliferative effects against GL261 targets in vitro absolutely required pretreatment of GL261 cells with IFN-γ. Although major histocompatibility complex (MHC) class II expression was undetectable on GL261 cells growing in vitro, pretreatment for 24 h with IFN-γ induced relatively high levels (Supplementary Figure 1A). Consistent with GL261 tumors being the target of direct CD4+ T-cell mediated inhibition of proliferation following Ad-CD40L treatment, GL261 tumors excised from the brainstems of mice treated with PBS had significantly lower levels of MHC class II expression than did GL261 tumors from brainstems of mice treated with Ad-CD40L (Supplementary Figure 1B). In addition, GL261 tumors showed significant induction of IFN-γ in Ad-CD40L-treated tumors compared with PBS or Ad-GFP-treated tumors (Supplementary Figure 1C), which is consistent with induction of MHC class II presented targets on tumors for CD4+ T-cell mediated recognition. We have not tested tumors directly from patients for MHC class II expression. However, although neither of the 2 diffuse midline glioma cell lines SJPDGF1 and SF7761 express detectable MHC class II molecules in normal cell culture, pretreatment for 24 h with human IFN-γ did induce class II expression (200-fold, SJPDGF1; 1000-fold, SF7761). So, although we do not know if patient tumors in situ express class II, patient-derived lines (cultured for multiple passages in vitro) have the capacity to do so.
Taken together, these data are consistent with a model in which Ad-CD40L induces a population of CD4+ T cells which exerts anti-proliferative effects on GL261 tumors growing in situ, in part through an in vivo upregulation of MHC class II on the tumor cells as a result of the pro-inflammatory tumor microenvironment induced by Ad-CD40L.
Therefore, CD4+ T cells were necessary to activate antigen-presenting cells, B cells and microglia, and were critical mediators of Ad-CD40L therapy—at least in part through a highly anti-proliferative activity against tumor cells.
Intratumoral Injection of Ad-CD40L Improves Survival of Glioma-Bearing Mice
Next, we assessed efficacy and toxicity of Ad-CD40L in an intratumoral injection model of established brainstem tumors. Direct injection of Ad-GFP at 106 pfu did not prolong survival of mice, all of which died of tumor growth (Figure 4A). When mice were treated with 106 pfu of Ad-CD40L, significant acute toxicity was observed, with loss of up to 20% body weight (Figure 4B) and some deaths (Figure 4A) within the first 30 days post treatment. However, with supportive care (fluid treatment), about 40% of these mice recovered and survived long term (Figure 4A, B). Treatment with Ad-CD40L was dose dependent as neither 5 × 104 nor 105 pfu significantly extended survival through tumor treatment (Figure 4A), but neither did they induce high-level toxicity (Figure 4B). Significant numbers of long-term tumor cures could be induced by increasing the dose of Ad-CD40L as high as 5 × 106 or 107 pfu, compared with mice treated with Ad-GFP at 107 pfu (Figure 4C), when fluids and nutrient support were provided over a period of 2 weeks (Figure 4D).
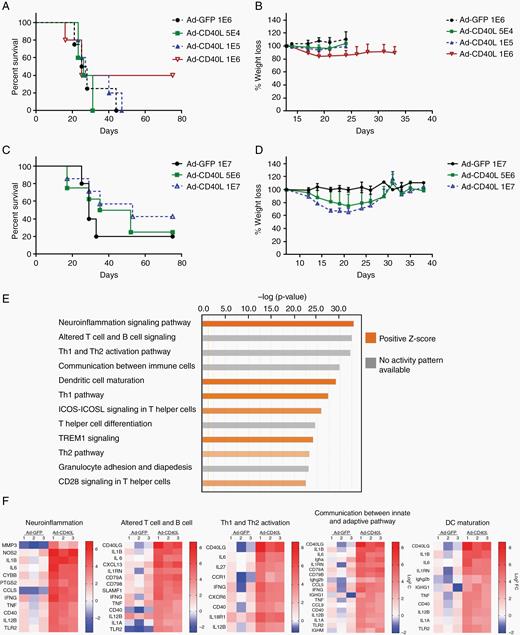
Intratumoral injection of Ad-CD40L results in tumor clearance with acute toxicity. (A, C) Survival curves of tumor-bearing mice treated with viruses. (B, D) Percent body weight loss of GL261-bearing mice treated with Ad-CD40L. Tumor bearing mice were intratumorally injected with viruses. On day 8, total RNA was isolated for RNA-seq analysis. (E) Top 12 canonical pathways compared between Ad-GFP and Ad-CD40L. (F) Heat map of genes expressed in Ad-GFP or Ad-CD40L over PBS (expression log ratio ≥3) in top 5 canonical pathways.
RNA-seq transcriptional profiling 8 days post-injection of tumor-bearing mice treated with PBS, Ad-GFP, or Ad-CD40L using Ingenuity Pathway Analysis (https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/) showed that Ad-CD40L induced genes involved in signaling pathways of neuroinflammation, T and B cell signaling, Th1 and Th2 activation, communication between innate and adaptive immune cells, and dendritic cell maturation (Figure 4E). Among genes which were most differentially expressed in each pathway (log2FC≥ 3), IL-1β, IL-6, IFN-γ, tumor necrosis factor alpha (TNF-α), CD40, IL-12-β, and Toll-like receptor 2 were shared between each of the top 5 pathways (Figure 4F). Consistent with Figure 2, genes responsible for B- and T-cell recruitment were also strongly upregulated in Ad-CD40L treatment. Thus, Ad-CD40L activated pro-inflammatory cytokines/chemokines, which are candidates as mediators of efficacy, toxicity, or both. Consistent with the ability of Ad-CD40L to reverse the immune suppressive tumor microenvironment, treatment with Ad-CD40L induced both significant reductions in the expression of immune suppressive factors, such as transforming growth factor beta and forkhead box P3, as well as increased expression of other cytokines, including IFN-γ and IL-6, compared with PBS or Ad-GFP treatment (Supplementary Figure 1).
Ad-CD40L Therapy Is Associated with Tertiary Lymphoid Structure-Like Formation
Brains of tumor-bearing mice 9 days post treatment showed reproducibly smaller, or the absence of, tumors following treatment with Ad-CD40L (Figure 5A, left panel) along with consistently higher levels of CD4+ T cells (Figure 5A, middle panel). B and CD3+ T cells were present along the meninges of Ad-CD40L-treated brains, organized in a manner that resembled TLS as found in inflamed or tumoral tissue22 (Figure 5A, right panel). In this respect, levels of claudin-5, a marker of vessel formation in the brain,23 were significantly upregulated following Ad-CD40L treatment compared with PBS or Ad-GFP (Supplementary Figure 1G) The formation of these TLS and increased vascularization are compatible with a wound-healing type of response to the inflammatory effects of Ad-CD40L. Brains harvested on days 2, 3, and 8 post-infection showed significantly increased numbers of CD45+ BILs following treatment with Ad-CD40L compared with Ad-GFP, as early as 72 hours post-infection (Figure 5B). Ad-CD40L also significantly increased CD11b+CD11c+, CD11b+Gr1+, CD11c+II+, CD45midCD11b+CD40+, and CD45midCD11b+II+ cells (Supplementary Figure 2). Similar to the pre-infection model, the majority of total cells in Ad-CD40L–treated mice were CD4+ T, CD8+ T, and B cells (Figure 5C–E). Interestingly, when Ad-CD40L was intracranially injected without tumor present, total BILs or CD45+ cells were not significantly different in mice injected with Ad-GFP or Ad-CD40L (Supplementary Figure 3A, B). Notably, we detected a marginal enhancement in CD8+ T and B cells (Supplementary Figure 3D, E), whereas CD4+ T and natural killer cells were decreased in Ad-CD40L-treated tumor-free animals (Supplementary Figure 3C, F). Therefore, the specific profile of immune cells depends on the presence of tumors in the brain.
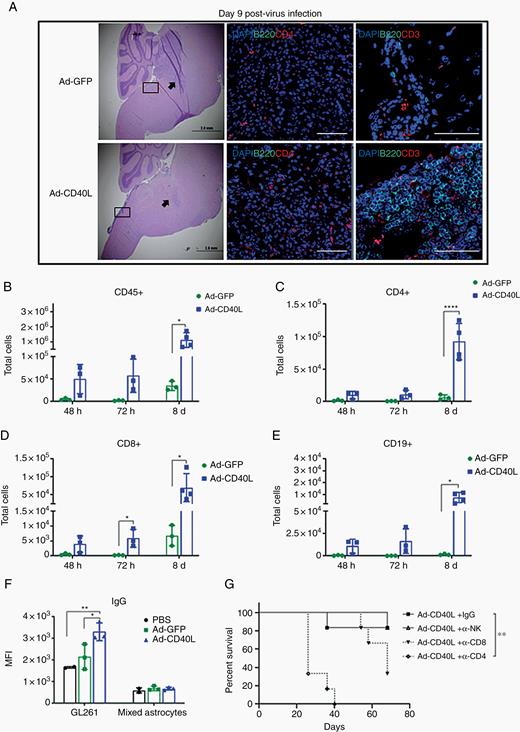
Intratumoral injection of Ad-CD40L induces immune cell infiltration and antitumor antibody. (A) Representative images (40x) of tumor-bearing mice treated with viruses. Left panel, hematoxylin/eosin staining; middle panel (arrow), brain tissues stained with DAPI, B220, and CD4 (scale bar, 100 µm); right panel (inset), brain tissues stained with dapi, B220, and CD3 (scale bar, 50 µm). (B–F) Flow analysis of brains treated with PBS, Ad-GFP, or Ad-CD40L. Numbers of total (B) CD45+, (C) CD4+ T, (D) CD8+ T, (E) CD19+ cells. Error bars ± SD (n = 3). Student’s t-test; *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001. (F) Average of median fluorescent intensity of IgG levels. Error bars ± SD (n = 3). Student’s t-test; *P ≤ 0.05, **P ≤ 0.01. (G) Mice injected with GL261 brainstem tumors on day 1 were injected with control IgG or with depleting antibodies against CD4+T, CD8+T, or natural killer cells on days 6, 8, 10, and 12. Mice (n = 6) were injected intratumorally with PBS or with 1 × 106 pfu of Ad-CD40L on day 10. Survival with time is shown.
Sera from tumor-bearing mice treated with Ad-CD40L significantly increased IgG levels specifically against GL261, but not against normal brain-derived mixed astrocyte cells24 as a negative control (Figure 5F), showing that Ad-CD40L induces both adaptive and humoral antitumor immune responses. However, the role of these tumor-specific antibodies remains unclear, since depletion of B cells had no significant effects on therapy (Figure 3G).
Finally, consistent with the results of Figure 3G with the pre-transduced model, depletion of CD4+ T cells completely abolished in vivo therapy following direct delivery of Ad-CD40L virus to established tumors (Figure 5G). In this model, however, depletion of CD8+ T cells also reduced therapeutic efficacy compared with CD8+ intact mice, suggesting that effector CD8+ T cell responses may also play a role in therapy of direct virus delivery (Figure 5G).
Replication-Competent Adenovirus Expressing CD40L Improves Antitumor Efficacy While Minimizing Off-Tumor Toxicity
We hypothesized that off-target expression of Ad-CD40L in nontumor brain tissue led to pro-inflammatory cytokine/chemokine production and the acute toxicity of Figure 4. Therefore, for clinical translation of CD40L therapy for diffuse midline gliomas, it will be important to restrict CD40L expression to tumor cells in vivo. To achieve this goal, we exploited the tumor-specific targeting conferred upon the replication competency of adenoviruses afforded by the incorporation of a 24 bp deletion in the E1A viral gene (rAd-Δ24) responsible for retinoblastoma protein (pRB) binding.25 Since many gliomas, including diffuse midline gliomas, have defects in the p16/retinoblastoma (Rb)/E2F pathway,26,27 the 24 bp deletion in the E1A gene confers rAd-Δ24 with high levels of selectivity to glioma cells. Therefore, we constructed rAd-Δ24-CD40L in which expression of CD40L will ultimately be under the control of the highly tumor selective replication of the virus.
To validate the replication and antitumor efficacy of this virus in human tumors, the patient-derived diffuse midline glioma cell line SJPDGF1 was infected with rAd-Δ24 expressing luciferase (rAd-Δ24-luc). Virus infection and transgene expression were observed within 3 days, which decreased with time due to the cytotoxicity associated with replication and oncolysis (Figure 6A). Consistent with this, rAd-Δ24-luc and rAd-Δ24-CD40L killed almost 60% of SJPDGF1 cells by day 6 (Figure 6B), results which were reproduced in a second human patient–derived diffuse midline glioma cell line, SF7761 (Figure 6B).
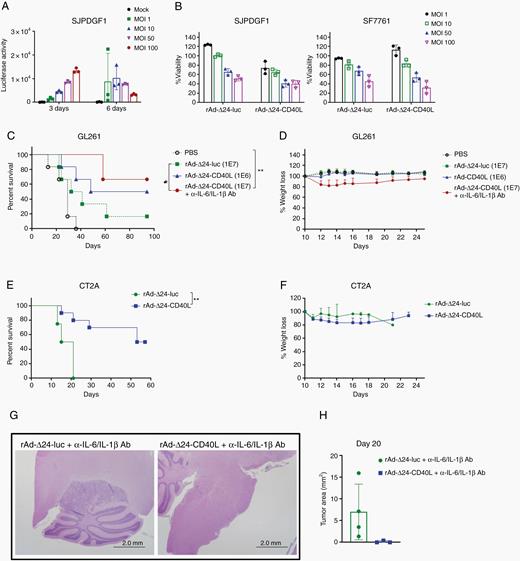
rAd-Δ24-CD40L treats brainstem tumors. (A) SJPDGF1 cells were infected with rAd-Δ24-luc at different MOIs and luciferase activity measured. (B) SJPDGF1 or SF7761 cells were infected with rAd-Δ24-luc or rAd-Δ24-CD40L and cell viability assessed by MTT assay. (C) Survival curves of GL261 tumor-bearing mice injected with PBS, rAd-Δ24-luc or rAd-Δ24-CD40L ± anti-IL-6 and anti-IL-1β neutralizing antibodies. *P ≤ 0.05, **P ≤ 0.01. (D) Percent body weight loss of GL261-bearing mice treated with rAd-Δ24s. (E) Survival curves of CT2A tumor-bearing mice injected with PBS, rAd-Δ24-luc or rAd-Δ24-CD40L. **P ≤ 0.01. (F) Percent body weight loss of CT2A-bearing mice treated with rAd-Δ24s. (G) Representative images of hematoxylin/eosin staining. (H) Tumor area of GL261-bearing mice treated with rAd-Δ24s. Error bars ± SD (n = 3).
Assessing the activity of rAd-Δ24-CD40L in patient-derived xenografts would require immunocompromised mice, thereby eliminating the immune-mediated effects of CD40L expression. Therefore, we tested the antitumor efficacy of rAd-Δ24-CD40L in 2 different murine immune competent models where either GL261 or CT2A cells were implanted into the brainstem on day 0, and rAd-Δ24 was intratumorally injected on day 10. While these murine cells only partially support adenoviral replication, they represent the effects of intact immune subsets on both antitumor efficacy and toxicity. Injection of rAd-Δ24-CD40L significantly improved survival of tumor-bearing mice over PBS or rAd-Δ24-luc in both immune-competent GL261 and CT2A scenarios, and led to at least 50% cure rates in these aggressive models (Figure 6C, E). Most importantly, rAd-Δ24-CD40L did not induce weight loss to the same degree as replication-defective Ad-CD40L (Figure 4). GL261-bearing mice treated with rAd-Δ24-CD40L lost less than 25% body weight 2 days post-infection, and recovered within 14 days (Figure 6D). Similarly, CT2A-bearing mice treated with rAd-Δ24-CD40L had weight loss less than 20% (Figure 6F).
To investigate the relative toxicity of the rAd-Δ24-CD40L virus to tumor and normal brain tissue, mixed astrocyte/glial cultures from normal brains of C57BL/6 mice were infected with increasing MOI of the rAd-Δ24-CD40L virus. Despite the relatively poor ability of replication competent adenoviruses to replicate in murine cells, both CT2A and GL261 glioma cell lines showed significant cytotoxicity upon infection with rAd-Δ24-CD40L (Supplementary Figure 4), consistent with the cytotoxicity observed against human diffuse midline glioma cell lines (Figure 6B). In contrast, similar MOI of rAd-Δ24-CD40L used to infect normal astrocyte/glial cultures did not cause significant reduction in cell numbers compared with mock infection (Supplementary Figure 4). These data confirm that the Δ24 mutation confers a tight degree of targeting of virus replication upon transformed glioma cell types, as opposed to normal brain cells.
Our data in Figure 4 demonstrated that both IL-6 and IL-1β were upregulated in Ad-CD40L treated tumors which were associated with high levels of toxicity. Therefore, we hypothesized that blocking these cytokines during therapy would reduce the toxicity associated with CD40L treatment while not necessarily diminishing therapy. In this respect, blockade of IL-6 and IL-1β using neutralizing antibodies—a strategy which is directly translatable to clinical studies28—allowed us to escalate the intratumoral treatment dose of rAd-Δ24-CD40L as high as 107 pfu in the GL261 model with manageable and nontoxic levels of body weight loss (Figure 6D). Treatment with neither anti-IL-6, anti-IL-1β, nor a combination of the two had any effect on therapy of GL261 tumors. Consistent with these results, histopathologic studies of the mouse brains on day 20 post-injection demonstrated that rAd-Δ24-CD40L treated mice exhibited smaller tumors than control mice (Figure 6G, H).
Discussion
New, aggressive therapies for diffuse midline gliomas and other pediatric brainstem tumors are urgently required. CD40/CD40L engagement has generated potent antitumor responses in multiple preclinical models in anatomic locations where high levels of inflammation can be readily tolerated.6,7 Therefore, in this study, we sought to address two major questions. First, we hypothesized that direct delivery of adenovirus expressing CD40L (Ad-CD40L) to brainstem tumors would induce immune-mediated antitumor clearance. Second, we tested whether such therapy would be associated with manageable toxicity due to immune-mediated inflammation in the brainstem.
To investigate the twin components of immune-mediated efficacy and toxicity of the Ad-CD40L strategy, immune-competent preclinical models are essential. Unfortunately, we were unable to grow explanted cell lines derived from the replication competent avian-like sarcoma (RCAS) spontaneous model of diffuse midline glioma29 reproducibly in our mice. Therefore, for the current studies, we used GL261 and CT2A murine gliomas. Although these lines are not derived from diffuse midline gliomas, we have shown that they can be reproducibly grown as transplantable tumors in the pons in the anatomic location that mirrors diffuse midline glioma growth in patients.5 Therefore, although the models of therapy and delivery that we have used here are not perfect replicates of the human situation, we believe that CD40L-mediated treatment of these realistically located tumors will reflect the efficacy and, importantly, the immune-mediated toxicity associated with immunotherapy for diffuse midline gliomas.
Although Ad-CD40L did not induce a direct cytopathic effect (Figure 1A), pre-infection of tumor cells before implantation into the brainstem induced complete tumor rejection and long-lasting antitumor memory without overt toxicity (Figure 1C, D). Our data show that CD4+ T cells were absolutely required for Ad-CD40L therapy, as depletion of these cells abolished APC activation and abrogated tumor rejection (Figure 3G). The data of Figure 3I, J show that, consistent with other reports of the therapeutic role of antitumor cytotoxic CD4+ T cells,20,21 this dependence upon CD4+ T cells for antitumor therapy is, at least in part, due to the generation of CD4+ T cells which exert a direct anti-proliferative effect upon target tumor cells. Central to this mechanism of action is the conversion of the tumor microenvironment from an immune quiescent phenotype to an immune inflamed phenotype which allows for upregulation of MHC class II molecules on the target tumor (Supplementary Figure 1A–C). Moreover, the cumulative data of Figures 2, 3 suggest that CD40L-activated CD4+ T cells were central in maintaining and licensing APCs to relay CD4+ T-cell help to cytotoxic T lymphocytes which have at least some role in directly delivered antitumor therapy (Figure 5G). These data are consistent with reports showing that the interaction of CD40/CD40L on APCs and activated CD4+ T cells promotes priming of potent antitumor T cells6,30,31 and helps to maintain neuroprotection through MHC class II+ APCs.32 In addition, we observed structures reminiscent of TLS around the meninges near the tumor areas in mice treated with Ad-CD40L—structures which could be a positive prognostic factor33 acting as privileged sites for local priming, activation, and proliferation of long-lasting memory Th, cytotoxic cells, memory B cells, and antibody-producing plasma cells.34–36 It may, therefore, be that these lymphoid-like aggregates found in Ad-CD40L–treated brains are local sites of antigen presentation for B- and T-cell activation.37 However, further studies are required to understand the role of these structures and the generation of the antiglioma immune responses in our model. In addition, ongoing phenotyping studies of the effector CD4+ and CD8+ T cells induced by CD40L treatment suggest that additional combination with immune checkpoint blockade may add further therapeutic potential to this approach—although additional toxicities may also be induced.
The interaction of follicular CD4+ T cells and B cells promotes B cell differentiation into antibody-secreting cells.38 In this respect, sera from tumor-bearing mice treated with Ad-CD40L significantly increased IgG levels specifically against GL261, but not against normal brain-derived astrocyte cells.24 Although it is tempting to speculate that CD40L-induced antitumor antibody contributed to therapy, the depletion of B cells had no significant effects on tumor treatment and so the role of these antibodies remains to be elucidated (Figure 3G).
Our data from the pre-infection model showed that tumor-restricted expression of CD40L represents an excellent candidate to induce immune-mediated rejection of tumor cells growing in the brainstem without necessarily inducing catastrophic toxicity. However, direct delivery of Ad-CD40L to established tumors did induce acute toxicity before day 30. Mice which survived that toxicity with fluid and nutrient support were cured of tumor (Figure 4A, C). We believe that this toxicity was a result of viral infection of nontumor tissue following direct injection, which did not occur with pre-infection of tumor cells with the replication-defective Ad-CD40L. Consistent with this, intracranial injection of Ad-CD40L into mice without tumors induced neurologic symptoms as early as 5 days post injection, almost certainly associated with CD40L expression in nontumor brain tissue leading to pro-inflammatory cytokines/chemokine production and acute toxicity, as supported by our RNA-seq transcriptional profile data (Figure 4).
Clinically, our ultimate goal will be to restrict CD40L expression to tumor cells in vivo. Therefore, we exploited the tumor specific targeting conferred upon the replication competency of adenoviruses afforded by the incorporation of a 24-bp deletion in the E1A viral gene (rAd-Δ24) responsible for pRB binding.25 The abnormalities of p16/Rb/E2F pathway allow oncolytic adenovirus with 24-bp deletion in E1A gene to preferentially replicate in gliomas26 which have disrupted Rb function, and confers antiglioma responses.25,39 Moreover, the backbone of this virus (without CD40L) has entered clinical trials for diffuse midline glioma.39 As expected of these tumor-specific, replication competent, oncolytic viruses, both the control rAd-Δ24-luc and the rAd-Δ24-CD40L viruses were cytolytic for patient-derived diffuse midline glioma cell lines SJPDGF1 and SF7761. Therefore, although it would be possible to assess the direct oncolytic activity of rAd-Δ24-CD40L in patient-derived xenografts, these experiments would have to be performed in immune deficient mice and both viruses would likely achieve similar antitumor effects. However, tumor therapy experiments in immune deficient mice would not reflect the immune-mediated effects of CD40L expression. Therefore, to assess the combined oncolytic and immunotherapeutic effect of CD40L expressed from the virus, we tested the antitumor efficacy of rAd-Δ24-CD40L in 2 different murine immune competent models (GL261 and CT2A). While these murine cells only partially support adenoviral replication, they do reflect the effects of intact immune subsets on both antitumor efficacy and toxicity. Importantly, the rAd-Δ24-luc virus could not prolong the survival of brainstem tumor–bearing mice (Figure 6). In contrast, incorporation of CD40L into the E1A mutant Δ24 adenovirus resulted in long-term cures of 50‒70% of GL261 or CT2A brainstem tumor–bearing mice (Figure 6). Because of the very limited replication of adenoviruses in these murine cells, our results here likely underestimate the levels of CD40L transgene spread and expression (and potentially therapy) that could be achieved in a human tumor where replication is more efficient. Importantly, however, the inclusion of the transgene into this vector whose replication is restricted to tumor cells greatly ameliorated the toxicity that we had observed with the Ad-CD40L vector, and this could be entirely rescued by nutrient and fluid support. We used RNA-seq transcriptional profiling analysis (Figure 4) to improve the safety profile still further. These data showed that IL-6 and IL-1β were consistently among the most differentially expressed cytokines between all 5 pathways upregulated by CD40L treatment of brainstem tumors (Figure 4F). Since antibody neutralization of both of these cytokines is clinically useful in other scenarios,40 we hypothesized that blocking these cytokines during therapy would reduce the toxicity associated with CD40L treatment. Consistent with this, blockade of IL-6 and IL-1β allowed us to escalate the intratumoral treatment dose of rAd-Δ24-CD40L as high as 107 pfu in the GL261 model with manageable and nontoxic levels of body weight loss (Figure 6D, G, H).
In conclusion, we show here that CD40L expressed from an adenoviral platform can potentially reverse the immunosuppressive environment in glioblastoma by recruiting and maintaining innate and adaptive antitumor immune responses in the brainstem. This is the first report, to our knowledge, showing that CD4+ T cells play a critical role in Ad-CD40L therapy for brainstem tumors. We also show that the tumor-restricted, replication-competent rAd-Δ24-CD40L has cytotoxicity against human patient–derived diffuse midline glioma cell lines in vitro, generates tumor cures against orthotopic brainstem gliomas in 2 different, fully immune-competent, murine models, and can be used in combination with cytokine neutralization at high intratumoral titers with readily manageable toxicity. Therefore, we believe that by building on the rAd-Δ24 backbone which is already in clinical trials for diffuse midline glioma,39 rAd-Δ24-CD40L represents a promising therapeutic for clinical evaluation as a treatment of brainstem tumors in the context of direct intratumoral injection with careful dose escalation.
Funding
This work was supported by the European Research Council, the Richard M. Schulze Family Foundation, Mayo Foundation, Cancer Research UK, National Institutes of Health (R01CA175386 to R.V., R01CA108961 to R.V.), the Shannon O’Hara Foundation, and Hyundai Hope on Wheels.
Authorship statement
Conception and design: P. Wongthida, R. Vile. Development of methodology: P. Wongthida, M. Schuelke, L. Evgin. Data acquisition: P. Wongthida, M. Schuelke, J. Thompson, T. Kottke, J. Tonne, C. Stone, J. Davies, A. Parker, C. Wetmore. Analysis and interpretation of data: P. Wongthida, M. Schuelke, L. Evgin, T. Kottke, C. Driscoll, A. Huff, R. Vile. Writing, review and/or revision of the manuscript: P. Wongthida, L. Evgin, M. Schuelke, T. Kottke, J. Thompson, C. Driscoll, A. Huff, J. Tonne, C. Stone, J. Davies, A. Parker, C. Wetmore, R. Vile. Study supervision: R. Vile
Acknowledgments
We thank Toni Woltman for expert editorial assistance, Dr Katayoun Ayasoufi for analysis of immune infiltrates, Christina McCarthy for mixed astrocytes, and the Genome Analysis and Bioinformatics Core Facilities for RNA-seq.
Conflict of interest statement
None.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}