





Abstract
Emerging data suggest that a subset of patients with diffuse isocitrate dehydrogenase (IDH)-mutant low-grade glioma (LGG) who receive adjuvant temozolomide (TMZ) recur with hypermutation in association with malignant progression to higher-grade tumors. It is currently unclear why some TMZ-treated LGG patients recur with hypermutation while others do not. MGMT encodes O6-methylguanine-DNA methyltransferase, a DNA repair protein that removes cytotoxic and potentially mutagenic lesions induced by TMZ. Here, we hypothesize that epigenetic silencing of MGMT by promoter methylation facilitates TMZ-induced mutagenesis in LGG patients and contributes to development of hypermutation at recurrence.
We utilize a quantitative deep sequencing assay to characterize MGMT promoter methylation in 109 surgical tissue specimens from initial tumors and post-treatment recurrences of 37 TMZ-treated LGG patients. We utilize methylation arrays to validate our sequencing assay, RNA sequencing to assess the relationship between methylation and gene expression, and exome sequencing to determine hypermutation status.
Methylation level at the MGMT promoter is significantly higher in initial tumors of patients that develop hypermutation at recurrence relative to initial tumors of patients that do not (45.7% vs 34.8%, P = 0.027). Methylation level in initial tumors can predict hypermutation at recurrence in univariate models and multivariate models that incorporate patient age and molecular subtype.
These findings reveal a mechanistic basis for observed differences in patient susceptibility to TMZ-driven hypermutation. Furthermore, they establish MGMT promoter methylation level as a potential biomarker to inform clinical management of LGG patients, including monitoring and treatment decisions, by predicting risk of hypermutation at recurrence.
MGMT promoter methylation facilitates TMZ-induced mutagenesis and contributes to hypermutation.
MGMT promoter methylation level in newly diagnosed LGG predicts hypermutation at recurrence.
MGMT promoter methylation may serve as a biomarker to inform clinical decision making in LGG.
Optimal clinical management remains controversial for patients diagnosed with diffuse IDH-mutant LGG as tumors inevitably recur following surgical resection and eventually cause death. While the chemotherapeutic agent TMZ extends survival in patients with glioblastoma, its clinical benefits in LGG are less clear and must be weighed against potential treatment-related risks. TMZ is potently mutagenic and drives hypermutation in association with malignant progression in a subset of patients with an initial diagnosis of LGG. Here, we identify promoter methylation at MGMT in initial LGG tumors as a predictor of hypermutation at recurrence. Our findings reveal a mechanistic basis for observed differences in susceptibility of LGG patients to TMZ-driven hypermutation. As optimal clinical management of LGG continues to be defined, our findings establish MGMT promoter methylation at diagnosis as a potential biomarker to inform monitoring and treatment decisions by stratifying patients based upon risk of developing hypermutation at recurrence.
Diffuse isocitrate dehydrogenase (IDH)-mutant low-grade gliomas (LGGs) are slow-growing brain tumors that include astrocytomas and oligodendrogliomas. Both LGG subtypes are characterized at the molecular level by mutations in IDH1/2, with astrocytomas showing additional mutations in ATRX and TP53, and oligodendrogliomas showing combined whole-arm losses of chromosome 1p and 19q.1–3 The infiltrative nature of LGGs precludes complete surgical resection and these tumors inevitably recur, albeit at highly variable intervals. Standard of care treatment of LGG is controversial, with post-surgical treatment regimens ranging from observation to aggressive treatment with radiotherapy in combination with chemotherapy.4 Temozolomide (TMZ) is an oral chemotherapeutic agent with pharmacological properties that enable crossing of the blood–brain barrier. Although it is commonly administered to LGG patients as adjuvant therapy following surgery, its clinical benefits remain unclear.5,6 For example, there was no significant difference in progression-free survival in a phase III study of LGG patients treated with either TMZ alone or radiotherapy alone.7 In another phase II study of LGG patients treated with adjuvant TMZ, only 6% of patients showed a partial radiographic response and none showed a complete response.8
LGGs arise in young, otherwise healthy patients with good prognosis relative to glioblastoma (GBM), a high-grade brain tumor that is rapidly fatal if left untreated. The potential benefits of treatment for LGG patients must therefore be carefully weighed against potential treatment-related risks. Like many chemotherapeutic agents, TMZ is potently mutagenic.9,10 TMZ alkylates the O6 position of guanine which, during DNA replication, mispairs with thymine instead of cytosine and results in G:C > A:T transition mutations. In a pilot study of TMZ-treated patients with an initial diagnosis of astrocytic LGG, we identified a hypermutator phenotype in 6 out of 10 recurrences.11 These recurrences harbored an order of magnitude of more mutations than non-hypermutated recurrences, nearly all showing the TMZ-mutagenesis signature of G:C > A:T transitions occurring predominantly at CpC and CpT dinucleotides. Importantly, all hypermutated recurrences harbored TMZ-induced mutations in glioma driver genes and underwent malignant progression to GBM. However, it has been unclear from this study and others why some patients develop hypermutation following TMZ treatment while others do not.
Understanding the risk of developing TMZ-driven hypermutation may inform clinical decision making regarding monitoring and treatment decisions for patients diagnosed with LGG. O6-methylguanine-DNA methyltransferase (MGMT) is a DNA repair protein that counters the cytotoxic and mutagenic activity of TMZ by removing alkyl groups from the O6 position of the guanine.12,13 As MGMT is irreversibly inactivated following removal of a single alkyl group, the amount of MGMT protein in a cell is a limiting factor in the repair of TMZ-induced lesions. In previous studies of recurrences from TMZ-treated LGGs, we have observed elevated levels of MGMT promoter methylation in hypermutated recurrences relative to non-hypermutated recurrences.11,14 Other groups have also observed co-occurrence of MGMT promoter methylation and hypermutation in recurrent tumors from LGG and GBM patients who received TMZ.15–17 Based on these observations, we hypothesize that epigenetic silencing of MGMT by promoter methylation facilitates TMZ-induced mutagenesis and contributes to the development of hypermutation. Here, we conduct a longitudinal cohort study of LGG patients who received adjuvant TMZ with the aim to determine whether MGMT promoter methylation level in initial tumors is a predictor of hypermutation at recurrence.
Materials and Methods
Sample Acquisition
All initial and recurrent tumor samples were collected during surgical resection and were either snap frozen and stored in liquid nitrogen, or formalin fixed and paraffin embedded (FFPE). In cases where more than one sample from a tumor was investigated, those samples were independent, spatially distinct pieces. DNA was extracted and processed by a standard phenol chloroform extraction as previously described11 or with the Qiagen FFPE DNA extraction kit (Qiagen) following manufacturer instructions. Samples were obtained from the Neurosurgery Tissue Bank at the University of California San Francisco (UCSF). Additional samples were obtained from The Brain Tumour Research Centre at McGill University. Sample use was approved by the Committee on Human Research at UCSF, and research was approved by the institutional review board at UCSF. All patients provided informed written consent.
Evaluation of Hypermutation Status
Exome-sequencing of the initial tumor and matched recurrence, along with germline (blood) DNA was conducted using either Agilent (SureSelect Human All Exon 50MB, SureSelect Human All Exon v4) or NimbleGen (SeqCap EZ Exome v3) exome capture kits according to manufacturer’s protocol. Exome-sequencing data has been previously published for 11 patients11,14,18,19 and is newly generated for 26 patients (Supplementary Table 1). Paired-end sequencing data from exome capture libraries were aligned to the reference human genome (hg19) and mutations were called as previously described.11 The software package deconstructSigs20 was used to conduct mutational signature analysis yielding proportionate contributions of each signature to individual exomes. Signature numbers correspond to those previously described.21 TMZ-induced hypermutation was defined by >40% contribution from mutational signature 11, which is associated with TMZ treatment.
Quantitative Sequencing–Based Assay of MGMT Promoter Methylation Level
Bisulfite amplicon sequencing (BSAS), a method that couples bisulfite conversion of genomic DNA with PCR enrichment of targeted regions and next-generation sequencing, was conducted as previously described22 using 250 ng–1 µg of genomic DNA isolated from fresh frozen or FFPE tissues. Quality control and quantification of BSAS methylation levels were conducted utilizing the Bismark software package.23 For validation, genome-scale methylation datasets were utilized from Infinium 450K or 850K (EPIC) Beadchip Arrays.24 These datasets have been previously published for 10 patients18,19 and are newly generated for 19 patients (Supplementary Table 1). Methylation at cg12981137 in the MGMT promoter region was calculated from Infinium array datasets using the MGMT-STP27 software package.25,26
Quantification of MGMT Gene Expression
RNA was isolated from tumor tissue samples with Trizol (Invitrogen) according to manufacturer’s instructions. Transcriptome sequencing libraries were prepared as previously described.11 Transcriptomes have previously been published for 6 patients18,19 and are newly generated for 12 patients (Supplementary Table 1). Read summarization was performed with featureCounts.27 Features with low read counts (row sums <1) were removed and the remainder were Variance-Stabilizing Transformed (VST) normalized with DESeq2.28 Batch correction was performed with ComBat.29 Transcripts assigned to ENSG00000170430.9 were considered as quantitation of MGMT gene expression.
Logistic Regressions and Predictive Modeling
Univariate and multivariate logistic regression models were used to assess the associations of clinical and methylation variables with hypermutation status. In addition, recursive partitioning via the partDSA algorithm,30 was employed to assess associations and explore cutoffs of continuous covariates. Models were evaluated via likelihood ratio tests and misclassification prediction error rates.
Results
Patient Cohort and Course After TMZ Treatment
Patient inclusion in this cohort was dependent upon (i) an initial diagnosis of grade II LGG, (ii) adjuvant TMZ treatment, and (iii) availability of at least one tissue sample each from the initial untreated tumor and a posttreatment recurrence. Histological grading was performed by a clinical neuropathologist, across multiple samples when available, and classification incorporated molecular genetic features as described in the 2016 World Health Organization guidelines.2 Out of the 37 patients included in the study, 23 had an initial diagnosis of grade II astrocytoma and 14 had an initial diagnosis of grade II oligodendroglioma (Table 1). Of the astrocytomas, 6 (26.1%) maintained grade II features while 6 (26.1%) progressed to grade III anaplastic astrocytoma and 11 (47.8%) to grade IV GBM. Of the oligodendrogliomas, 3 (21.4%) maintained grade II features while the remaining 11 (78.6%) underwent malignant progression to grade III anaplastic oligodendroglioma.
Description of patient cohort
| Astrocytoma (n = 23) | Oligodendroglioma (n = 14) | Overall (n = 37) | |
|---|---|---|---|
| Age at diagnosis | |||
| Median [interquartile range (IQR)] | 31.0 [27.5, 38.5] | 37.0 [32.25, 44] | 33.0 [28.0, 39.0] |
| Sex | |||
| Female | 11 (47.8%) | 6 (42.9%) | 17 (45.9%) |
| Male | 12 (52.2%) | 8 (57.1%) | 20 (54.1%) |
| TMZ cycles | |||
| Median [IQR] | 12.0 [11.75, 15.25] | 21.5 [12.0, 24.0] | 12.0 [12, 22.5] |
| Grade at recurrence | |||
| II | 6 (26.1%) | 3 (21.4%) | 9 (24.3%) |
| III | 6 (26.1%) | 11 (78.6%) | 17 (45.9%) |
| IV | 11 (47.8%) | 11 (29.7%) | |
| HM status at recurrence | |||
| HM | 9 (39.1%) | 6 (42.9%) | 15 (40.5%) |
| Mixed | 2 (8.7%) | 1 (7.1%) | 3 (8.1%) |
| nonHM | 12 (52.2%) | 7 (50.0%) | 19 (51.4%) |
| Astrocytoma (n = 23) | Oligodendroglioma (n = 14) | Overall (n = 37) | |
|---|---|---|---|
| Age at diagnosis | |||
| Median [interquartile range (IQR)] | 31.0 [27.5, 38.5] | 37.0 [32.25, 44] | 33.0 [28.0, 39.0] |
| Sex | |||
| Female | 11 (47.8%) | 6 (42.9%) | 17 (45.9%) |
| Male | 12 (52.2%) | 8 (57.1%) | 20 (54.1%) |
| TMZ cycles | |||
| Median [IQR] | 12.0 [11.75, 15.25] | 21.5 [12.0, 24.0] | 12.0 [12, 22.5] |
| Grade at recurrence | |||
| II | 6 (26.1%) | 3 (21.4%) | 9 (24.3%) |
| III | 6 (26.1%) | 11 (78.6%) | 17 (45.9%) |
| IV | 11 (47.8%) | 11 (29.7%) | |
| HM status at recurrence | |||
| HM | 9 (39.1%) | 6 (42.9%) | 15 (40.5%) |
| Mixed | 2 (8.7%) | 1 (7.1%) | 3 (8.1%) |
| nonHM | 12 (52.2%) | 7 (50.0%) | 19 (51.4%) |
Description of patient cohort
| Astrocytoma (n = 23) | Oligodendroglioma (n = 14) | Overall (n = 37) | |
|---|---|---|---|
| Age at diagnosis | |||
| Median [interquartile range (IQR)] | 31.0 [27.5, 38.5] | 37.0 [32.25, 44] | 33.0 [28.0, 39.0] |
| Sex | |||
| Female | 11 (47.8%) | 6 (42.9%) | 17 (45.9%) |
| Male | 12 (52.2%) | 8 (57.1%) | 20 (54.1%) |
| TMZ cycles | |||
| Median [IQR] | 12.0 [11.75, 15.25] | 21.5 [12.0, 24.0] | 12.0 [12, 22.5] |
| Grade at recurrence | |||
| II | 6 (26.1%) | 3 (21.4%) | 9 (24.3%) |
| III | 6 (26.1%) | 11 (78.6%) | 17 (45.9%) |
| IV | 11 (47.8%) | 11 (29.7%) | |
| HM status at recurrence | |||
| HM | 9 (39.1%) | 6 (42.9%) | 15 (40.5%) |
| Mixed | 2 (8.7%) | 1 (7.1%) | 3 (8.1%) |
| nonHM | 12 (52.2%) | 7 (50.0%) | 19 (51.4%) |
| Astrocytoma (n = 23) | Oligodendroglioma (n = 14) | Overall (n = 37) | |
|---|---|---|---|
| Age at diagnosis | |||
| Median [interquartile range (IQR)] | 31.0 [27.5, 38.5] | 37.0 [32.25, 44] | 33.0 [28.0, 39.0] |
| Sex | |||
| Female | 11 (47.8%) | 6 (42.9%) | 17 (45.9%) |
| Male | 12 (52.2%) | 8 (57.1%) | 20 (54.1%) |
| TMZ cycles | |||
| Median [IQR] | 12.0 [11.75, 15.25] | 21.5 [12.0, 24.0] | 12.0 [12, 22.5] |
| Grade at recurrence | |||
| II | 6 (26.1%) | 3 (21.4%) | 9 (24.3%) |
| III | 6 (26.1%) | 11 (78.6%) | 17 (45.9%) |
| IV | 11 (47.8%) | 11 (29.7%) | |
| HM status at recurrence | |||
| HM | 9 (39.1%) | 6 (42.9%) | 15 (40.5%) |
| Mixed | 2 (8.7%) | 1 (7.1%) | 3 (8.1%) |
| nonHM | 12 (52.2%) | 7 (50.0%) | 19 (51.4%) |
Hypermutation Status at Recurrence
In order to determine whether recurrent tumor samples had undergone hypermutation, we utilized new and previously reported11,14,18,19 whole-exome sequencing data to characterize the mutational burden of all tissue samples from recurrent tumors. We then conducted mutational signature analysis to specifically detect the presence of G:C > A:T transitions consistent with TMZ-induced damage.21 Hypermutated recurrence samples harbored a median of 2120 mutations (range, 411–3980) in the exome, while non-hypermutated recurrence samples harbored a median of 59 mutations (range, 20–138) (Supplementary Table 2). All recurrence samples analyzed were hypermutated for 15 patients (HM, 40.5%), while no recurrence samples analyzed were hypermutated for 19 patients (nonHM, 51.4%). The remaining 3 patients showed hypermutation in some recurrence samples, but not others (Mixed HM status, 8.1%). This is consistent with intratumoral heterogeneity, which is a key feature of glioma that reflects evolutionary dynamics of cancer.31,32 TMZ-induced hypermutation likely occurs only in few cells, which then undergo clonal expansion. If initial surgical resection is incomplete, for example, residual tumor may continue to grow and be resampled at recurrence as a non-hypermutated component of tumor.
Patients that underwent hypermutation at recurrence generally progressed to higher-grade tumors: 14/15 (93.3%) HM patients progressed to grade III or IV tumors, while only 12/19 (63.2%) nonHM patients progressed (P = 0.039 by chi-square test; Fig. 1A). Clinical follow-up of the only HM patient in our cohort with a grade II recurrence (patient 337) reveals that the patient progressed within 2 years of the first recurrence to a second recurrence with grade III features. We sought to determine whether differences in hypermutation status at recurrence could be explained by known patient or treatment-related factors. We found no association between HM status and patient age at diagnosis (Fig. 1B) or number of TMZ cycles received (Fig. 1C). While patients received a median of 12 cycles of adjuvant TMZ treatment, one nonHM patient (patient 163) received an outlier 51 cycles of TMZ treatment over the span of 5 years. This patient had one posttreatment recurrence, which was not hypermutated and had maintained grade II features. Collectively, these data suggest that these otherwise important prognostic factors are not predictive of risk for developing hypermutation.
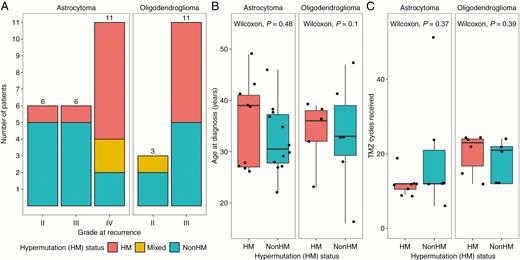
Patient hypermutation status and association with clinical characteristics. (A) Hypermutation (HM) status by grade at recurrence. (B) HM status by patient age at diagnosis. (C) HM status by number of TMZ cycles received prior to recurrence.
Assaying Methylation at the MGMT Promoter
To test our hypothesis of a role for MGMT promoter methylation in the development of TMZ-driven hypermutation, we characterized methylation levels in the MGMT promoter region in a quantitative and high-throughput manner using BSAS.22 We targeted a 273 bp region of interest (chr10: 129 467 210‒129 467 482; hg38) containing 27 cytosine-phosphate-guanine (CpG) sites. This region spans that interrogated by our previous study14 and that which is commonly interrogated by methylation-specific PCR (MSP), a clinically used method that provides a binary indicator of methylation status.33 Notably, the region encompasses an enhancer located at the first exon/intron boundary34 as well as regions at which methylation at CpG sites is linked to silencing of MGMT gene expression.35–37 Sequence motif-analysis38 and examination of ENCODE ChIP-Seq binding profiles39 indicate that the region is likely bound and acted upon by several transcriptional regulators (Supplementary Figure 1).
We characterized methylation by BSAS at an average of 28 635x coverage on 109 total initial tumor and recurrence samples from all 37 patients in the cohort (Supplementary Table 4). We utilized new and previously reported18,19 data from Infinium arrays24 to perform independent validation of BSAS results on 75 of the tumor samples. Infinium arrays include two CpG sites in the MGMT promoter region that are linked to MSP-determined MGMT methylation status and MGMT gene expression.25,26 One of these CpG sites (cg12981137, located +174 bp from the transcription start site [TSS]) is within the region assayed by BSAS. Comparison of methylation measures at this particular CpG site from both methods revealed a high degree of concordance across samples (Fig. 2A).
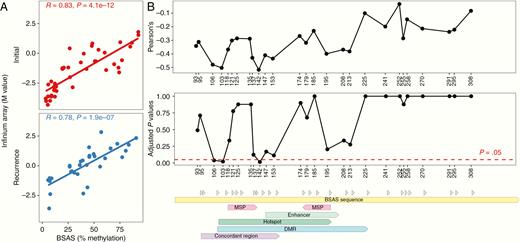
Validation of bisulfite amplicon sequencing (BSAS) method for assaying MGMT promoter methylation level. (A) Association between methylation level at CpG site +174 bp from TSS assayed by BSAS and by Infinium Array across samples. (B) Association between methylation level at each CpG site assayed by BSAS and MGMT mRNA level across samples. Pearson correlations and P-values adjusted for multiple hypothesis testing are shown for each CpG site labeled by distance from TSS. Sequence annotations are shown for regions assayed by methylation-specific PCR (MSP), an enhancer, and regions for which associations between methylation and transcription have previously been shown (hotspot, DMR, concordant region).
As methylation of the MGMT promoter is associated with gene silencing, we sought to directly investigate the relationship between methylation in the region assayed by BSAS and MGMT gene expression. We utilized new and previously reported18,19 data from whole transcriptome profiling on 41 samples from initial tumors and recurrences. We examined correlations between methylation levels at each of the 27 CpG sites in the region and MGMT mRNA level. Adjusting for multiple-hypothesis testing by the Holm–Bonferroni method, 3 CpG sites (+106, +113, +142 bp from TSS) showed strong, inverse relationships between methylation levels and gene expression that were statistically significant (Fig. 2B). All 3 CpG sites were within regions where methylation has been previously linked to MGMT gene expression: “hotspot,” 35 “concordant region,” 36 and “differentially methylated region” (DMR).37
MGMT Promoter Methylation Level at Recurrence Is Associated with Hypermutation Status
To investigate the relationship between MGMT promoter methylation and hypermutation, we began by characterizing methylation profiles of individual recurrent tumor samples in which we had detected the presence or absence of hypermutation. We conducted hierarchical clustering of MGMT methylation profiles using Ward’s minimum variance method, finding that hypermutated recurrence samples generally showed increased methylation levels across CpG sites and clustered separately from those that were not hypermutated (Fig. 3A). A similar separation of clusters was observed when this analysis was restricted to only the 3 CpG sites that showed a significant inverse relationship with MGMT gene expression (+106, +113, +142 bp from TSS; Supplementary Figure 2). Levels of MGMT gene expression were significantly reduced in hypermutated recurrences relative to non-hypermutated recurrences (Fig. 3B). A negative relationship was observed between average MGMT promoter methylation level and MGMT gene expression level (Pearson R = −0.59, P = 0.002), indicating that MGMT was epigenetically silenced in hypermutated recurrences by promoter methylation (Fig. 3C).
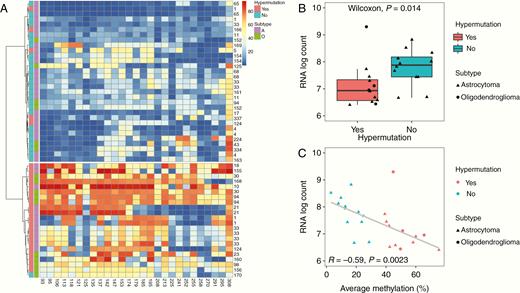
Hypermutation in individual recurrence samples is associated with increased MGMT promoter methylation and reduced expression. (A) Hierarchical clustering by Ward’s minimum variance method of methylation levels at 27 CpG sites for individual recurrence samples labeled by patient ID number, presence or absence of hypermutation (HM), and subtype (A: astrocytoma, O: oligodendroglioma). (B) MGMT expression level by RNA log count for individual recurrence samples by presence of absence of hypermutation. (C) MGMT expression level by RNA log count and average methylation level at the MGMT promoter for each individual recurrence sample.
While individual recurrence samples from HM and nonHM patients showed similar levels of methylation at the MGMT promoter, samples from patients with Mixed HM status showed greater variability (Fig. 4A). We used a bootstrapping-based methodology to quantify this, randomly taking one sample from each patient, and then resampling 100 times to generate a distribution of methylation levels. We found a wider variance of methylation levels for samples from patients with Mixed HM status (mean 37.3, SD 8.600) relative to patients that were HM (mean 45.7, SD 0.973) or nonHM (mean 20.5, SD 0.387) (Fig. 4B ). We next examined individual recurrence samples for each of the 3 patients with Mixed HM status. For each patient, we found that samples from the same tumor that were hypermutated showed higher methylation levels at the MGMT promoter than samples that were not hypermutated (Fig. 4C). Thus, a link between MGMT promoter methylation level and hypermutation is supported not only by interpatient analysis of recurrent tumors, but also by intratumoral analysis of individual samples from recurrent tumors.
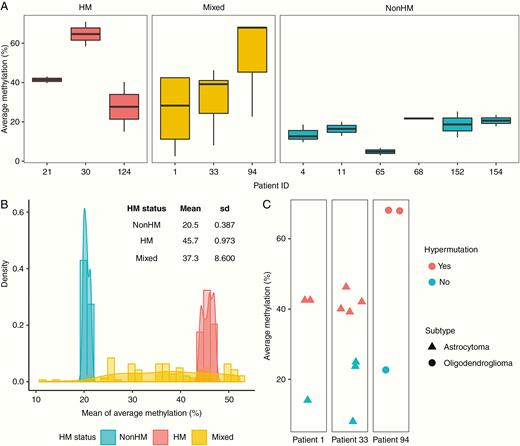
Intratumoral analysis of MGMT promoter methylation and hypermutation. (A) Average methylation level at the MGMT promoter methylation for individual recurrence samples from the patient, classified by HM status. (B) Quantification of variation in MGMT promoter methylation levels by bootstrapping. (C) Average methylation level and presence of hypermutation in individual recurrence samples of patients with Mixed HM status.
MGMT Promoter Methylation Level in Initial Tumors Predicts Hypermutation at Recurrence
We next sought to determine whether MGMT promoter methylation levels in initial, untreated tumors differed between patients who developed hypermutation at recurrence and patients who did not. We found higher methylation at nearly every CpG site in the region evaluated in initial tumors from HM patients than those from nonHM patients (Fig. 5A). Average methylation level was significantly higher in initial tumors of HM patients than nonHM patients (46.0% vs 34.8%, P = 0.027 by Wilcoxon rank-sum test, Fig. 5B). Average methylation level was also significantly higher in recurrence tumors of HM patients than nonHM patients (44.0% vs 20.5%, P < 0.001 by Wilcoxon rank-sum test), confirming observations from our previous study.14 For HM patients, methylation levels did not significantly change between initial tumors and recurrences (46.0% vs 45.6%, P = 0.86 by Wilcoxon signed-rank test; Fig. 5C). In contrast, for nonHM patients, methylation levels significantly decreased between initial tumors and recurrences (34.8% vs 20.5%, P < 0.001 by Wilcoxon signed-rank test; see also Fig. 5D).
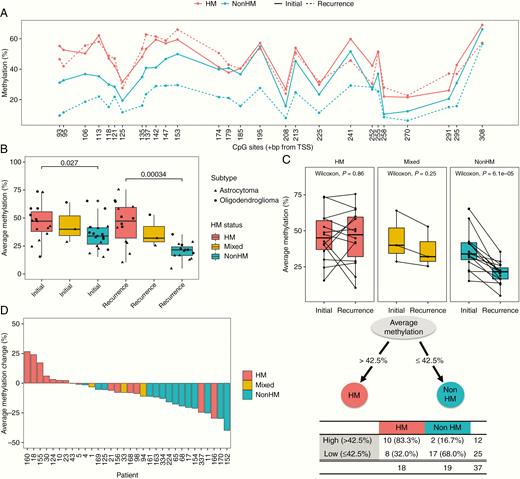
MGMT promoter methylation level in initial tumors predicts hypermutation status at recurrence. (A) Methylation level at each CpG site for initial tumors and recurrences of patients by HM status. (B) Average methylation level for initial tumors and recurrences of patients by HM status. (C) Paired analysis of average methylation level between initial tumors and recurrences. (D) Change in average MGMT promoter methylation level from initial tumor to recurrence. (E) Regression tree identifying average methylation level in initial tumors as a significant predictor of HM status at recurrence.
Given differences in MGMT promoter methylation level between initial tumors of patients that did and did not develop hypermutation, we evaluated logistic regression models to test the association of methylation levels in initial tumors with hypermutation status at recurrence. To enable binary classification of hypermutation status, we followed precedent from neuropathology practice where the most malignant component of the tumor defines clinical grade, even if remnants of low-grade tumor are present alongside transformation. We therefore classified any patient with at least one hypermutated recurrence sample as hypermutated. A univariate logistic model revealed that average methylation level in initial tumors was associated with hypermutation at recurrence (odds ratio, 1.06; 95% CI [1.01, 1.12], P = 0.04). Average methylation level remained significant after adjusting for molecular subtype (astrocytoma vs oligodendroglioma) and age of diagnosis (odds ratio, 1.09; 95% CI [1.02, 1.18], P = 0.02). We tested other clinical variables including sex and number of TMZ cycles received in univariate and multivariate models, but did not find significant associations with hypermutation at recurrence. Evaluation of individual CpG sites revealed that methylation at the CpG site +113 bp from TSS in the initial tumor was most associated with hypermutation at recurrence (odds ratio, 1.04; 95% CI [1.01, 1.08], P = 0.01). Notably, this site has been associated with silencing of MGMT gene expression by this study and others.35–37 Finally, we conducted regression tree analyses,30 which identified average methylation level in an initial tumor as a significant predictor of hypermutation at recurrence. An average methylation level of greater than 42.5% was associated with hypermutation (P < 0.01 by Fisher’s exact test) (Fig. 5E, Supplementary Table 5).
Discussion
This study establishes MGMT promoter methylation level in initial untreated LGG tumors as a significant predictor of TMZ-driven hypermutation at recurrence. This study also confirms our previous observation14 that MGMT promoter methylation levels at recurrence are higher in patients who develop hypermutation than in patients who do not. Collectively, these results provide evidence in support of our hypothesis that epigenetic silencing of MGMT by promoter methylation facilitates TMZ-induced mutagenesis and contributes to the development of hypermutation. They reveal a mechanistic basis for observed differences in propensity of TMZ-treated patients with LGG to develop hypermutation at recurrence. Furthermore, they establish the potential of MGMT promoter methylation to serve as a clinically useful biomarker for LGG patients by predicting risk of developing TMZ-driven hypermutation at recurrence.
MGMT promoter methylation is associated with chemosensitivity and is prognostic for longer overall survival in GBM patients and in patients with astrocytic LGG that are classified as “high-risk” due to patient age, tumor size and invasion into corpus callosum, and/or preoperative neurological deficits.40,41 In longitudinal studies that have been conducted of TMZ-treated GBM patients, recurrences show downward shifts in levels of MGMT promoter methylation consistent with increased TMZ resistance in less methylated clones.42–45 In our study of LGG patients, we find that patients who do not develop hypermutation at recurrence also show a downward shift in MGMT promoter methylation level. However, patients that develop hypermutation show increased MGMT promoter methylation levels in initial tumors that are maintained at recurrence. Taken together, the data support a model whereby MGMT promoter methylation in LGG patients confers sensitivity both to TMZ-induced cytotoxicity and to TMZ-induced mutagenesis, which is prerequisite to the generation and selection of hypermutated tumor clones. MGMT promoter methylation level in initial tumors may thus be used to anticipate future aggressive clonal outgrowths of hypermutated and malignantly transformed tumor cells.
Standard of care for LGG patients remains controversial, with post-surgical treatment regimens ranging from observation to aggressive treatment with radiotherapy in combination with chemotherapy.4 Knowledge of MGMT promoter methylation level may provide insight into risk for developing TMZ-driven hypermutation leading to appropriate patient counseling. For example, the aforementioned patient (patient 163) who received 51 cycles of TMZ treatment had a low average MGMT promoter methylation level of 28.3% in the initial tumor and may have been predicted to be of low risk for developing hypermutation. Patients with higher levels of MGMT promoter methylation may be considered candidates for alternative treatments such as IDH inhibitors currently in development.46,47 If treated with TMZ, these patients may be monitored more closely by imaging surveillance. Notably, while hypermutation may contribute to malignancy, the radically altered genome of hypermutated tumors may also present new opportunities for therapeutic exploitation.6,48 For example, the mutational burden and clonal mutational architecture of hypermutated tumors may increase neoantigen load, thereby conferring sensitivity to immunotherapy. Hypermutated gliomas that arise due to heritable or somatic defects in DNA repair have demonstrated clinically significant responses to immunotherapy by checkpoint blockade.49,50 A phase II clinical trial has recently been launched to test immune checkpoint inhibitor nivolumab in IDH-mutant glioma patients with and without hypermutation (NCT03718767). As optimal adjuvant management of LGG continues to be defined, future and ongoing studies should examine MGMT promoter methylation as a predictive biomarker to confirm its clinical utility for patient monitoring and treatment.
Funding
This work was supported by the National Institutes of Health (1F32CA239472–01 and 5T32CA151022–09 to R.M., R01CA169316 to J.F.C., and P50CA097257 to J.F.C. and S.M.C.). Additional support was provided by a gift from the Dabbiere family.
Data Availability
All relevant sample information, patient annotations, and data from BSAS, Infinium Array, exome and RNA sequencing are provided in Supplementary Table 3 and Supplementary Table 4. Previously published datasets are available at the European Genome-Phenome Archive EGAS00001000579 [refs 11,14,18], EGAS00001001255 [ref 18], and EGAS00001001854 [ref 19]. New datasets are available at EGAS00001003956. Code is available on Github (radhikamathur/MGMT-Project/MGMT_Code.R).
Conflict of interest statement. No conflicts of interest to declare.
Acknowledgments
We thank the staff at the UCSF Brain Tumor Research Center Tissue Core for timely and significant contributions of key samples. We thank the Center for Advanced Technology at UCSF and the Targeted DNA Methylation & Mitochondrial Heteroplasmy Core at the University of Oklahoma Health Science Center for services in genomic sequencing and analysis.
Authorship statement. Experimental design: R.M., J.F.C; implementation: R.M., Y.Z., M.R.G., C.H., M.Z., S.B. J.J.P, A.M.M.; data analysis and interpretation: R.M., Y.Z., M.R.G., M.Z., S.B., K.P., J.C., M.S.B., J.J.P, N.A.O., A.M.M., S.M.C., J.F.C; writing: R.M. with input and approval from all authors.
References



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}