




Abstract
The DNA damage response (DDR) entails reorganization of proteins and protein complexes involved in DNA repair. The coordinated regulation of these proteomic changes maintains genome stability. Traditionally, regulators and mediators of DDR have been investigated individually. However, recent advances in mass spectrometry (MS)-based proteomics enable us to globally quantify changes in protein abundance, post-translational modifications (PTMs), protein localization, and protein-protein interactions (PPIs) in cells. Furthermore, structural proteomics approaches, such as crosslinking MS (XL-MS), hydrogen/deuterium exchange MS (H/DX-MS), Native MS (nMS), provide large structural information of proteins and protein complexes, complementary to the data collected from conventional methods, and promote integrated structural modeling. In this review, we will overview the current cutting-edge functional and structural proteomics techniques that are being actively utilized and developed to help interrogate proteomic changes that regulate the DDR.
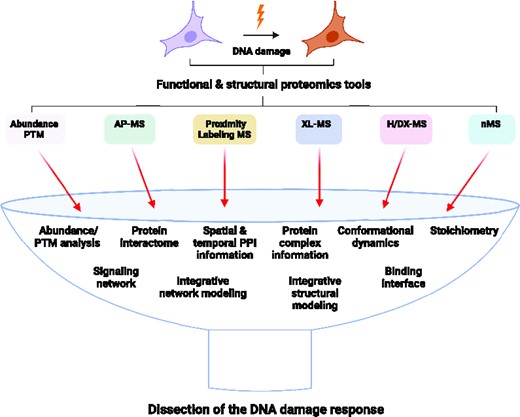
INTRODUCTION
Liquid chromatography followed by tandem mass spectrometry (LC–MS/MS) is being widely used to quantitatively measure both protein levels and post translational modifications. LC–MS/MS approaches can be performed in two different ways: ‘top-down’ approaches profile intact proteins and protein complexes, whereas ‘bottom-up’ or shotgun approaches profile proteins that have been digested to peptides. Peptides are then separated over a column (e.g. reversed phase C18) using increasing concentrations of organic solvent (e.g. acetonitrile), and as peptides elute from the column, they go through electrospray ionization (ESI) and the intact mass to charge ratio (m/z) of precursor or MS1 ions are measured. Precursor MS1 ions are then subjected to MS/MS or MS2 analysis, involving fragmentation of the peptide ion and subsequent measurement of fragment or product ion m/z (1,2). Through this tandem MS process, peptides in the samples are identified by comparing the collected data to a species-specific proteome database of in silico-digested proteins and scoring the resulting matches (3,4).
LC–MS/MS allows simultaneous identification and quantification of thousands of proteins or PTM sites on peptides in a single run. Abundance of each peptide across samples can be quantified and compared in several different ways: sample comparison can be assessed by using stable isotope labeling of amino acids in cells (SILAC) prior to tryptic digestion (5). Similarly, samples can be labeled post-digestion using isobaric tags (e.g. iTRAQ, TMT) (6–8) and multiple samples can be combined (up to 18 for TMT and 8 for iTRAQ) (9–11) for multiplexed quantification. Each tag possesses identical chemical structures and is differentiated via isotopes at different positions that generate peptides with the same m/z in MS1. However, upon MS2 fragmentation the tags produce different mass reporter ions, allowing relative quantification of peptide abundance. Alternatively, the relative abundance of each peptide in two or more biological samples can be directly measured without utilizing any labeling or special growth conditions (label-free).
Regarding MS data acquisition, there are two different acquisition modes for collecting MS data. In Data-Dependent Acquisition (DDA) mode, MS instrument selects the most abundant (typically top 10–20) peptide ions from MS1 scan in each column retention time window, and then these selected precursor peptide ions are fragmented and analyzed in MS2. However, the stochastic precursor selection of DDA could result in inconsistent detection and undersampling of less abundant peptides, which cause higher variation across replicates. Alternatively, in Data-Independent Acquisition (DIA) mode, MS instrument gathers MS2 data from all precursor ions in a narrow mass window (e.g. 400–425 m/z), and then repeats this process across the entire mass range (e.g. 400–1200 m/z) to systematically collect MS2 from all detected peptide ions in each retention time window (e.g. 2–4 s). MS instrument continues collecting MS2 data across the entire retention time range, thus generating highly multiplexed and comprehensive maps of fragment ions that elute on the chromatography. DIA uses a spectral library to extract peptide information from MS2 data, which leads to more accurate peptide quantification, lower variation across replicates, and fewer missing peptide identification, compared to DDA. But DDA is preferred for targeted analysis and library generation as it offers more accurate identification than DIA due to longer acquisition time per data point and more robust database-based search methods (12–14).
Interrogating the structure-function correlation of proteins and protein complexes has been crucial for understanding several cellular processes and disease mechanisms and developing various therapies. It is established that protein conformations are not just single specific three-dimensional quaternary structures but have multiple transient conformations with dynamic properties (15). Protein dynamics facilitate its functions, such as macromolecular associations and other downstream processes in cellular pathways (16). Conventional structural biology tools such as X-ray crystallography (XRC), cryo-electron microscopy (cryoEM), and nuclear magnetic resonance (NMR) solve structures of important protein complexes but need to consistently deal with dynamics and heterogenous assemblies and require large quantities of protein, which limits their application. Protein chemistry combined with various mass spectrometry-based structural proteomics approaches, including XL-MS, H/DX-MS, and nMS, provide large structural information of proteins and protein complexes, in part due to their wide applicability to many different types of proteins, including structured, disordered, monomeric, and multimeric proteins (17). Furthermore, structural proteomics methods enable studying transient interactions of proteins and their complexes up to the megadalton range in vitro and in vivo under physiological conditions (18–22). Although structural proteomics tools alone do not solve structures, they provide invaluable information complementary to conventional methods, promoting integrated structural modeling (23). This review covers a repertoire of various proteomics approaches that can further our understanding of dynamic changes in proteome and mechanistic basis of PPIs that regulate DNA repair (Figure 1; Table 1).
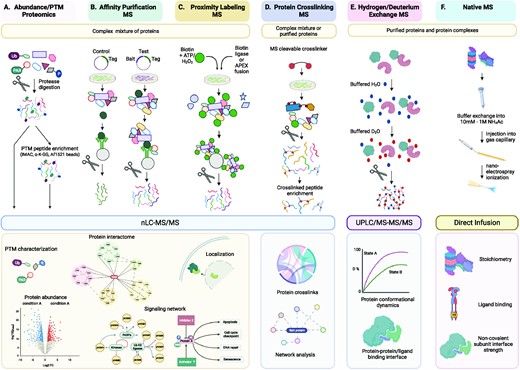
Schematic representation of functional and structural proteomics methods. (A) Proteins in cell lysates are digested and peptides are subsequently ionized and fragmented through nLC–MS/MS either directly (abundance proteomics) or after enrichment of PTM peptides (PTM proteomics). (B) Affinity Purification-MS (AP-MS) is to systematically identify PPIs by expressing and purifying affinity-tagged ‘bait’ proteins in cells and detecting stably bound protein interactors by MS. (C) Proximity labeling MS relies on a promiscuous biotin protein ligase (BirA/BioID/TurboID) or an engineered ascorbic acid peroxidase (APEX) fused to a protein of interest (bait) to label proteins in close proximity to the bait protein through covalent transfer of biotin or biotin derivatives in a distance-dependent manner. Upon addition of biotin, the proximal proteins are biotinylated, enriched with streptavidin beads, and identified by quantitative MS. These MS datasets from functional proteomics are integrated by network modeling and analysis to extract biological insight. (D) Protein crosslinking MS (XL-MS) involves crosslinking or surface modification of protein complexes followed by proteolysis and MS identifications of amino acid residue pairs that are within close spatial proximity, thus providing structural insights into proteins and protein assemblies. (E) Hydrogen/Deuterium exchange MS (H/DX-MS) measures dynamic changes in protein structures over a specified time scale by measuring the relative uptake of protein backbone amide hydrogen for deuterium (and vice versa). Residue-level information can be derived from gas-phase fragmentation of the deuterated peptides. (F) Native MS (nMS) involves analyzing and characterizing protein complexes, while retaining their inter and intramolecular interactions. The stoichiometry of a protein complex can be deduced from the calculated mass, and structural preference can be informed by the number of charges it carries. Data from the diverse structural proteomics methodologies can be combined to obtain maximum structural insight through integrative modeling. nLC, nano liquid chromatography; UPLC, ultra-performance liquid chromatography. Edges and arrows connecting proteins to other proteins and/or biological processes in the interactomes and networks are not only based on MS-driven proteomics data but also inferred from network modeling and analysis.
Exemplary Proteomic Studies of the DNA Damage Response
| Proteomic method | Pros | Cons | Study | Reference |
|---|---|---|---|---|
| Abundance proteomics | Quantitatively measure changes in abundance of thousands of proteins simultaneously | Proteome coverage could be limited for high complexity samples, in which fractionation before LC-MS/MS is recommended | Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment | (28) (28) |
| Phospho-proteomics | Determine the identity of phosphorylated proteins and the amino acid residues (and their level) which hold the phosphate group | Phospho-peptide enrichment step may not recover low abundant peptides and some enrichment beads (IMAC, TiO2) have higher efficiency for Ser/Thr than Tyr | Global changes in protein level and phosphorylation site profiles bySILAC with cisplatin treatment ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response Analyzing the phospho-proteome regulated by Spinophilin (PP1 regulatory subunit) that interacts with BRCA1 | (29,30) (43) (44) (46) |
| Ub-proteomics | Enables the large-scale analysis of ubiquitinated proteins and amino acid residues in cells | K-GG remnant profiling after trypsin digestion fails to recognize -GG- modified protein N-termini; loses Ub chain topology information; and may recognizes ISG15- and NEDD8-modified proteins, which also yield K-GG remnant peptides upon cleavage by trypsin | Quantitative proteomic assessment of Ubiquitination and Acetylation in the DNA Damage Response Profiling of Proteome and Ubiquitome Changes in Human Lens Epithelial Cell Line after Ultraviolet-B Irradiation | (47) (49) |
| ADPr-proteomics | Provides global information of ADPr proteins and amino acid residues | Numerous modifiable amino acid residues and the labile nature of ADPr bond pose a challenge for site-specific localization of ADPr Requires electron transfer dissociation (ETD)-based methods to preserve labile ADPr | Systems-wide analysis of Serine ADP-Ribosylation in response to oxidative stress Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses | (62) (63,64) |
| AP-MS | Allows to identify in vivo PPIs by affinity purification using antibodies recognizing endogenous proteins of interest or affinity tags | Data could vary by antibodies employed Transient or weak PPIs may not be captured effectively Cannot distinguish between direct and indirect PPIs | Analyzing the effect of pathogenic mutations on the BRCA1 interactome TEADs associate with DNA repair proteins to facilitate recovery from DNA damage PPARγ interacts with the MRN complex and the E3 ubiquitin ligase UBR5 | (46) (76) (127) |
| iPOND, NCC | Enables to identify proteins specifically localized to chromosomal loci under active DNA replication | DNA labeling with EdU or biotin-dUTP and de/crosslinking steps are required NCC requires cell membrane permeabilization | Analysis of protein dynamics at active, stalled, and collapsed replication forks | (78,82–86) |
| ChIP-MS | Capture PPIs on crosslinked chromatin at specific gene loci and/or genome-wide | Relies on antibodies Requires de/crosslinking steps | Telomeres were immunoprecipitated and known and novel telomere-binding proteins were identified by MS | (79,80) |
| Proximity proteomics | Detects transient and/or weak interactions Biotin-streptavidin binding is compatible with purification under harsh denaturing conditions Offers spatial and temporal resolution of PPIs | Proteins within proximity of a bait are identified, some of which are not necessarily interacting proteins May have high backgrounds from abundant cellular proteins and/or endogenous biotinylation Biotinylation labeling time varies (1 min - 24 hr) depending on the enzyme used | APEX reveals Shieldin as a 53BP1 effector complex in DSB repair BioID identifies Ku70-associated proteins BioID analysis of ATRX reveals its association with SLF2 that helps inhibit telomere exchanges | (91) (96) (97) |
| XL-MS | Elucidates PPIs and protein complex information by providing distance constraints within a protein or between proteins Depending on application needs (target functional group, solubility, membrane permeability, MS-cleavability, etc), a correct crosslinker can be selected | Crosslinking efficiency is often low (1–5%), leading to marginal crosslinks Long crosslinking time may induce protein aggregates Hybrid dipeptides generated by crosslinkers could greatly expand the search space during spectra matching | PPARγ interact with the MRN complex via NBS1 Mapping the regions involved in the Timeless–Tipin interaction | (127) (128) |
| H/DX-MS | Offers to analyze protein structures, dynamics, folding, and interactions Not limited by the size of proteins or protein complexes Requires few microliters of low micromolar concentration of proteins Detects co-existing protein conformations | Deuterium/hydrogen scrambling effect (redistribution of the exchanged protons within a peptide) could complicate the analysis Back exchange of deuterium label during chromatographic separation due to intrinsic amino acid hydrogen exchange rate differences could lead to loss and alteration of signal and biased measurement | Dynamic changes in DNA-binding domains of RPA upon ssDNA binding is revealed Helical subdomain (HD) of the PARP1 catalytic domain undergoes rapid unfolding when PARP1 encounters a DNA break, relieving the autoinhibition | (149) (150) |
| nMS | Widely applicable to samples varying in mass, degree of flexibility, symmetry, and polydispersity Multiple oligomeric states of proteins and complexes can be analyzed simultaneously Requires few microliters of low micromolar concentration of proteins Does not require samples to be chemically labeled or crosslinked. | The relative abundance of detected complexes may deviate from that in solution because of different complexes' distinct ionization, transmission, and detection probabilities In the gas phase, hydrophobic interactions weaken, and electrostatic interactions become stronger than in-solution making detecting specific assemblies impossible without prior crosslinking | A MutS intermediate state that is simultaneously bound to a DNA mismatch and a nucleoside triphosphate is detected The effect of mutations in Staphylococcus aureus uracil-DNA glycosylase inhibitor on its association with uracil-DNA glycosylase is analyzed | (160) (161) |
| Integrative structural modeling | Computationally integrates structural data from multiple sources to enable structure determination of macromolecules and their complexes | Accuracy of output models relies on quantity and resolution of the input information | Structures of protein complexes are derived by integrative modeling using structural proteomics data as well as data collected from conventional methods | (167,170,174,176) |
| Proteomic method | Pros | Cons | Study | Reference |
|---|---|---|---|---|
| Abundance proteomics | Quantitatively measure changes in abundance of thousands of proteins simultaneously | Proteome coverage could be limited for high complexity samples, in which fractionation before LC-MS/MS is recommended | Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment | (28) (28) |
| Phospho-proteomics | Determine the identity of phosphorylated proteins and the amino acid residues (and their level) which hold the phosphate group | Phospho-peptide enrichment step may not recover low abundant peptides and some enrichment beads (IMAC, TiO2) have higher efficiency for Ser/Thr than Tyr | Global changes in protein level and phosphorylation site profiles bySILAC with cisplatin treatment ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response Analyzing the phospho-proteome regulated by Spinophilin (PP1 regulatory subunit) that interacts with BRCA1 | (29,30) (43) (44) (46) |
| Ub-proteomics | Enables the large-scale analysis of ubiquitinated proteins and amino acid residues in cells | K-GG remnant profiling after trypsin digestion fails to recognize -GG- modified protein N-termini; loses Ub chain topology information; and may recognizes ISG15- and NEDD8-modified proteins, which also yield K-GG remnant peptides upon cleavage by trypsin | Quantitative proteomic assessment of Ubiquitination and Acetylation in the DNA Damage Response Profiling of Proteome and Ubiquitome Changes in Human Lens Epithelial Cell Line after Ultraviolet-B Irradiation | (47) (49) |
| ADPr-proteomics | Provides global information of ADPr proteins and amino acid residues | Numerous modifiable amino acid residues and the labile nature of ADPr bond pose a challenge for site-specific localization of ADPr Requires electron transfer dissociation (ETD)-based methods to preserve labile ADPr | Systems-wide analysis of Serine ADP-Ribosylation in response to oxidative stress Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses | (62) (63,64) |
| AP-MS | Allows to identify in vivo PPIs by affinity purification using antibodies recognizing endogenous proteins of interest or affinity tags | Data could vary by antibodies employed Transient or weak PPIs may not be captured effectively Cannot distinguish between direct and indirect PPIs | Analyzing the effect of pathogenic mutations on the BRCA1 interactome TEADs associate with DNA repair proteins to facilitate recovery from DNA damage PPARγ interacts with the MRN complex and the E3 ubiquitin ligase UBR5 | (46) (76) (127) |
| iPOND, NCC | Enables to identify proteins specifically localized to chromosomal loci under active DNA replication | DNA labeling with EdU or biotin-dUTP and de/crosslinking steps are required NCC requires cell membrane permeabilization | Analysis of protein dynamics at active, stalled, and collapsed replication forks | (78,82–86) |
| ChIP-MS | Capture PPIs on crosslinked chromatin at specific gene loci and/or genome-wide | Relies on antibodies Requires de/crosslinking steps | Telomeres were immunoprecipitated and known and novel telomere-binding proteins were identified by MS | (79,80) |
| Proximity proteomics | Detects transient and/or weak interactions Biotin-streptavidin binding is compatible with purification under harsh denaturing conditions Offers spatial and temporal resolution of PPIs | Proteins within proximity of a bait are identified, some of which are not necessarily interacting proteins May have high backgrounds from abundant cellular proteins and/or endogenous biotinylation Biotinylation labeling time varies (1 min - 24 hr) depending on the enzyme used | APEX reveals Shieldin as a 53BP1 effector complex in DSB repair BioID identifies Ku70-associated proteins BioID analysis of ATRX reveals its association with SLF2 that helps inhibit telomere exchanges | (91) (96) (97) |
| XL-MS | Elucidates PPIs and protein complex information by providing distance constraints within a protein or between proteins Depending on application needs (target functional group, solubility, membrane permeability, MS-cleavability, etc), a correct crosslinker can be selected | Crosslinking efficiency is often low (1–5%), leading to marginal crosslinks Long crosslinking time may induce protein aggregates Hybrid dipeptides generated by crosslinkers could greatly expand the search space during spectra matching | PPARγ interact with the MRN complex via NBS1 Mapping the regions involved in the Timeless–Tipin interaction | (127) (128) |
| H/DX-MS | Offers to analyze protein structures, dynamics, folding, and interactions Not limited by the size of proteins or protein complexes Requires few microliters of low micromolar concentration of proteins Detects co-existing protein conformations | Deuterium/hydrogen scrambling effect (redistribution of the exchanged protons within a peptide) could complicate the analysis Back exchange of deuterium label during chromatographic separation due to intrinsic amino acid hydrogen exchange rate differences could lead to loss and alteration of signal and biased measurement | Dynamic changes in DNA-binding domains of RPA upon ssDNA binding is revealed Helical subdomain (HD) of the PARP1 catalytic domain undergoes rapid unfolding when PARP1 encounters a DNA break, relieving the autoinhibition | (149) (150) |
| nMS | Widely applicable to samples varying in mass, degree of flexibility, symmetry, and polydispersity Multiple oligomeric states of proteins and complexes can be analyzed simultaneously Requires few microliters of low micromolar concentration of proteins Does not require samples to be chemically labeled or crosslinked. | The relative abundance of detected complexes may deviate from that in solution because of different complexes' distinct ionization, transmission, and detection probabilities In the gas phase, hydrophobic interactions weaken, and electrostatic interactions become stronger than in-solution making detecting specific assemblies impossible without prior crosslinking | A MutS intermediate state that is simultaneously bound to a DNA mismatch and a nucleoside triphosphate is detected The effect of mutations in Staphylococcus aureus uracil-DNA glycosylase inhibitor on its association with uracil-DNA glycosylase is analyzed | (160) (161) |
| Integrative structural modeling | Computationally integrates structural data from multiple sources to enable structure determination of macromolecules and their complexes | Accuracy of output models relies on quantity and resolution of the input information | Structures of protein complexes are derived by integrative modeling using structural proteomics data as well as data collected from conventional methods | (167,170,174,176) |
Exemplary Proteomic Studies of the DNA Damage Response
| Proteomic method | Pros | Cons | Study | Reference |
|---|---|---|---|---|
| Abundance proteomics | Quantitatively measure changes in abundance of thousands of proteins simultaneously | Proteome coverage could be limited for high complexity samples, in which fractionation before LC-MS/MS is recommended | Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment | (28) (28) |
| Phospho-proteomics | Determine the identity of phosphorylated proteins and the amino acid residues (and their level) which hold the phosphate group | Phospho-peptide enrichment step may not recover low abundant peptides and some enrichment beads (IMAC, TiO2) have higher efficiency for Ser/Thr than Tyr | Global changes in protein level and phosphorylation site profiles bySILAC with cisplatin treatment ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response Analyzing the phospho-proteome regulated by Spinophilin (PP1 regulatory subunit) that interacts with BRCA1 | (29,30) (43) (44) (46) |
| Ub-proteomics | Enables the large-scale analysis of ubiquitinated proteins and amino acid residues in cells | K-GG remnant profiling after trypsin digestion fails to recognize -GG- modified protein N-termini; loses Ub chain topology information; and may recognizes ISG15- and NEDD8-modified proteins, which also yield K-GG remnant peptides upon cleavage by trypsin | Quantitative proteomic assessment of Ubiquitination and Acetylation in the DNA Damage Response Profiling of Proteome and Ubiquitome Changes in Human Lens Epithelial Cell Line after Ultraviolet-B Irradiation | (47) (49) |
| ADPr-proteomics | Provides global information of ADPr proteins and amino acid residues | Numerous modifiable amino acid residues and the labile nature of ADPr bond pose a challenge for site-specific localization of ADPr Requires electron transfer dissociation (ETD)-based methods to preserve labile ADPr | Systems-wide analysis of Serine ADP-Ribosylation in response to oxidative stress Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses | (62) (63,64) |
| AP-MS | Allows to identify in vivo PPIs by affinity purification using antibodies recognizing endogenous proteins of interest or affinity tags | Data could vary by antibodies employed Transient or weak PPIs may not be captured effectively Cannot distinguish between direct and indirect PPIs | Analyzing the effect of pathogenic mutations on the BRCA1 interactome TEADs associate with DNA repair proteins to facilitate recovery from DNA damage PPARγ interacts with the MRN complex and the E3 ubiquitin ligase UBR5 | (46) (76) (127) |
| iPOND, NCC | Enables to identify proteins specifically localized to chromosomal loci under active DNA replication | DNA labeling with EdU or biotin-dUTP and de/crosslinking steps are required NCC requires cell membrane permeabilization | Analysis of protein dynamics at active, stalled, and collapsed replication forks | (78,82–86) |
| ChIP-MS | Capture PPIs on crosslinked chromatin at specific gene loci and/or genome-wide | Relies on antibodies Requires de/crosslinking steps | Telomeres were immunoprecipitated and known and novel telomere-binding proteins were identified by MS | (79,80) |
| Proximity proteomics | Detects transient and/or weak interactions Biotin-streptavidin binding is compatible with purification under harsh denaturing conditions Offers spatial and temporal resolution of PPIs | Proteins within proximity of a bait are identified, some of which are not necessarily interacting proteins May have high backgrounds from abundant cellular proteins and/or endogenous biotinylation Biotinylation labeling time varies (1 min - 24 hr) depending on the enzyme used | APEX reveals Shieldin as a 53BP1 effector complex in DSB repair BioID identifies Ku70-associated proteins BioID analysis of ATRX reveals its association with SLF2 that helps inhibit telomere exchanges | (91) (96) (97) |
| XL-MS | Elucidates PPIs and protein complex information by providing distance constraints within a protein or between proteins Depending on application needs (target functional group, solubility, membrane permeability, MS-cleavability, etc), a correct crosslinker can be selected | Crosslinking efficiency is often low (1–5%), leading to marginal crosslinks Long crosslinking time may induce protein aggregates Hybrid dipeptides generated by crosslinkers could greatly expand the search space during spectra matching | PPARγ interact with the MRN complex via NBS1 Mapping the regions involved in the Timeless–Tipin interaction | (127) (128) |
| H/DX-MS | Offers to analyze protein structures, dynamics, folding, and interactions Not limited by the size of proteins or protein complexes Requires few microliters of low micromolar concentration of proteins Detects co-existing protein conformations | Deuterium/hydrogen scrambling effect (redistribution of the exchanged protons within a peptide) could complicate the analysis Back exchange of deuterium label during chromatographic separation due to intrinsic amino acid hydrogen exchange rate differences could lead to loss and alteration of signal and biased measurement | Dynamic changes in DNA-binding domains of RPA upon ssDNA binding is revealed Helical subdomain (HD) of the PARP1 catalytic domain undergoes rapid unfolding when PARP1 encounters a DNA break, relieving the autoinhibition | (149) (150) |
| nMS | Widely applicable to samples varying in mass, degree of flexibility, symmetry, and polydispersity Multiple oligomeric states of proteins and complexes can be analyzed simultaneously Requires few microliters of low micromolar concentration of proteins Does not require samples to be chemically labeled or crosslinked. | The relative abundance of detected complexes may deviate from that in solution because of different complexes' distinct ionization, transmission, and detection probabilities In the gas phase, hydrophobic interactions weaken, and electrostatic interactions become stronger than in-solution making detecting specific assemblies impossible without prior crosslinking | A MutS intermediate state that is simultaneously bound to a DNA mismatch and a nucleoside triphosphate is detected The effect of mutations in Staphylococcus aureus uracil-DNA glycosylase inhibitor on its association with uracil-DNA glycosylase is analyzed | (160) (161) |
| Integrative structural modeling | Computationally integrates structural data from multiple sources to enable structure determination of macromolecules and their complexes | Accuracy of output models relies on quantity and resolution of the input information | Structures of protein complexes are derived by integrative modeling using structural proteomics data as well as data collected from conventional methods | (167,170,174,176) |
| Proteomic method | Pros | Cons | Study | Reference |
|---|---|---|---|---|
| Abundance proteomics | Quantitatively measure changes in abundance of thousands of proteins simultaneously | Proteome coverage could be limited for high complexity samples, in which fractionation before LC-MS/MS is recommended | Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment | (28) (28) |
| Phospho-proteomics | Determine the identity of phosphorylated proteins and the amino acid residues (and their level) which hold the phosphate group | Phospho-peptide enrichment step may not recover low abundant peptides and some enrichment beads (IMAC, TiO2) have higher efficiency for Ser/Thr than Tyr | Global changes in protein level and phosphorylation site profiles bySILAC with cisplatin treatment ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response Analyzing the phospho-proteome regulated by Spinophilin (PP1 regulatory subunit) that interacts with BRCA1 | (29,30) (43) (44) (46) |
| Ub-proteomics | Enables the large-scale analysis of ubiquitinated proteins and amino acid residues in cells | K-GG remnant profiling after trypsin digestion fails to recognize -GG- modified protein N-termini; loses Ub chain topology information; and may recognizes ISG15- and NEDD8-modified proteins, which also yield K-GG remnant peptides upon cleavage by trypsin | Quantitative proteomic assessment of Ubiquitination and Acetylation in the DNA Damage Response Profiling of Proteome and Ubiquitome Changes in Human Lens Epithelial Cell Line after Ultraviolet-B Irradiation | (47) (49) |
| ADPr-proteomics | Provides global information of ADPr proteins and amino acid residues | Numerous modifiable amino acid residues and the labile nature of ADPr bond pose a challenge for site-specific localization of ADPr Requires electron transfer dissociation (ETD)-based methods to preserve labile ADPr | Systems-wide analysis of Serine ADP-Ribosylation in response to oxidative stress Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses | (62) (63,64) |
| AP-MS | Allows to identify in vivo PPIs by affinity purification using antibodies recognizing endogenous proteins of interest or affinity tags | Data could vary by antibodies employed Transient or weak PPIs may not be captured effectively Cannot distinguish between direct and indirect PPIs | Analyzing the effect of pathogenic mutations on the BRCA1 interactome TEADs associate with DNA repair proteins to facilitate recovery from DNA damage PPARγ interacts with the MRN complex and the E3 ubiquitin ligase UBR5 | (46) (76) (127) |
| iPOND, NCC | Enables to identify proteins specifically localized to chromosomal loci under active DNA replication | DNA labeling with EdU or biotin-dUTP and de/crosslinking steps are required NCC requires cell membrane permeabilization | Analysis of protein dynamics at active, stalled, and collapsed replication forks | (78,82–86) |
| ChIP-MS | Capture PPIs on crosslinked chromatin at specific gene loci and/or genome-wide | Relies on antibodies Requires de/crosslinking steps | Telomeres were immunoprecipitated and known and novel telomere-binding proteins were identified by MS | (79,80) |
| Proximity proteomics | Detects transient and/or weak interactions Biotin-streptavidin binding is compatible with purification under harsh denaturing conditions Offers spatial and temporal resolution of PPIs | Proteins within proximity of a bait are identified, some of which are not necessarily interacting proteins May have high backgrounds from abundant cellular proteins and/or endogenous biotinylation Biotinylation labeling time varies (1 min - 24 hr) depending on the enzyme used | APEX reveals Shieldin as a 53BP1 effector complex in DSB repair BioID identifies Ku70-associated proteins BioID analysis of ATRX reveals its association with SLF2 that helps inhibit telomere exchanges | (91) (96) (97) |
| XL-MS | Elucidates PPIs and protein complex information by providing distance constraints within a protein or between proteins Depending on application needs (target functional group, solubility, membrane permeability, MS-cleavability, etc), a correct crosslinker can be selected | Crosslinking efficiency is often low (1–5%), leading to marginal crosslinks Long crosslinking time may induce protein aggregates Hybrid dipeptides generated by crosslinkers could greatly expand the search space during spectra matching | PPARγ interact with the MRN complex via NBS1 Mapping the regions involved in the Timeless–Tipin interaction | (127) (128) |
| H/DX-MS | Offers to analyze protein structures, dynamics, folding, and interactions Not limited by the size of proteins or protein complexes Requires few microliters of low micromolar concentration of proteins Detects co-existing protein conformations | Deuterium/hydrogen scrambling effect (redistribution of the exchanged protons within a peptide) could complicate the analysis Back exchange of deuterium label during chromatographic separation due to intrinsic amino acid hydrogen exchange rate differences could lead to loss and alteration of signal and biased measurement | Dynamic changes in DNA-binding domains of RPA upon ssDNA binding is revealed Helical subdomain (HD) of the PARP1 catalytic domain undergoes rapid unfolding when PARP1 encounters a DNA break, relieving the autoinhibition | (149) (150) |
| nMS | Widely applicable to samples varying in mass, degree of flexibility, symmetry, and polydispersity Multiple oligomeric states of proteins and complexes can be analyzed simultaneously Requires few microliters of low micromolar concentration of proteins Does not require samples to be chemically labeled or crosslinked. | The relative abundance of detected complexes may deviate from that in solution because of different complexes' distinct ionization, transmission, and detection probabilities In the gas phase, hydrophobic interactions weaken, and electrostatic interactions become stronger than in-solution making detecting specific assemblies impossible without prior crosslinking | A MutS intermediate state that is simultaneously bound to a DNA mismatch and a nucleoside triphosphate is detected The effect of mutations in Staphylococcus aureus uracil-DNA glycosylase inhibitor on its association with uracil-DNA glycosylase is analyzed | (160) (161) |
| Integrative structural modeling | Computationally integrates structural data from multiple sources to enable structure determination of macromolecules and their complexes | Accuracy of output models relies on quantity and resolution of the input information | Structures of protein complexes are derived by integrative modeling using structural proteomics data as well as data collected from conventional methods | (167,170,174,176) |
FUNCTIONAL PROTEOMICS
Abundance proteomics
Profiling of total proteome in cells can be performed by digesting proteins to peptides with trypsin (and/or other peptidases) and applying them to LC–MS/MS. The relative abundance of each identified peptide is then measured and individual peptide data for each protein are combined to give protein-level quantification. This analysis allows us to quantitatively measure changes in abundance of thousands of proteins in cells and tissues simultaneously (Figure 1). To increase the proteome coverage (i.e. number of peptides and associated proteins identified in a given MS run), samples are often fractionated by ion exchange or reversed phase chromatography (24,25) and each fraction is individually analyzed by LC–MS/MS to maximize peptide identification. Furthermore, to globally map cellular macromolecular complexes, Complex-Centric Proteome Profiling method was recently developed (26). In this method, cell lysates are fractionated by size exclusion chromatography, and native protein complexes in each fraction are digested into peptides and analyzed by DIA MS. Quantification and composition of protein complexes are then determined based on protein patterns across the fractions by error-controlled, complex-centric analysis. This technique revealed the mitotic proteome reorganization, including disassembly of the nuclear pore complex, by quantitatively comparing human protein complexes during transition from interphase to mitosis (27).
Comparative proteomic analysis between BRCA1-deficient and BRCA1-proficient mouse tumors upon cisplatin treatment revealed that proteins involved in centrosome organization, chromosome condensation, HR repair and nucleotide metabolism were up-regulated in cisplatin-sensitive BRCA1-deficient tumors (28). However, several studies show that overall changes in protein abundance during or after DNA damage exposure are much less conspicuous than PTM changes (29,30), necessitating the profiling of PTMs of proteins in addition to protein levels.
Post-translational modification (PTM) proteomics
PTMs (e.g. phosphorylation, ubiquitylation, and ADP-ribosylation among others) of many DDR proteins are known to induce changes in protein stability, cellular localization, enzymatic activity, and/or interaction with DNA and/or other proteins (31–38). To properly profile and quantify PTMs of proteins by MS, digested peptides from lysates, bearing a given type of PTMs, need to be enriched prior to LC–MS/MS analysis (Figure 1). For phospho-peptide enrichment, Fe3+-based Immobilized Metal Affinity Chromatography (IMAC) beads are widely used and about 10 000–20 000 phosphorylated peptides (phospho-peptides) can be typically detected in a single LC–MS/MS run (39). Similarly, other metal-based (e.g. Ti4+, Zr4+) IMAC beads and titanium dioxide (TiO2) can be also utilized to enrich phospho-peptides (39,40). To enrich ubiquitylated peptides, an antibody-bead conjugate that recognizes a di-glycine remnant of ubiquitin left on Lysine residues (K-GG) of protein substrates after trypsin digestion is used (41,42). This enrichment followed by LC–MS/MS allows quantitative analysis of thousands of non-redundant phosphorylated or ubiquitinated peptides and exact sites of modification after searching MS spectra against putative protein sequences in a species-specific proteome database.
Phospho-proteomic studies reveal that there is a considerable variation in the kinetics and directionality of phospho-proteome changes following DNA damage (43,44), and that over one-third of the captured phospho-peptides are dephosphorylated within minutes of DNA damage (43), indicating that phosphatases are not only involved in counter-balancing DNA double-strand break (DSB)-induced phosphorylation events by resetting activated DDR factors to the initial homeostatic state following the repair of damaged DNA, but also play a primary role in initiating the repair process by removing constitutive phosphorylation that inhibits the function of DNA repair factors to activate these proteins (45,46). Ubiquitome analysis has been also successfully used to profile changes in the ubiquitome in response to DNA damage (47). Because ubiquitination often regulates protein stability, pairing ubiquitin proteomics with abundance proteomics and proteasome inhibitor treatments (e.g. MG132) is often used to identify proteins whose levels are regulated by ubiquitination (48,49).
ADP-ribose (ADPr) unit is transferred by poly(ADP-ribose) polymerases (PARPs) from NAD+ to a carboxylic acid residues (Glu, Asp) (50–52), but other residues containing a thiol group (Cys), an alcohol group (Ser, Thr, Tyr), a guanidinium group (Arg) or Lys are also found to serve as acceptors (53–63). Despite the role of PARylation in DDR, high-throughput profiling of ADPr-modified proteins has been hindered due to the difficulty of PAR enrichment. However, the Af1521 macrodomain, which has a strong ADPr-binding affinity, was shown to successfully enrich and identify ADP-ribosylated proteins from cell lysates induced under different genotoxic stress conditions (64). Interestingly, another study reveals that Ser residues are the major ADPr site in response to oxidative stress, and that significant portion of these Ser residues are co-targeted by mutually exclusive ADP-ribosylation and phosphorylation, indicating a crosstalk between the two PTMs (62) that remains to be further validated for their roles in cellular stress response.
For the MS analysis of ADPr proteins, numerous modifiable amino acid residues and the labile nature of the bond between ADPr and the amino acid residues pose a challenge for site-specific localization using the conventional collision-induced dissociation (CID) MS/MS method. Instead, the radical-driven electron transfer dissociation (ETD) method has proven to preserve labile ADPr and allow confident determination of the exact ADPr-modified amino acid residues (62). Recently, the activated ion ETD (AI-ETD) method that uses infrared photoactivation during the ETD reaction was developed, which allows to overcome the high degree of non-dissociative electron transfer (ETnoD) during regular ETD, especially for ADPr-peptide precursors with low m/z ratio (63). AI-ETD was shown to map significantly more ADPr sites than ETD and identify more physiological ADPr sites with less input materials, many of which were not previously reported.
Protein-protein interaction mapping by affinity purification MS
Coordinated regulation of protein complex formation is central to the DDR response. For example, BRCA1, a hereditary breast and ovarian cancer susceptibility gene (65–67) that plays a crucial role in the repair of DNA double-strand breaks (DSB) (68), carries out its repair functions in concert with a large number of proteins involved at distinct steps of homologous recombination (69). Affinity Purification-MS (AP-MS) is a proteomics method to systematically identify PPIs by expressing and purifying affinity-tagged ‘bait’ proteins in cells and detecting stably bound protein interactors (prey) by MS (46,70–72). Samples that express the affinity tag alone or repress the expression of tagged bait protein are often used in parallel as negative control for statistical PPI scoring (Figure 1). Alternatively, direct immunoprecipitation of the endogenous protein of interest may also be performed if a sensitive and specific antibody exists (73).
Recently, Kim et al. applied AP-MS to comprehensively analyze the BRCA1 interactome and how these interaction profiles change with pathogenic BRCA1 mutations (46). This AP-MS study revealed a number of previously unidentified BRCA1-interacting proteins in addition to known interactors and showed that these interactions are altered by mutations in different domains of BRCA1: C-terminal BRCT domain mutants (S1655F, 5382insC, M1775R) completely or significantly lost the interaction with multiple homologous recombination repair proteins (including FAM175A, BRIP1, RBBP8, UIMC, BRE), whereas N-terminal RING domain mutants (I26A, C61G, R71G) maintain these interactions although they are not as strong as with wild-type BRCA1. These results concur with previous suggestions that RING domain mutants may retain residual activity; for example, the C61G variant is only moderately sensitive to cisplatin and PARP inhibitors and becomes readily resistant to these drugs (74,75). AP-MS of transcriptional enhanced associate domain family members 1–4 (TEADs) also revealed their association with DNA repair proteins (XRCC5, XRCC6, PARP1, RIF1) to facilitate cellular recovery from DNA damage (76).
Protein–DNA interaction mapping
The DDR also involves choreographed interactions between DNAs and proteins and cataloging the dynamic spatial and temporal changes in DNA/chromatin-associated proteome is essential to thoroughly understand the DNA repair and replication processes. To identify proteins enriched at DNA replication forks, isolation of Proteins on Nascent DNA (iPOND) and Nascent Chromatin Capture (NCC) techniques were developed (77,78). Additionally, chromatin immunoprecipitation (ChIP) followed by MS (ChIP-MS) was devised to probe chromatin-bound proteins at specific gene loci as well as genome-wide (79,80).
In iPOND, newly synthesized DNAs are labeled with 5-ethynyl-2’-deoxyuridine (EdU) incorporation by the replisome DNA polymerases. After crosslinking proteins to DNAs with formaldehyde, biotin is covalently conjugated to an alkyne group of EdU using click chemistry, and 100–200 bp protein-bound chromatin fragments are generated by sonication. Finally, proteins bound on the EdU labeled chromatin fragments are captured by streptavidin beads and analyzed by quantitative MS after protein-DNA decrosslinking. Duration of EdU labeling (pulse) and subsequent chase after removal of EdU determines the spatial information of proteins relative to the replication fork. Furthermore, the pulse and chase samples are critical to distinguish proteins engaged in DNA replication from those that are simply associated with bulk chromatin (81). NCC is similar to iPOND except it utilizes biotin-dUTP instead of EdU, such that NCC does not require a biotin conjugation step. However, due to impermeability of biotin-dUTP, it is necessary to permeabilize cell membranes in a hypotonic buffer during biotin-dUTP labeling. Quantitative MS analysis of enriched proteins in iPOND and NCC has been performed by either label-free or SILAC/iTRAQ/TMT-based protocols (78,82–86) to identify new DNA replication proteins. In these studies, labeling protocols tend to provide more quantitatively precise and reproducible datasets than label-free methods (81). ChIP-MS is similar to AP-MS, in that both methods rely on antibody-based immunoprecipitation/purification, but ChIP-MS is used to specifically identify PPIs on crosslinked chromatin. Proteins in the male-specific lethal (MSL) complex on the chromatin template as well as proteins enriched in telomeres were monitored by ChIP-MS and successfully identified novel proteins relevant to the function of MSL complex and telomere maintenance, respectively (79,80).
Proximity-dependent labeling MS
Proximity-dependent labeling has emerged as a complementary method to AP-MS to study PPIs and relies on a promiscuous biotin protein ligase (BirA/BioID/TurboID) (87,88) or an engineered ascorbic acid peroxidase (APEX) (89) fused to a protein of interest (bait) to label proteins in close proximity to the bait protein through covalent transfer of biotin or biotin derivatives in a distance-dependent manner. Upon addition of biotin, the proximal proteins are biotinylated, enriched with streptavidin beads, and identified by quantitative MS (Figure 1). The covalent biotin labeling of proximal proteins is particularly useful to capture transient or weaker PPIs, compatible with purification under harsh cell lysis conditions (e.g. higher detergent and urea concentrations in lysis buffer), and offers spatial and temporal resolution of PPIs within various subcellular locations (87,89,90). APEX-based proximity proteomics characterized the endogenous network of BRCA1, 53BP1, and MDC1 and revealed Shieldin is a 53BP1 effector complex in DSB repair (91). Recently, biotin ligase enzymes have been engineered to split into two parts which enable contact-specific proximity-dependent labeling. While the two split enzyme parts remain inactive apart, they become active when bound together by PPIs. (92–94). These split enzyme-based approaches offer greater targeting specificity of biotinylation than full-length enzymes alone and provide powerful tools for validating and functionally characterizing high-confidence PPIs identified by AP-MS and/or other methods (95). Abbasi and Schild-Poulter applied BioID to the Ku interactome analysis and identified ∼250 Ku70-associated proteins in HEK293 cells, many of which are involved in RNA metabolism, chromatin remodeling, and microtubule dynamics as well as DNA repair and replication, revealing additional cellular roles of the Ku complex (96). Another BioID analysis of ATRX revealed its association with SLF2 that helps inhibit telomere exchanges (97).
MS data quantification, interpretation and integration using bioinformatics and network modeling
Once data is collected, they first pass through quality control. Specifically, one must ensure reproducibility in peptide detections and intensity quantifications between biological replicates. Replicates that fail quality control are discarded. Global proteomics data of whole cell lysates (e.g. abundance proteomics, phosphoproteomics, ubiquitin proteomics, or similar) can be subjected to relative quantification to compare each experimental condition to a control. One software, called MSstats, offers quantitative and comparative measurement of protein levels, PTMs, or PPIs between two biological states, such as diseased versus healthy or mutant versus wild-type (98). This data preprocessing step generates log2 fold changes and P-values, and thus defines differentially expressed or regulated proteins (DEP). For AP-MS data, additional algorithms are used to remove background signal; even after performing an affinity purification, some proteins detected may be non-specifically binding the tube or other reagents used during sample preparation. The analysis workflow typically utilizes software packages (e.g. compPASS, SAINT, MIST) (99–103) to assign quantitative scoring metrics (i.e. abundance, specificity, and reproducibility) to all pairwise PPIs and filter high-confidence interacting proteins (e.g. SAINTscore ≥ 0.9, Bayesian false discovery rate ≤ 0.05). These quantitative, differential, preprocessed data are subsequently interpreted using more sophisticated computational algorithms, which enable the integration of new data with prior knowledge.
A highly-used, and often first-line, approach to data interpretation is gene enrichment analysis, which enables the assignment of biological terms to differentially abundant proteins. Gene sets have been defined by several groups, many of which are distributed by the Molecular Signatures Database (MSigDB, www.gsea-msigdb.org). Commonly used databases include Reactome (104), KEGG (105), Wikipathways (106) and Gene Ontology (107). One enrichment analysis algorithm, called Gene Set Enrichment Analysis (GSEA), creates a ranked list of proteins derived from experiment (often ranked by log2 fold change) and compares it to annotated gene sets (e.g. cellular pathways) to determine whether genes in each gene set occur toward the top or bottom (large or small fold changes) of the list. If so, this indicates the gene set is ‘correlated’ with proteins under regulation in the experiment (108). A similar approach uses a Fisher's exact or hypergeometric test to assess the overlap of DEP to each gene set (109). The end result is the same: significantly enriched gene sets represent cellular pathways that are regulated in the experiment. These results provide general biological understanding and often guide subsequent validation experiments. Importantly, these gene set databases possess many terms related to the DNA damage response. In Gene Ontology, there are over 40 terms that either possess ‘DNA repair’ or ‘DNA damage,’ spanning pathways such as ‘double strand break repair via non-homologous end joining’ and ‘single strand break repair via break induced replication’. Gene enrichment analysis is a good way to gain a high-level summary of the data but is often insufficient to more deeply interrogate the data to reveal mechanistic hypotheses for further experimental testing.
Network modeling, a branch of computational modeling that uses pre-defined networks of gene-gene interactions, can be used to integrate MS datasets and extract biological insight at the interface between distinct proteomics datasets. Several freely-available, large (often genome-wide scale) biological networks exist [e.g. PathwayCommons (110), STRING (111), ReactomeFI (112)], which capture decades of experimental knowledge detailing how genes and proteins physically and/or functionally interact in protein complexes or functional pathways. Data from an experiment can be overlaid onto these networks and analyzed using network modeling algorithms to understand biological interconnectivity across multiple datasets (Figure 1).
One approach is called network propagation (113). Briefly, proteins of interest from the experiment (‘nodes’) are labeled in the network. This signal is then propagated through each proteins’ molecular interactions (‘edges’) to nearby nodes in an iterative manner, often using a random walk or heat kernel mathematical process. A permutation test is subsequently performed by shuffling node labels or randomizing network structure in order to assign a p-value to each node, which represents the network proximity, and functional connectivity, of each gene to the initial labeled nodes. Network propagation can be used to identify the extent of intersection between two or more gene lists by evaluating their shared neighborhoods within the network. This approach is particularly useful when two gene lists have poor overlap at the gene level but high potential overlap at the pathway level. We and others have previously used this approach to discover links between the human papillomavirus (HPV) virus-host physical interactome and the mutational landscape of HPV-negative tumors, revealing converging routes to oncogenesis (114), and to integrate genome-wide CRISPR hits from human cells infected with distinct coronaviruses, revealing host pathway targets for pan-coronavirus therapeutics (115).
Data integration using network propagation can be especially useful when a single dataset is insufficient to extract the level of mechanistic insight desired. It can also be used as an exploratory tool to screen for interesting connections between distinct data types. Furthermore, network propagation is useful when data is sparse in nature, which can be the case for proteomics datasets; in these cases, network propagation can be used to explore how proteins coalesce into the same complex or pathway. However, because network propagation provides a wealth of information, the results can be difficult to interpret. Additional approaches are needed to make it easier for users to pinpoint the most relevant mechanisms from the resulting network.
Network modeling tools could be transformative for discovering new mechanisms underlying DDR signaling. For example, one might be interested in understanding the mechanisms underlying how a specific cancer driver point mutation (e.g. BRCA1 mutation) drives cancer progression. To study this, one could perform global proteomics (abundance proteomics and PTM analysis) comparing wild-type cells to cells that possess the mutation of interest. Additionally, one could perform differential AP-MS or proximity-dependent labeling to quantitatively compare how a protein-coding point mutant alters PPIs. The protein-level overlap between these datasets might be small; indeed, proteins detected to be differentially regulated in the global proteomics data may reflect gene expression responses several steps downstream the mutant, and thus may not be contained within the physical interactome. Network propagation could be used here to reveal the molecular networks that ‘connect’ the differential PPIs to the downstream signaling events. Specifically, network propagation could be performed in two parts. First, label and propagate nodes corresponding to differentially interacting PPIs. Second, label and propagate nodes corresponding to differentially regulated proteins from the global proteomics studies. Genes that are significant (P < 0.05) in both propagation analyses represent molecular networks that lie at the interface between BRCA1 mutant protein interactions and the downstream signaling effects of the BRCA1 mutation. Such an analysis may delineate a novel cellular pathway linking a BRCA1 mutation to its signaling ramifications, and thereby suggest subsequent validation experiments to further understand BRCA1 function. This approach is specifically applicable to DDR signaling because one important question in the field is how specific mutants (especially variants of uncertain significance of key DDR proteins) impact protein complex formation and DNA repair outcomes, and may provide a more comprehensive view of proteomic changes and signaling pathways induced by a given mutation, which is often difficult to capture by any single proteomics attempt.
STRUCTURAL PROTEOMICS
Crosslinking MS (XL-MS)
XL-MS is a powerful technique that informs protein-protein interactions and provides structural insights into proteins and protein assemblies by identifying amino acid residue pairs that are within close spatial proximity (116–121). Typically, this approach involves crosslinking or surface modification of protein complexes followed by proteolysis and MS identifications (Figure 1). Applications of XL-MS vary from studying macromolecular complexes (122,123) to their structural dynamics (124,125).
Qiu et al. conducted a comprehensive investigation of DNA-binding proteins in vivo using XL-MS and identified >100 proteins, including those involved in DNA replication and repair (126). Another study found that peroxisome proliferator activated receptor γ (PPARγ) interacts with the MRE11-RAD50-NBS1 (MRN) complex and the E3 ubiquitin ligase UBR5 by AP-MS and that PPARγ promotes UBR5-mediated degradation of ATM interactor (ATMIN), leading to ATM activation and the initiation of DNA repair upon DNA damage (127). Subsequent XL-MS experiment identified PPARγ peptides crosslinked to NBS1, which are located in the DNA-binding domain and the ligand-binding domain of PPARγ, indicating that PPARγ interact with the MRN complex via NBS1, and that NBS1 binding may interfere with PPARγ transcription activator function. Holzer et al. elucidated the structural basis for the human Timeless–Tipin complex using various complementary tools including XL-MS as well as XRC and nMS. They used lysine-specific crosslinker and demonstrated that the Timeless–Tipin interaction is extensive, such that three distinct regions of Timeless between amino acids 400 and 1100 are crosslinked to three different sites in Tipin. Using this integrated approach they were able to show that Timeless forms a binding surface capable of surrounding the smaller protein Tipin (128).
There are a few inherent challenges of XL-MS: (i) the combinatorial nature of the crosslinks and mixture of unmodified peptides increases the complexity of the spectra; (ii) sequencing of crosslinked peptides often results in low-quality or uninterpretable MS/MS spectra because of insufficient or complex fragmentation; (iii) identification and filtering of nonspecific interactions are challenging especially in validating novel observed PPIs. Thus, high-throughput bioinformatic approaches are required to decipher complete structural information. Some of these challenges can be overcome by increasing data acquisition rates, higher sequencing capacity, and novel hybrid fragmentation techniques like electron transfer/higher-energy collision dissociation (EThcD) and ultraviolet photodissociation (UV-PD) (129). Regardless, XL-MS is a valuable tool for structural characterization of protein complexes by providing real-time insight into the PPIs and protein-DNA/RNA interactions that may undergo structural changes in response to DNA damage.
Hydrogen/deuterium exchange MS (H/DX-MS)
H/DX-MS measures dynamic changes in protein structures over a specified time scale by measuring the relative uptake of protein backbone amide hydrogen for deuterium (and vice versa) (Figure 1). The rate of this reaction is dependent on various factors like solvent accessibility, chemical properties of the underlying amino acid sequence, stability of hydrogen bonding networks, and inductive effect of the neighboring groups (130–133). It has been applied to explore protein/protein complex conformations (134), protein-ligand binding sites (135), allosteric effects of proteins (136), protein folding dynamics (137), intrinsically disordered proteins (138), and protein − membrane interactions (139). The resolution of data depends on the fragmentation technique adopted during the MS data acquisition. Residue-level information can be derived from gas-phase fragmentation of the deuterated peptides using ETD or electron-capture dissociation (ECD) (140,141). The vibrationally cold energy utilized in these techniques results in minimized hydrogen scrambling (i.e. redistribution of the exchanged protons within a peptide) as opposed to other methods like collision-induced dissociation (CID) (142–144). In 2013, Resetca et al. developed a method called time-resolved electrospray ionization mass spectrometry (TRESI-MS), which uses a microfluidic chip in-line with all the steps involved in a ‘bottom-up’ H/DX workflow (145). This development provided faster sample preparation times and improved reproducibility, making it feasible to characterize rapid structural transitions that occur during protein folding (146), ligand binding (147), or post-translational modification (148).
Ahmad et al. applied H/DX-MS to explore ssDNA-driven dynamics of the individual domains of single-strand DNA (ssDNA)-binding protein, replication protein A (RPA) (149). They observed changes in DNA binding domains (DBD) A-E of RPA upon ssDNA binding (149): DBD-A and B were shown to be dynamic and no protection was observed upon ssDNA binding, while DBD-C displayed extensive changes signifying a major role in stabilizing RPA on ssDNA binding. These H/DX-MS data proposed that DBD-A and -B of RPA serve as the dynamic half and DBD-C, -D and -E function as the less-dynamic half, such that ssDNA buried in DBD-A and B could be more accessible to RPA-interacting proteins (149). Dawicki-McKenna et al. monitored conformational dynamics of PARP1 using H/DX-MS and observed a >10 000-fold faster exchange within specific portions of the helical subdomain (HD) of the catalytic domain, which undergoes rapid unfolding when PARP1 encounters a DNA break (150). This result indicates that DNA damage detection by PARP1 relieves the autoinhibition posed by the HD.
Native MS (nMS) and intact protein analysis
nMS and intact protein MS involve analyzing and characterizing macromolecules, predominantly protein complexes, while retaining their inter and intramolecular interactions (Figure 1). To ionize protein complexes directly from aqueous solutions, buffers in which protein complexes are suspended during purification steps are exchanged into volatile buffers, such as ammonium acetate which has a pH range of 6–8 and evaporates readily during ionization. nMS resorts to a soft transfer of the analyte from the solution to the gas phase using nano-electrospray ionization (nESI). During ionization the macromolecules are charged, and the solvent molecules are stripped off before the ions reach the mass analyzer under (ultra)high-vacuum conditions. One may argue that proteins may not always retain a fully native state during nMS. However, under careful and optimized conditions, nMS provides gas-phase ions of proteins that retain many of their native features (151,152). The stoichiometry of a protein complex can be deduced from the calculated mass, and structural preference can be informed by the number of charges it carries (153). Structured protein conformations typically carry limited charges per unit mass due to the limited solvent accessibility owing to their compact nature. In contrast, unfolded/denatured proteins have a larger solvent-accessible surface area which enables them to accumulate higher charges (154–156). nMS combined with CID or surface-induced dissociation (SID) disrupts noncovalent interactions between protein subunits based on their strength and thus can help decipher the stoichiometry and topology of protein complexes (157,158). SID is a rapid dissociation that is almost a single-step energy deposition process when ions collide with a surface. No extensive unfolding of proteins and subunits is observed in this process (159).
nMS analysis of Escherichia coli MutS detected a MutS dimer state that is simultaneously bound to a DNA mismatch and a nucleoside triphosphate, which has been thought as an intermediate for faithful initiation of mismatch repair (160). Furthermore, this analysis could distinguish between binding of nucleoside diphosphate and nucleoside triphosphate in the two composite ATPase sites of MutS dimer, providing mechanistic and structural insight into how mismatched DNA binding and release are controlled by binding and hydrolysis of ATP. This study demonstrates that nMS is a powerful tool to detect mechanistically relevant reaction intermediates of allosteric enzyme complexes that cannot be easily addressed by other techniques. Besides, nMS was utilized to quantitatively measure the macromolecular association between uracil–DNA glycosylase and its inhibitor in Staphylococcus aureus and revealed that naturally occurring mutations in the inhibitor lead to appreciable changes in the dissociation constants for the complex (161).
Despite significant advances in nMS methods, there are some analytical challenges that remain to be addressed. For example, complexes expressed and purified from mammalian cells often carry PTMs and/or small ligands, which cause small mass differences and result in a heterogeneous mixture. It is challenging to resolve heterogeneous protein complexes, although charge changing experiments can sometimes help solve this problem (162,163). Additionally, biomolecules need to be prepared to be amenable to native electrospray ionization. But they often require nMS-incompatible components, including metal ions, small molecules, or lipids to maintain the integrity. Improving gas-phase desalting and adduct removal techniques would not only maintain the biomolecule in a ‘native-like’ environment but also reduce adduct formation and aid solvent removal at low activation energies, resulting in clean native spectra (164). Alternative ionization methods, such as desorption electrospray ionization (DESI) that enable biomolecules to directly ionize from biological tissues (165) may also help mitigate this problem. DESI involves directing a fast-moving charged solvent stream at an angle relative to the sample surface under atmospheric conditions to extract analytes from the surfaces resulting in the secondary ions being propelled toward the mass analyzer (166).
Integrative structural modeling
Data from the diverse structural proteomics methodologies discussed above can be combined to obtain maximum structural insight through integrative modeling (IM) (23,167,168). Structure determination of protein assemblies is vital for a mechanistic understanding of their function. IM takes experimental input data from structural, biochemical, proteomic, and genetic studies to optimize and compute a comprehensive model of protein complexes with properties that satisfy information within the uncertainty of the data (23,167,169,170). The primary sources of input information for IM are XRC, NMR spectroscopy or comparative modeling, XL-MS, H/DX-MS, stoichiometric data from nMS, solution scattering data from small-angle X-ray scattering (SAXS), and protein–protein interactions from AP-MS. Structures of several protein complexes have been derived using integrative methods to explain their architecture and evolutionary principles of large assemblies (167,171–173), rationalize the effect of disease mutations (167,174), and derive structural heterogeneity of flexible protein complexes (170,175,176).
Gutierrez et al. applied the IM approach to investigate in-solution architecture of CSN9-dependent structural changes and conformational dynamics of the COP9 signalosome (CSN) complex (170). Based on XL-MS data, XRC structures, and comparative models, they proposed that CSN9 binding triggers the CSN complex to adopt a configuration that facilitates CSN–CRL interactions, thereby enhancing CSN deneddylase activity. Most importantly, they observed additional conformations and configurations of CSN in solution that were absent in the static structure.
TFIIH is a conserved 10-subunit complex that plays a critical role in both RNA polymerase II transcription (174,177) and DNA repair (178). Mutations in TFIIH subunits lead to different cancers (179,180) and autosomal recessive disorders, such as xeroderma pigmentosum (XP), and trichothiodystrophy (TTD) (178). EM studies derived structural information of yeast and human TFIIH (181–184). By adopting an integrative approach using data from XL-MS, biochemical analyses, and previously reported EM maps, Luo et al. determined the molecular architecture of both human and yeast TFIIH (174). They identified four new conserved regions that function as hubs for TFIIH assembly and >35 conserved topological features within TFIIH, illuminating a network of interactions involved in TFIIH assembly and regulation of its activities.
FUTURE PERSPECTIVES
Development and application of myriads of tools over the past few years have helped in shaping our understanding of how protein complexes, PTM signaling, PPIs and protein structure/function regulate the DDR. This opens up an arena of DDR research in the future that can be decoded with integrated multidisciplinary approaches to gain a deeper mechanistic understanding of these genome maintenance processes both in vitro and in vivo.
There are several areas of mass spectrometry that are currently transforming the field. One is single-cell proteomics, which is a relatively new field focusing on analyzing the proteome of individual cells. This technique can provide important insights into cellular heterogeneity in signaling responses (185,186), which could reveal cell-to-cell variability in DDR.
Ion mobility mass spectrometry (IM-MS) is another powerful technique that is used to further separate ions based on their collisional cross section, affected by their size, shape, and charge (187). IM-MS is particularly useful for deconvolving complex mixtures where traditional mass spectrometry may struggle to differentiate between closely related species (188). Furthermore, IM-MS can be used in combination with other mass spectrometry-based techniques, such as native MS and top-down proteomics, to obtain structural information about proteins and protein complexes (189,190).
Additionally, data independent acquisition (DIA) methods such as SWATH and diaPASEF allow for the simultaneous quantification of thousands of proteins in complex mixtures (191–194). DIA methods are particularly useful for large-scale proteomics studies, where traditional DDA methods may be limited by their dynamic range and sensitivity. SWATH and diaPASEF are both highly reproducible and can provide accurate and precise quantitative measurements, making them valuable tools for proteome profiling.
Although further innovation and addressing limitations in existing technologies remain crucial, we are about to unravel the complexity of regulatory mechanisms in DNA repair at an extraordinary level of detail and at a rapid pace.
DATA AVAILABILITY
No new data were generated or analysed in support of this research.
ACKNOWLEDGEMENTS
We would like to thank members of the Krogan lab for critical reading of the manuscript. Figure and graphical abstract were created with Biorender.com.
FUNDING
National Institutes of Health [U54CA274502, P50AI150476, U19AI135990 to N.J.K.]; Martha and Bruce Atwater Breast Cancer Research Fund; Benioff Initiative for Prostate Cancer Research; UTHSCSA Startup Research Fund (to M.K.); F. Hoffmann-La Roche and Vir Biotechnology and gifts from The Ron Conway Family (to N.J.K.).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
Comments