





Abstract
Skull base meningiomas are considered to be difficult for surgical treatment. We wondered whether genetic alterations recently identified in benign non-NF2-mutated World Health Organization (WHO) grade I meningiomas are related to clinical features of skull base meningiomas and whether druggable signaling pathways are activated.
We analyzed 93 skull base meningiomas (82 WHO grade I, 11 WHO grade II) for mutations of hot spots or the most relevant exons of AKT1, KLF4/TRAF7, SMO, PI3K, and the TERT promoter.
The AKT1E17K mutation was present in 31% of patients and was related to meningothelial histology. AKT1E17K had a negative effect on the time to tumor recurrence. Analyses of activated signaling proteins revealed among AKT1E17K tumors a significantly higher rate of phospho–mammalian target of rapamycin (mTOR) and phospho-p70S6K+ tumors. AKT1E17K tumors with immunoexpression of phospho–extracellular signal-regulated kinase 1 or 2 (ERK1/2) were characterized by significantly shorter time to tumor recurrence compared with AKT1wt tumors expressing phospho-ERK1/2 (P = .046). KLF4 mutations (K409Q) were present in 11.8% of cases, with significant association to the secretory/transitional subtype (P < .001). The presence of the KLF4 K409Q mutation was associated with favorable outcome. One phosphatidylinositol-3 kinase (PI3K) mutation but no SMO or TERT promoter mutation was found.
AKT1E17K mutation is frequent in skull base meningiomas, results in activation of the mTOR and ERK1/2 signaling pathways, and has negative impact on tumor recurrence. Patients with skull base meningiomas with AKT1E17K mutation might benefit from additional treatment targeting the mTOR pathway. Generally, the PI3K-Akt-mTOR axis might be a potential target for kinase inhibitors in these tumors.
The study reveals that in a selected cohort of skull base meningiomas, the frequency of the AKT1E17K mutation is rather high (~30%) compared with previous studies analyzing meningioma cohorts, including other tumor locations. This result is in accordance with the known enrichment of this mutation in skull base meningiomas. We also show that the presence of the AKT1E17K mutation indicates shorter time to tumor recurrence compared with wild-type tumors and that the AKT1E17K mutations activate mTOR and ERK1/2 signaling pathways. Therefore, especially the mTOR pathway might be a promising target in skull base meningioma patients suffering from tumors with the AKT1E17K mutation and needing additional treatment.
Meningiomas represent the most frequent intracranial tumors, with about 80% being categorized as World Health Organization (WHO) grade I meningiomas.1,2 Sites preferentially affected by intracranial meningiomas are the meninges of the convexity of the brain and the skull base, while intraventricular meningiomas are rare. Collectively, meningiomas can be separated into non–skull base and skull base meningiomas, the latter being further separated into anterior, middle, and posterior fossa meningiomas.3 The localization of the skull base meningioma is important for surgical decision making and further clinical course.4,5
Despite tremendous improvement in the microsurgical treatment of these tumors in recent years, they still represent a surgically challenging entity. Feasibility of radical resection at the skull base depends mainly on the anatomical location and involvement of neighboring neurovascular structures. Neurovascular compression or infiltration is relatively common, which may hinder radical surgical resection in order to prevent severe postoperative neurological deficits.4 Even some skull base meningiomas may undergo only partial surgical resection and require further adjuvant nonsurgical treatment options. Although radiotherapy and stereotactic radiosurgery nowadays belong to the modern armamentarium for the treatment of difficult skull base meningiomas, no effective medical treatment options exist so far which may be helpful in the treatment of extensive otherwise untreatable meningiomas.
Besides the extent of resection and the histological grade of malignancy, no clear molecular characteristics have been identified to affect the outcome of skull base meningiomas. It has been well established that about 50% of meningiomas have alterations of the NF2 tumor suppressor gene.6 Recently, whole genome sequencing approaches have identified additional molecular alterations. These alterations occur in wild-type NF2 meningiomas and can be found preferentially at the skull base. These genetic alterations affect mainly the genes SMO, AKT1, TRAF7, KLF4, and POLR2A, respectively.7–9
In order to evaluate the impact of molecular alterations on the clinical course, we correlated clinicopathologic features of skull base meningiomas with mutational status and the expression of druggable target proteins in a series of 93 patients suffering from skull base–located meningiomas.
Materials and Methods
Tumor Material
The study included 93 tumors from 93 individual patients with skull base meningiomas. All tumors have been reviewed twice for histological diagnosis according to the 2007 WHO classification of brain tumors by 2 experienced neuropathologists (C.H., C.M.).2 The median follow-up time was 40.0 months (range: 6–103 mo). The study was approved by the local institutional review board at Hannover Medical School (#2715-2015). With regard to the recently launched update of the WHO classification,10 the histological diagnoses remained unchanged. Based on representative hematoxylin and eosin (H&E) sections, presence of brain invasion, necroses, and mitotic activity were assessed (Ü.Y., C.M.). The following clinical data were retrieved from the files of the Department of Neurosurgery, Hannover Medical School: tumor localization separated into central skull base (including olfactory groove, planum sphenoidale, tuberculum sellae, optic sheath/canal, cavernous sinus, cerebello-pontine angle, petroclival, bony foramina, foramen magnum) and lateral skull base (including middle and lateral sphenoid wing, orbitosphenoid, orbital roof, lateral tentorial and temporal bone),11 presence or absence of peritumoral edema, extent of edema according to the Steinhoff classification,12 extent of resection (Simpson grade13), time to tumor recurrence, additional radiotherapy, signs of neurofibromatosis type 2 (NF2), and involvement of the cavernous sinus. Preoperative tumor embolization was not reported in any of the meningioma samples studied.
DNA Extraction and Direct Sequencing
Tumor DNA was extracted from formalin-fixed paraffin embedded blocks after selection of tissue parts without bone or dural tissue parts using the NucleoSpin Tissue Kit (Macherey-Nagel). Based on the corresponding H&E-stained slides, the tumor tissue was marked by a circle, comprising approximately 80% or more of the total tumor tissue. Paired normal samples were not sequenced. For all genes studied, tumor samples with known present or absent mutations were used as positive and negative controls, respectively. For AKT1, malignant meningioma cell lines, stably transfected with human AKT1 with or without the mutation E17K, served as additional controls. The genes AKT1 and KLF4 were selectively screened for the hot spot mutations E17K and K409Q, respectively. TERT promoter was selectively analyzed for the activating mutations C228T and C250T and SMO for the most frequent missense mutation, W535L. For TRAF7 the most relevant exons, which harbored mutations in meningiomas in published studies, were sequenced with the primers listed in Supplementary Table S1. BigDye terminator cycle sequencing was performed on an ABI-310C capillary sequencer (Applied Biosystems).
Fragments spanning codons 545 and 1047 of PIK3CA were amplified. For PCR, 20 ng of DNA and TaqPlus 2× PCR Master Mix (Qiagen) were employed. PCR was performed in a total volume of 30 µL and included initial denaturation at 95°C for 180 s, followed by 35 cycles with denaturation at 95°C for 30 s, annealing at 56°C for 25 s, and extension at 72°C for 40 s. Two microliters of the amplification product was submitted to bidirectional sequencing using the BigDye Terminator v3.1 Sequencing Kit (Applied Biosystems). Sequences were determined using an ABI-3500 Genetic Analyzer (Applied Biosystems) and Sequence Pilot v4.0.1 software (JSI-Medisys).
Construction of a Tissue Microarray
Using the formalin-fixed paraffin embedded blocks, we constructed 2 tissue microarrays (TMAs), as previously described.14 The first TMA contained 43 tumors and the second TMA contained 42 tumors. Not all patient samples could be used for construction of TMAs due to technical limitations. In some cases the tumor areas of the embedded material could not be reached by the needle of the punching device and sometimes the tumor texture was too firm. To minimize a bias by the small tissue areas of TMA samples, each tumor sample was represented in a total of 3 TMA spots derived from different regions of the original paraffin embedded sample. The following antibodies were used for immunostaining of the TMA: phosphorylated (phospho-)Akt (Ser473, rabbit, polyclonal, dilution 1:400; Santa Cruz), phospho–mammalian target of rapamycin (mTOR) (Ser2448, rabbit, polyclonal, 1:100; Cell Signaling), phospho–p70 ribosomal protein S6 kinase (p70S6K) (rabbit, monoclonal, dilution 1:100; Abcam), phospho–eukaryotic initiation factor 4E binding protein 1 (4EBP1) (rabbit, polyclonal, dilution 1:100; Cell Signaling), and phospho–extracellular signal-regulated kinase 1 or 2 (ERK1/2) (Thr202, Thr204, rabbit, polyclonal, dilution 1:100; Cell Signaling). All stainings were run with both appropriate positive and negative controls as reported previously.14,15 Intensity of the immunoexpression was graded by 2 observers (Ü.Y., C.M.) as follows: 0 = no staining, 1 = weak staining, and 2 = strong staining. However, it should be noted that due to technical limitations, not all tumor spots on the TMA could be evaluated for each immunostaining, giving rise to differences between the numbers of cases analyzed for each staining, as well as for lower overall numbers in the immunostaining results compared with the sequencing numbers. Additionally, the analysis is potentially biased because if spots were negative for a given tumor, we did not repeat the staining using complete sections. Immunoexpression was graded as 0 = no immunostaining, 1 = focal or weak immunostaining, and 2 = strong immunostaining. This scoring was applied for comparison of frequencies between genotypes (wild-type or mutation). For analysis of time to tumor recurrence, expression data were summarized into 0 = no immunostaining and 1 = positive immunostaining.
Statistics
The statistical analysis used 93 samples representing 93 individual patients. Besides descriptive analyses, the presence of mutations and protein immunoexpression were cross-tabulated with each other and with other patient characteristics. To determine associations between protein immunoexpression and AKT1 or KLF4 mutation status, a chi-square test was applied. Furthermore, the recurrence time was analyzed and compared with respect to mutation status and protein immunoexpression (or both combined) in Kaplan–Meier analyses, a log-rank test, a Fleming–Harrington test,16 or a Cox proportional hazards regression including the calculation of concordance indices.17 The analyses were carried out with the statistical packages SPSS v22 (IBM) and SAS v9.4. All tests use an error level of .05 at an exploratory claim.
Results
We analyzed a total of 93 skull base meningiomas (82 WHO grade I, 11 WHO grade II). The demographic data of the cohort are given in Table 1. The distribution of histological subtypes is given in Table 2. Brain invasion was present in 3 patients (3.2%). In 9 patients, tumor necroses were present in H&E sections (9.7%). In 75 cases, no mitoses were identified, while in 15 cases (16.1%) 1–3 and in 3 cases (3.2%) 4 or more mitoses per 10 high-power field were counted. Regarding tumor localization, 57 tumors (61.3%) were located centrally and 36 (38.7%) laterally. Thirty-three patients (36.9%) showed peritumoral edema which could be separated into 12 grade 1, 16 grade 2, and 5 grade 3 tumors according to the Steinhoff classification12 (3 missing classifications due to lacking data).
Demographic characteristics of 93 patients with skull base meningiomas
| Age, y, mean ± SD/median/range | 54.3 ± 12.2/54.0/24–85 |
| Sex | |
| Female | 68 (73.1%) |
| Male | 25 (26.9%) |
| Localization | |
| Central skull base | 57 (61.3%) |
| Lateral skull base | 36 (38.7%) |
| Primary tumors | 82 (88.2%) |
| Primary tumors with recurrence | 7 (8.5%) |
| Recurrent tumors$ | 11 (11.8%) |
| Simpson grade# | |
| 1 | 45 (48.4%) |
| 2 | 17 (18.3%) |
| 3 | 11 (11.3%) |
| 4 | 13 (14.0%) |
| 5 | 7 (7.5%) |
| Postoperative irradiation | 24 (25.8%) |
| Tumor edema (Steinhoff classification)*§ | |
| 0 | 57 (63.3%) |
| 1 | 12 (13.3%) |
| 2 | 16 (17.8%) |
| 3 | 5 (5.6%) |
| Age, y, mean ± SD/median/range | 54.3 ± 12.2/54.0/24–85 |
| Sex | |
| Female | 68 (73.1%) |
| Male | 25 (26.9%) |
| Localization | |
| Central skull base | 57 (61.3%) |
| Lateral skull base | 36 (38.7%) |
| Primary tumors | 82 (88.2%) |
| Primary tumors with recurrence | 7 (8.5%) |
| Recurrent tumors$ | 11 (11.8%) |
| Simpson grade# | |
| 1 | 45 (48.4%) |
| 2 | 17 (18.3%) |
| 3 | 11 (11.3%) |
| 4 | 13 (14.0%) |
| 5 | 7 (7.5%) |
| Postoperative irradiation | 24 (25.8%) |
| Tumor edema (Steinhoff classification)*§ | |
| 0 | 57 (63.3%) |
| 1 | 12 (13.3%) |
| 2 | 16 (17.8%) |
| 3 | 5 (5.6%) |
*Data available for 90 patients. #Simpson data were taken from the intraoperative surgery records. §Steinhoff scores were derived from preoperative MRIs. $Recurrence was defined as MRI visible tumor or volume increase according to MRI in case of subtotal resections. Median follow-up time was 40.0 months (range: 6–103 mo).
Demographic characteristics of 93 patients with skull base meningiomas
| Age, y, mean ± SD/median/range | 54.3 ± 12.2/54.0/24–85 |
| Sex | |
| Female | 68 (73.1%) |
| Male | 25 (26.9%) |
| Localization | |
| Central skull base | 57 (61.3%) |
| Lateral skull base | 36 (38.7%) |
| Primary tumors | 82 (88.2%) |
| Primary tumors with recurrence | 7 (8.5%) |
| Recurrent tumors$ | 11 (11.8%) |
| Simpson grade# | |
| 1 | 45 (48.4%) |
| 2 | 17 (18.3%) |
| 3 | 11 (11.3%) |
| 4 | 13 (14.0%) |
| 5 | 7 (7.5%) |
| Postoperative irradiation | 24 (25.8%) |
| Tumor edema (Steinhoff classification)*§ | |
| 0 | 57 (63.3%) |
| 1 | 12 (13.3%) |
| 2 | 16 (17.8%) |
| 3 | 5 (5.6%) |
| Age, y, mean ± SD/median/range | 54.3 ± 12.2/54.0/24–85 |
| Sex | |
| Female | 68 (73.1%) |
| Male | 25 (26.9%) |
| Localization | |
| Central skull base | 57 (61.3%) |
| Lateral skull base | 36 (38.7%) |
| Primary tumors | 82 (88.2%) |
| Primary tumors with recurrence | 7 (8.5%) |
| Recurrent tumors$ | 11 (11.8%) |
| Simpson grade# | |
| 1 | 45 (48.4%) |
| 2 | 17 (18.3%) |
| 3 | 11 (11.3%) |
| 4 | 13 (14.0%) |
| 5 | 7 (7.5%) |
| Postoperative irradiation | 24 (25.8%) |
| Tumor edema (Steinhoff classification)*§ | |
| 0 | 57 (63.3%) |
| 1 | 12 (13.3%) |
| 2 | 16 (17.8%) |
| 3 | 5 (5.6%) |
*Data available for 90 patients. #Simpson data were taken from the intraoperative surgery records. §Steinhoff scores were derived from preoperative MRIs. $Recurrence was defined as MRI visible tumor or volume increase according to MRI in case of subtotal resections. Median follow-up time was 40.0 months (range: 6–103 mo).
Histological subtypes and mutation frequencies in skull base meningiomas
| Histological Subtype | AKT1E17K(n [%]) | KLF4K409Q(n [%]) | TRAF7 (n [%]) |
|---|---|---|---|
| Meningothelial (n = 54 [58.1%]) | 18 [33.3] | 4 [7.4] | 1 [1.8] |
| Fibroblastic (n = 9 [9.7%]) | 2 [22.2] | 0 | 0 |
| Transitional (n = 11 [11.8%]) | 3 [27.3] | 2 [18.2] | 0 |
| Secretory (n = 3 [3.2%]) | 0 | 3 [100] | 1 [33.3] |
| Psammomatous (n = 5 [5.4%]) | 3 [60] | 2 [40] | 0 |
| Atypical grade II (n = 11 [11.8%]) | 3 [27.3] | 0 | 0 |
| Histological Subtype | AKT1E17K(n [%]) | KLF4K409Q(n [%]) | TRAF7 (n [%]) |
|---|---|---|---|
| Meningothelial (n = 54 [58.1%]) | 18 [33.3] | 4 [7.4] | 1 [1.8] |
| Fibroblastic (n = 9 [9.7%]) | 2 [22.2] | 0 | 0 |
| Transitional (n = 11 [11.8%]) | 3 [27.3] | 2 [18.2] | 0 |
| Secretory (n = 3 [3.2%]) | 0 | 3 [100] | 1 [33.3] |
| Psammomatous (n = 5 [5.4%]) | 3 [60] | 2 [40] | 0 |
| Atypical grade II (n = 11 [11.8%]) | 3 [27.3] | 0 | 0 |
Histological subtypes and mutation frequencies in skull base meningiomas
| Histological Subtype | AKT1E17K(n [%]) | KLF4K409Q(n [%]) | TRAF7 (n [%]) |
|---|---|---|---|
| Meningothelial (n = 54 [58.1%]) | 18 [33.3] | 4 [7.4] | 1 [1.8] |
| Fibroblastic (n = 9 [9.7%]) | 2 [22.2] | 0 | 0 |
| Transitional (n = 11 [11.8%]) | 3 [27.3] | 2 [18.2] | 0 |
| Secretory (n = 3 [3.2%]) | 0 | 3 [100] | 1 [33.3] |
| Psammomatous (n = 5 [5.4%]) | 3 [60] | 2 [40] | 0 |
| Atypical grade II (n = 11 [11.8%]) | 3 [27.3] | 0 | 0 |
| Histological Subtype | AKT1E17K(n [%]) | KLF4K409Q(n [%]) | TRAF7 (n [%]) |
|---|---|---|---|
| Meningothelial (n = 54 [58.1%]) | 18 [33.3] | 4 [7.4] | 1 [1.8] |
| Fibroblastic (n = 9 [9.7%]) | 2 [22.2] | 0 | 0 |
| Transitional (n = 11 [11.8%]) | 3 [27.3] | 2 [18.2] | 0 |
| Secretory (n = 3 [3.2%]) | 0 | 3 [100] | 1 [33.3] |
| Psammomatous (n = 5 [5.4%]) | 3 [60] | 2 [40] | 0 |
| Atypical grade II (n = 11 [11.8%]) | 3 [27.3] | 0 | 0 |
AKT1E17K Mutation
AKT1E17K mutation was detected in 29 of the 93 skull base meningiomas (31.2%). The mutation was preferentially found in tumors of meningothelial and psammomatous subtype, as well as in atypical meningiomas (Table 2). Figure 1A shows an example for the AKT1E17K mutation. As shown in Fig. 1B, the presence of the AKT1E17K mutation showed a trend to be associated with shorter time to tumor recurrence, but this could not be statistically proven (P = .094 [log-rank test]). To assess whether late-occurring recurrences might affect progression-free survival significantly, a Fleming–Harrington test for late events (with parameters P = 0 and q = 1) was performed, but did not show a significant effect (P = .223). However, in a multivariate analysis (Table 3) using a Cox regression model including AKT1 mutational status, WHO grade, Simpson score, and tumor edema, AKT1E17K mutation and WHO grade were significantly associated with recurrence-free survival (P = .047 and P = .007, respectively), while extent of resection (Simpson score) and edema were not (P = .415 and P = .861, respectively). Due to the small number of events, this analysis has to be understood as exploratory. For the same reason, the levels of the 2 variables—Simpson score and tumor edema—have been considered as linear factors so that the reported hazard rates correspond to an increase of the score by one level. Nevertheless, one can see that the concordance index of the multivariate model is distinctly larger than from any of the corresponding univariate analyses, which have been repeated here in the framework of the Cox model for better comparison.
Results of Cox regression models with factors AKT1E17K, WHO grade, Simpson score, and Steinhoff classification
| P | Hazard ratio | 95% CI | Concordance index | |
|---|---|---|---|---|
| Multivariate model | 0.786 | |||
| AKT1E17K | .047 | 3.936 | (1.020–15.186) | |
| WHO grade | .007 | 7.488 | (1.727–32.471) | |
| Simpson score | .415 | 1.216 | (0.760–1.946) | |
| Steinhoff classification | .861 | 0.947 | (0.517–1.736) | |
| Univariate model | ||||
| AKT1E17K | .107 | 2.549 | (0.817–7.957) | 0.609 |
| WHO grade | .048 | 3.930 | (1.010–15.295) | 0.644 |
| Simpson score | .947 | 1.014 | (0.679–1.512) | 0.530 |
| Steinhoff classification | .847 | 0.944 | (0.525–1.969) | 0.623 |
| P | Hazard ratio | 95% CI | Concordance index | |
|---|---|---|---|---|
| Multivariate model | 0.786 | |||
| AKT1E17K | .047 | 3.936 | (1.020–15.186) | |
| WHO grade | .007 | 7.488 | (1.727–32.471) | |
| Simpson score | .415 | 1.216 | (0.760–1.946) | |
| Steinhoff classification | .861 | 0.947 | (0.517–1.736) | |
| Univariate model | ||||
| AKT1E17K | .107 | 2.549 | (0.817–7.957) | 0.609 |
| WHO grade | .048 | 3.930 | (1.010–15.295) | 0.644 |
| Simpson score | .947 | 1.014 | (0.679–1.512) | 0.530 |
| Steinhoff classification | .847 | 0.944 | (0.525–1.969) | 0.623 |
The multivariate analysis (upper part) is completed by the univariate analyses with the single factors. Simpson grade and Steinhoff classification are considered here as linear factors in the hazard model. The concordance index represents the degree of concordance between expected time to event giving the covariables and observed time (1 = perfect concordance, 0.5 = only random degree of concordance).
Results of Cox regression models with factors AKT1E17K, WHO grade, Simpson score, and Steinhoff classification
| P | Hazard ratio | 95% CI | Concordance index | |
|---|---|---|---|---|
| Multivariate model | 0.786 | |||
| AKT1E17K | .047 | 3.936 | (1.020–15.186) | |
| WHO grade | .007 | 7.488 | (1.727–32.471) | |
| Simpson score | .415 | 1.216 | (0.760–1.946) | |
| Steinhoff classification | .861 | 0.947 | (0.517–1.736) | |
| Univariate model | ||||
| AKT1E17K | .107 | 2.549 | (0.817–7.957) | 0.609 |
| WHO grade | .048 | 3.930 | (1.010–15.295) | 0.644 |
| Simpson score | .947 | 1.014 | (0.679–1.512) | 0.530 |
| Steinhoff classification | .847 | 0.944 | (0.525–1.969) | 0.623 |
| P | Hazard ratio | 95% CI | Concordance index | |
|---|---|---|---|---|
| Multivariate model | 0.786 | |||
| AKT1E17K | .047 | 3.936 | (1.020–15.186) | |
| WHO grade | .007 | 7.488 | (1.727–32.471) | |
| Simpson score | .415 | 1.216 | (0.760–1.946) | |
| Steinhoff classification | .861 | 0.947 | (0.517–1.736) | |
| Univariate model | ||||
| AKT1E17K | .107 | 2.549 | (0.817–7.957) | 0.609 |
| WHO grade | .048 | 3.930 | (1.010–15.295) | 0.644 |
| Simpson score | .947 | 1.014 | (0.679–1.512) | 0.530 |
| Steinhoff classification | .847 | 0.944 | (0.525–1.969) | 0.623 |
The multivariate analysis (upper part) is completed by the univariate analyses with the single factors. Simpson grade and Steinhoff classification are considered here as linear factors in the hazard model. The concordance index represents the degree of concordance between expected time to event giving the covariables and observed time (1 = perfect concordance, 0.5 = only random degree of concordance).
AKT1E17K mutated tumors were preferentially located in the anterior/middle skull base. The AKT1E17K mutation was associated with a significantly higher rate of phospho-mTOR+ tumors compared with AKT1wt tumors (chi-square test: P = .008) (Table 4, Fig. 1C). The AKT1E17K mutation was additionally associated with positive phospho-p70S6K immunoexpression (chi-square test: P = .029) compared with AKT1wt tumors. However, no significant relation between AKT1E17K and expression of phospho-4EBP1 was found. As expected for the nuclear protein Krüppel-like factor 4 (KLF4), we observed no association between KLF4 mutation and phospho-mTOR/phospho-p70S6K expression, suggesting a specific activation of the mTOR signaling pathway solely by AKT1E17K (ie, by activation of a kinase known to be involved in the mTOR pathway). We next wondered whether the association between mTOR pathway activation and AKT1E17K status affects the time to tumor recurrence. As shown in Fig. 1D, patients with tumors harboring the AKT1E17K mutation and immunopositivity for phospho-mTOR (upper figure) or phospho-4EBP1 (lower figure) had the shortest time to tumor recurrence, with statistically significant differences between AKT1E17K phospho-4EBP1 positive tumors and AKT1wt phospho-4EBP1 negative tumors (P = .046) but also between AKT1E17K phospho-4EBP1 positive tumors and AKT1E17K phospho-4EBP1 negative tumors (P = .036). There was a nonsignificant difference (P = .095) between AKT1E17K phospho-p70S6K positive tumors and AKT1wt phospho-p70S6K positive tumors (data not shown).
Immunoexpression in relation to AKT1 mutation status
| p-Akt (N = 72)# | p-p70S6K (N = 77)# | p-mTOR (N = 75)# | p-4EBP1 (N = 73)# | p-ERK1/2 (N = 73)# | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | |
| AKT1wt | 10 | 14 | 24 | 22 | 15 | 15 | 27 | 18 | 5 | 29 | 11 | 8 | 22 | 9 | 17 |
| AKT1E17K | 4 | 5 | 15 | 5 | 15 | 5 | 8 | 7 | 10 | 15 | 7 | 3 | 7 | 3 | 15 |
| p-Akt (N = 72)# | p-p70S6K (N = 77)# | p-mTOR (N = 75)# | p-4EBP1 (N = 73)# | p-ERK1/2 (N = 73)# | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | |
| AKT1wt | 10 | 14 | 24 | 22 | 15 | 15 | 27 | 18 | 5 | 29 | 11 | 8 | 22 | 9 | 17 |
| AKT1E17K | 4 | 5 | 15 | 5 | 15 | 5 | 8 | 7 | 10 | 15 | 7 | 3 | 7 | 3 | 15 |
0 = no immunoexpression, 1 = weak-moderate immunoexpression, 3 = strong immunoexpression.
Number of patients in each group is given. #Total number of cases available for evaluation for the given immunostaining.
Immunoexpression in relation to AKT1 mutation status
| p-Akt (N = 72)# | p-p70S6K (N = 77)# | p-mTOR (N = 75)# | p-4EBP1 (N = 73)# | p-ERK1/2 (N = 73)# | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | |
| AKT1wt | 10 | 14 | 24 | 22 | 15 | 15 | 27 | 18 | 5 | 29 | 11 | 8 | 22 | 9 | 17 |
| AKT1E17K | 4 | 5 | 15 | 5 | 15 | 5 | 8 | 7 | 10 | 15 | 7 | 3 | 7 | 3 | 15 |
| p-Akt (N = 72)# | p-p70S6K (N = 77)# | p-mTOR (N = 75)# | p-4EBP1 (N = 73)# | p-ERK1/2 (N = 73)# | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | |
| AKT1wt | 10 | 14 | 24 | 22 | 15 | 15 | 27 | 18 | 5 | 29 | 11 | 8 | 22 | 9 | 17 |
| AKT1E17K | 4 | 5 | 15 | 5 | 15 | 5 | 8 | 7 | 10 | 15 | 7 | 3 | 7 | 3 | 15 |
0 = no immunoexpression, 1 = weak-moderate immunoexpression, 3 = strong immunoexpression.
Number of patients in each group is given. #Total number of cases available for evaluation for the given immunostaining.
![AKT1E17K mutation and mTOR signaling in skull base meningiomas. (A) Example for the detection of the AKT1E17K mutation by Sanger sequencing. (B) Kaplan–Meier plot of recurrence-free survival for patients with skull base meningiomas harboring AKT1E17K (N = 29, 6 events) or AKT1wt (N = 64, 6 events; P = .094 [log rank test]) (see also results of Cox regression analysis in text). (C) Examples for immunoexpression of phospho-mTOR, phospho-p70S6K, and phospho-4EBP1 in relation to the AKT1 mutation status (AKT1wt [left panel] or AKT1E17K [right panel]). Scoring of immunostaining is given below [each scale bar = 100 µm]. (D) Kaplan–Meier plots with separation for immunoexpression of phospho-mTOR (upper figure) or phospho-4EBP1 (lower figure). Case numbers and events are given in Supplementary Table S2.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/neuro-oncology/19/8/10.1093_neuonc_nox018/2/m_nox01801.jpeg?Expires=1750227245&Signature=PqaQZg75tSW6v14JGYDtai7Q6tMB95O9b-vOFruakOe4nqzxZgwKTUHmMUf0RiUwr2xg2W~govQ72BN4PZ5uIG~rXZnVFxD1I0N0CYaUe1Wh3aWLvzxEOEcZKpkBOyZq6nF-q8OOAC1R0AsHBUCFII~ggKSOVScqxeSjwJiLxVwPMetVamw8daJNVnpHJGXPzppDJr2mPmfKw9Xe98sU1-iAsT9Jvm2kAX~rUjJU24P0CeoDn0ROdq6B0NFHi-gjgAU-Y7b-G7M1yS4ItDPrxBCcHM4mEC1bSuAKBiuo2nasVfzJowJkKypPRc8WZdJK6-afuw0~LuRyNXwfyLye1g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AKT1E17K mutation and mTOR signaling in skull base meningiomas. (A) Example for the detection of the AKT1E17K mutation by Sanger sequencing. (B) Kaplan–Meier plot of recurrence-free survival for patients with skull base meningiomas harboring AKT1E17K (N = 29, 6 events) or AKT1wt (N = 64, 6 events; P = .094 [log rank test]) (see also results of Cox regression analysis in text). (C) Examples for immunoexpression of phospho-mTOR, phospho-p70S6K, and phospho-4EBP1 in relation to the AKT1 mutation status (AKT1wt [left panel] or AKT1E17K [right panel]). Scoring of immunostaining is given below [each scale bar = 100 µm]. (D) Kaplan–Meier plots with separation for immunoexpression of phospho-mTOR (upper figure) or phospho-4EBP1 (lower figure). Case numbers and events are given in Supplementary Table S2.
We also found a significant effect of the AKT1 mutation on tumor recurrence in relation to the immunoexpression of phospho-ERK1/2 (Fig. 2A). Significant differences in recurrence-free survival occurred between AKT1wt tumors with and without phospho-ERK1/2 expression (P = .036), AKT1wt phospho-ERK1/2 positive and AKT1E17K phospho-ERK1/2 negative tumors (P = .046) and between AKT1wt phospho-ERK1/2 positive tumors and tumors with AKT1E17K mutation and immunopositivity for phospho-ERK1/2 (P = .018; Fig. 2B). No significant association was found between the presence of AKT1E17K mutation and extent of resection, presence and size of peritumoral edema, and histological subtype.
![AKT1E17K mutation and ERK1/2 signaling in skull base meningiomas. (A) Examples for immunoexpression of phospho-ERK1/2 in relation to the AKT1 mutation status (AKT1wt [left panel, scale bar = 100 µm] or AKT1E17K [right panel, scale bar = 50 µm]). Scoring of immunostaining is given below. (B) Kaplan–Meier plots showing differences between phospho-ERK1/2 positive versus negative tumors in relation to the AKT1 mutation status (AKT1wt or AKT1E17K). Case numbers and events are given in Supplementary Table S1.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/neuro-oncology/19/8/10.1093_neuonc_nox018/2/m_nox01802.jpeg?Expires=1750227245&Signature=s0ZS71qvtjIxw~KMe8Z~2u43iL3sAr9X8MUDTT8saU8R~fb12Li7S0IOfawr7oS98i68IYxgXAmloPuEyZbZyQlVxFMkEa7KEzjykUvF-kOA4V6F8jPQVtnZAVXvFg90USjwd~EvdS1E7fjeThpj439POU0d0d86bqKNLTMVXwp04jGdclQigz2QoiM88TP7TMEo7DdHflq4ucWyKOl4L9aUlt~XlRRP1eJ3HBhiVeB6y-joyYoVa2xrOQqxKZ28nohx6IMIXRVbemDII3Zv5IPrFHqDhwr~7KOLjAw5lOh8ZRr46TIMVbfK1Ru~JgGpeKGsfu9W0iWezI4DvF1s7A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AKT1E17K mutation and ERK1/2 signaling in skull base meningiomas. (A) Examples for immunoexpression of phospho-ERK1/2 in relation to the AKT1 mutation status (AKT1wt [left panel, scale bar = 100 µm] or AKT1E17K [right panel, scale bar = 50 µm]). Scoring of immunostaining is given below. (B) Kaplan–Meier plots showing differences between phospho-ERK1/2 positive versus negative tumors in relation to the AKT1 mutation status (AKT1wt or AKT1E17K). Case numbers and events are given in Supplementary Table S1.
KLF4 Mutation
The known hot spot mutation K409Q in the KLF4 gene was detected in 11 patients (11.8%; Fig. 3A). The distribution of the mutations favored the median skull base region. Among the 11 KLF4-mutated tumors, 2 (18.2%) had simultaneously the known mutation N520S in the TRAF7 gene (combined KLF4/TRAF7 mutation). KLF4 mutations were significantly associated with secretory and transitional histological subtype (P < .001). Interestingly, none of the 11 KLF4-mutated patients with available follow-up showed tumor recurrence, although the difference to the limited recurrence-free survival of patients without mutation was not statistically significant (P = .280 [log rank test]; Fig. 3B). The 2 patients with known NF2 had no other genetic alteration tested.
![KLF4 mutation in skull base meningioma. (A) Example for detection of the KLF4 mutation. (B) Kaplan–Meier plots showing differences in terms of tumor recurrence depending on the KLF4 mutation (KLF4wt: N = 82, 12 events; KLF4K409Q: N = 11, 0 events [log-rank test]).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/neuro-oncology/19/8/10.1093_neuonc_nox018/2/m_nox01803.jpeg?Expires=1750227245&Signature=RkOLKxRJ3ZMf7cG-6nKpmL6s8ID8dBGgsxhxqSeXzBWDcRPZ4bcSvviQXef4~9ji~7mrpuDw4RWvYStJLAuluv4VqGXH2QEdkxusuTpfBI5waCdKiV6IQyoYIyJVacI0MbEcTGsXYnlgo8MMw96DWyXT8g-DfeE32qrzCkvYYMdWb7mxZbJXcgHh3DhnZLyp1yyIeTwGaa8FZXeorHxjV0hDXb~U2pwXyfwma2SFs1QO12kDSyW0~o5UatToIQWFtzZwiOZWgYt7PFQK8MUR-1YJCjrAvnyDi-HWxEeEQ-6WVcKFs3~NzGXqPtzv6XSf0dYjRV4YVxUS5wO2uxMwMg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
KLF4 mutation in skull base meningioma. (A) Example for detection of the KLF4 mutation. (B) Kaplan–Meier plots showing differences in terms of tumor recurrence depending on the KLF4 mutation (KLF4wt: N = 82, 12 events; KLF4K409Q: N = 11, 0 events [log-rank test]).
The intracranial distribution of the mutations in both AKT1 and KLF4/TRAF7 is depicted in Fig. 4, while Supplementary Fig. S1 shows the affected domains of the proteins. One WHO grade I meningioma showed a PI3K mutation (not shown). No mutation in SMO was detected in all 93 cases. No TERT promoter mutation was present among 85 cases with suitable tumor DNA available.
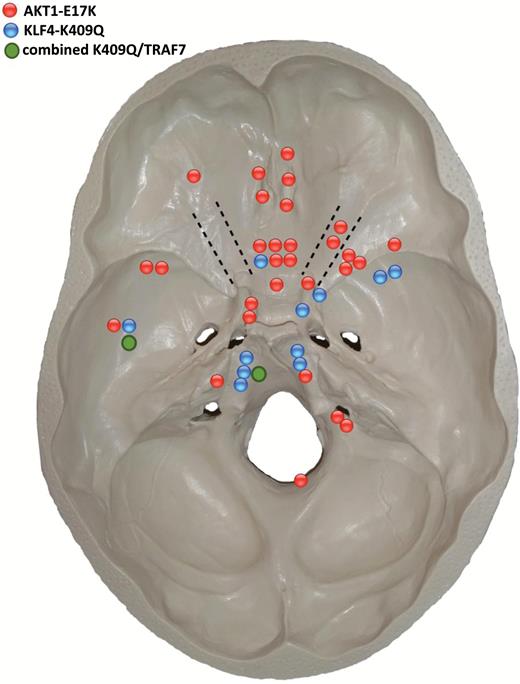
Distribution of AKT1E17K (red dots), KLF4 (blue dots), and combined KLF4/TRAF7 mutations (green dots) at the skull base.
Analysis of Druggable Signaling Pathways in Skull Base Meningiomas
For clinical practice it might be difficult to assess the individual mutation status, while immunohistochemistry could be applied more easily. Therefore, we analyzed this skull base meningioma series for altered phospho-protein expression, irrespective of the mutational status for AKT1 or KLF4/TRAF7. We observed that phospho-Akt was positive in 58/72 (80.6%), phospho-mTOR in 40/75 (53.3%), phospho-p70S6K in 50/77 (65.0%), phospho-4EBP1 in 29/73 (39.8%), and phospho-ERK1/2 in 44/73 (60.2%) tumors. There was no significant association between the histological subtype and expression of any of the signaling proteins. When we analyzed the immunoexpression in relation to tumor grading, comparing the WHO grade I meningiomas with atypical WHO grade II meningiomas, we found that atypical meningiomas had a significantly higher rate of tumors with strong immunoexpression of phospho-4EBP1 (WHO grade I: 7/62, WHO grade II: 4/11, P = .029). It should be noted that none of the proteins showed any significant effect on time to tumor recurrence if analyzed without consideration of the mutation status.
Discussion
We have analyzed a large series of skull base meningiomas, mostly WHO grade I tumors, for the role of mutations in AKT1, KLF4/TRAF7, SMO, and the TERT promoter genes in relation to activated signaling pathways which can potentially serve as treatment targets. The major findings are a high frequency of the AKT1E17K mutations and the association of this mutation with shorter time to tumor recurrence, as well as an activation of signaling proteins of the mTOR and ERK1/2 pathway.
Our finding of about 30% AKT1E17K mutated tumors is much higher than previously reported. The AKT1E17K mutation activates the PI3K/Akt signaling pathway18 (Fig. 5). This somatic mutation occurs in breast, ovarian, and colorectal cancers.18,19 In meningioma, this mutation was found nearly exclusively in 7%–12% of WHO grade I meningiomas but is rare in grade II meningiomas and absent in grade III meningiomas.7,20,21 Moreover, the AKT1E17K mutation was predominantly found in the meningothelial or transitional subtype of grade I tumors, and meningiomas harboring the AKT1E17K mutation were of NF2 wild type. We also observed a clear predominance of the meningothelial subtype in our series. However, the lower frequency of the AKT1E17K mutation in previous reports can be explained by the analysis of meningiomas from different locations. Skull base–located meningiomas can cause substantial therapeutic problems due to the anatomical location and the involvement of neighboring neurovascular structures. This might in turn result in incomplete resection and tumor recurrence. So far, the therapeutic options (ie, surgery and irradiation) are limited.22,23 Recently, sunitinib has been shown to have modest activity in aggressive meningiomas.24 We had previously shown in mouse models that inhibition of the mTOR signaling pathway by everolimus or temsirolimus reduces meningioma growth.14 It was recently reported that the combination of bevacizumab and everolimus might represent an additional treatment option.25 Here we show that indeed skull base meningiomas characterized by the AKT1E17K mutation show evidence for an activation of the mTOR signaling pathway. Rare mTOR mutations, together with activation of mTOR-associated proteins, were already reported in the series from Brastianos et al.9 Based on the present data, it could be suggested for testing skull base meningiomas as a precondition for the administration of mTOR inhibitors as a secondary treatment option.
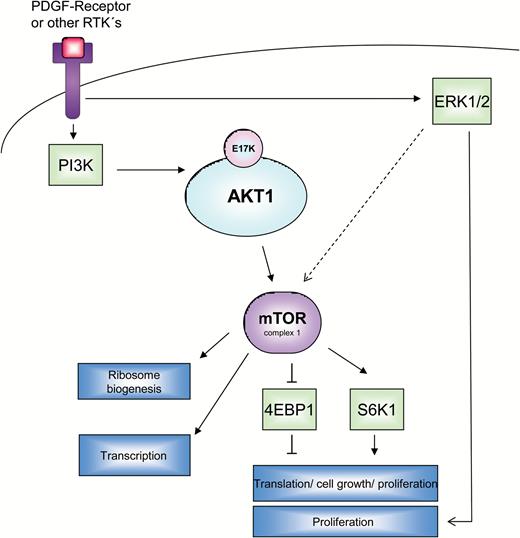
The network of kinase reactions (phosphorylations), which transmit pro-proliferative signals from receptor tyrosine kinases (RTKs) along the PI3K-Akt-mTOR or ERK1/2 pathways is drafted in this scheme. A very important RTK in meningiomas is platelet-derived growth factor receptor (PDGFR), especially the predominant form, PDGFR-ß. Proliferation of meningioma cells is stimulated by PDGF-mediated activation of the receptor and subsequent kinase reactions of PI3K and AKT1. The latter partially exerts its effects via a downstream pathway involving mTOR complex 1 (mTORC1) and its direct targets p70S6K and 4EBP1. Activation of AKT1, which may occur either by upstream kinases or eventually by the mutation E17K, leads to an activation of the downstream mTOR pathway, detectable as an enhanced expression of the phosphorylated forms of mTOR and p70S6K, while the activation is reflected in a lower phosphorylation of 4EBP1. An alternative route of mitogenic signal transduction via ERK1/2 is possible and is reflected by a higher abundance of phosphorylated ERK1/2. This pathway can potentially be crosslinked with the first one (activation of mTORC1 by ERK1/2).
Another finding of our study was related to the KLF4 mutation. This mutation was highly associated with secretory subtype of grade I meningiomas as previously reported.26 However, we could show that patients with KLF4-mutated tumors never showed tumor recurrence within the available follow-up time, which indicates a favorable biological role of this mutation.
Surprisingly, we did not detect any SMO mutation in our series. SMO mutations occur in 4%–5% of grade I meningiomas and are restricted to the medial anterior skull base near the midline.7,9 Presumably this finding is due to our rather small cohort of 93 tumors derived from this location. This might be at least partly explained by the restriction of SMO mutations to the olfactory groove region,7 while our cohort comprised several skull base locations.
During preparation of the manuscript an interesting paper described oncogenic PI3K mutations in meningiomas.21 In this series, PI3CA mutations were detected in 7% of non–NF2-mutant meningiomas and were associated with the skull base. We detected only one PI3K gene mutation in our series, suggesting that in skull base meningiomas PI3K mutations are rare events.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
The study was supported by a grant (2014) from the Niedersächsische Krebsgesellschaft (to M.N. and C.M.). The project was further supported by grants from the German Research Foundation (DFG) (grants #MA2530/6-1 and MA2530/8-1), Wilhelm Sander-Stiftung (grant #2014.092.1), and the Deutsche Krebshilfe (grant #111853) (all to C.M.).
Conflict of interest statement. No conflicts of interest to be stated (on behalf of all authors).
Acknowledgments
We highly appreciate the skillful technical assistance of Ines Schellhase and Gaby Schulze. Sandra Hartmann (all from Magdeburg) helped with the TMA construction.
References
Author notes
Corresponding Author: Christian Mawrin, MD, Department of Neuropathology, Otto-von-Guericke University Magdeburg, Leipziger Strasse 44, D-39120 Magdeburg, Germany ([email protected]).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}