




Abstract
The exonuclease domain of DNA polymerases epsilon's catalytic subunit (POLE) removes misincorporated nucleotides, called proofreading. POLE-exonuclease mutations cause colorectal- and endometrial cancers with an extreme burden of single nucleotide substitutions. We recently reported that particularly the hereditary POLE exonuclease mutation N363K predisposes in addition to aggressive giant cell glioblastomas. We knocked-in this mutation homozygously into human cell lines and compared its properties to knock-ins of the likewise hereditary POLE L424V mutation and to a complete proofreading-inactivating mutation (exo-null). We found that N363K cells have higher mutation rates as both L424V- or exo-null mutant cells. In contrast to L424V cells, N363K cells expose a growth defect, replication stress and DNA damage. In non-transformed cells, these burdens lead to aneuploidy but macroscopically normal nuclei. In contrast, transformed N363K cells phenocopy the enlarged and disorganized nuclei of giant cell glioblastomas. Taken together, our data characterize a POLE exonuclease domain mutant that not only causes single nucleotide hypermutation, but in addition DNA damage and chromosome instability, leading to an extended tumor spectrum. Our results expand the understanding of the polymerase exonuclease domain and suggest that an assessment of both the mutational potential and the genetic instability might refine classification and treatment of POLE-mutated tumors.
INTRODUCTION
Hypermutated cancers have an extreme mutational burden of 10 to over 100 single nucleotide substitutions per Mb (1). The majority of these cancers are caused by the inactivation of the mismatch repair system (MMR) that removes misincorporated nucleotides post-replicative. MMR deficiency destabilizes particularly microsatellite sequences and thus these cancers are denominated microsatellite instable (MSI). The Lynch-syndrome comprises probably the best characterized group of such cancers (for review, (2)). MMR deficiency causes mainly colon- and endometrial cancers. A second, even higher mutated group of cancers was identified more recently, caused by mutations in the exonuclease-domain of the catalytic active subunit (POLE) of polymerase epsilon (Polϵ) and –delta (POLD1, Polδ respectively), compromising their proofreading activity (1) (for review (3)). Polϵ and Polδ are the two polymerases that replicate the bulk of the genome in each S-phase. Whereas Polδ is thought to replicate primarily the lagging strand, Polϵ is the polymerase of the leading strand, replicating up to 100 and more kb of DNA without pausing or disassembly from its template (for review (4,5)).
The striking fidelity of POLE and POLD1 towards the DNA template is firstly achieved by a very high nucleotide selectivity, incorporating a non-matching nucleotide only every 105–106 nucleotides. Secondly, both polymerases can proofread the newly incorporated nucleotide. If a mismatch is detected, the polymerase hands the newly synthesized strand containing the mismatch over to its exonuclease domain, which removes the faulty nucleotide. Only after mismatch removal, the nascent strand is then handed back to the polymerase domain, resuming replication. This proofreading activity increases replication fidelity by a factor of hundred, boosting polymerase fidelity to only one mismatched nucleotide for every ten million correctly replicated nucleotides. As mentioned above, the MMR system finally decreases this error-rate even further, remarkably down to only one misincorporated nucleotide for every 109–1010 correct nucleotides, which is approximately only one faulty incorporated nucleotide per replication of the whole genome (4) (for review (6)).
All so far characterized POLE/D exonuclease domain mutants were reported to increase exclusively the single nucleotide substitution rate, similarly as MMR defects do. Thus, the tumor spectrum of both MMR defects and polymerase proofreading mutations are alike, giving rise mainly to colon- and endometrial cancers. However, polymerase proofreading mutated cancers do not show microsatellite instability and thus are, in contrast to MMR deficient cancers, labeled microsatellite stable (MSS).
In the past, for the ease of their functional characterization and justified by the high conservation of POLE and POLD1, most of the exonuclease domain mutations found in human cancers were modeled and studied in yeast. Such studies permitted not only to estimate the mutagenic potential of a given cancer mutation, but also to directly compare it with the mutagenicity of a designed mutant without any residual exonuclease activity (exo-null) (7). Strikingly, in yeast, several mutations homologous to cancer-associated mutations show a much higher mutagenic potential than the blunt exo-null mutant. Furthermore, such highly mutagenic mutations are found with a much higher frequency in cancers than less mutagenic mutants. Particularly, the most frequently found cancer associated POLE mutation, P286R, modelled in yeast (P301R), was shown to have a mutagenic potential around fifty times higher than the (yeast) exo-null D275A/E277A mutation (8). These facts suggested that defects in the exonuclease activity of at least some mutants could not explain their whole mutagenic potential (3,8,9).
Upgrading the model organisms, Albertson et al. introduced and characterized in 2009 an exo-null mutant of POLE in mice. Homozygous mutant mice showed a strongly reduced lifespan due to the development of mainly intestinal tumors (10). Finally, the aforementioned P286R exonuclease domain mutation was introduced in mice by Li et al. (11). Heterozygous P286R mutated mice were strongly cancer prone, and mouse embryonic fibroblasts (MEFs) showed a massively increased mutation rate of calculated 1.6 nucleotide substitutions per Mb and cell cycle, around thousand times higher than the wildtype mutation rate. As expected for POLE proofreading deficiency, the authors found basically exclusively single nucleotide substitutions in cultured cells and tumors of these mice. Concomitantly, the authors did not detect DNA damage or signs of DNA breaks in the P286R mutant. Further in depth biochemical and structural work of other groups showed that the P286R mutation hinders entry of the nascent DNA strand into the exonuclease active site and generates not only a proofreading compromised enzyme, but a hyperactive polymerase domain, which did not any more discriminate properly between mismatched termini or secondary structures that would slow down or pause wildtype POLE (12,13).
From a clinical perspective, tumors with somatic POLE (or POLD1) exonuclease domain mutation have a rather good prognosis. Due to their hypermutated nature, these tumors expose a high number of neoantigens and are therefore generally good responders to immune checkpoint therapy (14–16). However, prognosis is fundamentally different for patients with germline POLE exonuclease mutations. Even though only a small percentage of exonuclease mutations are present in the germline, such families and patients suffer from young age on from repeatedly occurring colon- and endometrial tumors, a syndrome denominated polymerase proofreading-associated polyposis syndrome (PPAP) (17). Most PPAP families carry the POLE c.1270C > G; p.(Leu424Val) mutation (hereafter L424V), which entails like other proofreading mutations mainly a predisposition to colon- and endometrial cancers (18). Rohlin et al. reported a family carrying a so far unknown POLE c.1089C > A; p.(Asn363Lys) mutation (hereafter N363K), which—besides colon and endometrial cancers—predisposes to extra-intestinal tumors, mainly ovarian- and brain tumors. Of the two reported brain tumors, one was a glioblastoma of the rare giant cell glioblastoma type at age 28 (19).
In 2019, we reported the second family carrying this N363K mutation with at the time three N363K carrier patients presenting with glioblastoma (20). Since this publication in 2019, the fourth patient with glioblastoma in this family was verified as a N363K carrier (IV.19 in Vande Perre et al. 2019). Furthermore, in the meantime, a new case of glioblastoma was diagnosed in yet another N363K carrier of this family at age 33 (IV.6 in Vande Perre et al. 2019), thus becoming the fifth case of glioblastoma with the N363K mutation in this family (sixth in the literature). Thus, there is a complete genotype-phenotype correlation between N363K and glioblastoma in this family even though some of the carriers are distant up to the fifth degree of kinship (reducing the probability for an interfering unknown genetic factor). Mean age at diagnosis is early (37.5 years age, combining the six cases from the two families, compared to 65 years of general glioblastoma (21)). As a reminder, the incidence of glioblastoma is only around 3–4/100 000 inhabitants in Europe.
Even more intriguing, all these N363K glioblastomas belonged to the rare giant-cell subtype, which represents only 2–5% of all glioblastomas (19–22). Gliomas are cancers with very high, if not the highest, level of DNA damage and chromosome instability seen in cancers (23,24). Giant cell glioblastomas are characterized by cells with bizarrely enlarged nuclei or multiple nuclei per cell and obvious chromosomal instability and aneuploidy and such cells show a particular high DNA damage (25).
Due to these facts, we hypothesized that the N363K mutation not only entails single nucleotide substitutions but might cause in addition replication stress and concomitant DNA damage. We used CRISPR/Cas9 to knock-in into different cell lines the point mutations to generate an exo-null-, the PPAP L424V (whose tumor spectrum is poorly associated with glioblastoma) and the PPAP N363K mutation in the POLE locus. We first assessed their mutational potential by whole genome sequencing and then characterized their growth properties and quantified DNA damage and chromosomal instability. Our data show that the N363K mutation constitutes a new class of cancer-associated POLE exonuclease mutants whose mutagenic potential not only exceeds the exo-null and other exonuclease mutations, but causes in addition DNA damage, chromosomal instability, and aneuploidy. Strikingly, we found that the single POLE N363K mutation is sufficient to induce in transformed cells the specific giant cell nuclear phenotype of giant cell glioblastomas. These findings question the paradigm that mutations in the exonuclease-domain of POLE (and likely in POLD1, too) do not cause DNA damage and do not obstruct the processivity of the polymerase function. From a clinical point of view, our data suggest that cancer-associated POLE (and POLD1) exonuclease mutants need to be scrutinized not only for their SNV frequency, but also for DNA damage and the genetic instability they cause. Such a refined classification might be necessary for the design of adapted therapies relying either primarily on the existence of neoantigens or considering an elevated amount of replication stress, chromosome breaks and aneuploidy, features more susceptible to classic DNA damaging chemotherapy.
MATERIALS AND METHODS
Cell culture
hTERT RPE-1 and HCT-116 cell lines (ATCC CRL-4000 and CCL-247 respectively) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (A3160402, Thermo Scientific) and antibiotic antimycotic solution (A5955, Merck) under standard conditions. To quantify long term cellular growth, cells were counted at each passage (every three days) and each time re-seeded at a fixed density. Cumulative cell count was then calculated for each cell line and plotted. To generate clonal populations, cells were resuspended with trypsin in complete medium, centrifuged and resuspended at a concentration of 5–10 million cells in 0.5 ml of PBS supplemented with 5% FBS. Finally, single cells were sorted in 96 well plates using a FACS Melody™ Cell Sorter (BD Biosciences) and subsequently amplified until having enough biological material for further analysis. Brightfield photos were generated with a ZOE Fluorescent Cell Imager (BioRad). To silence MLH1, cells were transfected with smart pool siRNA (Dharmacon) directed against MLH1 (L-003906-00-0005) or a non-targeting pool (D-001810-10-05) as control using Lipofectamin™ RNAiMax (13778075, Thermo Fisher) in six-well plates.
POLE genome editing
To generate POLE mutated cell lines (Knock-In), double strand break (DSB) at loci of interest were introduced using the programmable nuclease system CRISPR/Cas9 pre-assembled in ribonucleoprotein complexes (RNP). To this end, recombinant Alt-R® S.p. Cas9 Nuclease V3 (1081059, IDT) and custom in vitro transcribed single guide RNAs (sgRNA, IDT) were mixed following manufacturer's instructions. 200 000 RPE-1 or HCT-116 cells were nucleofected with these preassembled RNP complexes using Amaxa4D 20μl Nucleocuvette™ Strips (Lonza, kit SE using protocol CM-150 and kit P3 using protocol EA-104, respectively) with or without (as control) single stranded oligonucleotide donor (ssODN) as homologous directed reparation (HDR) matrix. ssODN were ordered as PAGE purified ultrameres (IDT), with two thiosulfate modified bonds at each extremity (to increase stability), containing the mutation(s) of interest and in addition a PAM mutation to avoid Cas9 re-cutting after repair. Detailed sequences of sgRNA and ssODN donor sequences are listed below. To avoid NHEJ-mediated unintended repair events of CRISPR/Cas9 induced DSB (e.g. insertions/deletions) cells were pre-treated 4 h before RNP nucleofection with the DNA-PK inhibitor Nu7441 (R&D systems-bio-techne – #3712) at a final concentration of 2 μM for a total of 24 h. Additionally, to remove unedited cells, an additional step of scarless co-selection editing strategy was performed as described previously (26). Briefly, during the POLE editing process, a point mutation in the ATP1A1 gene locus was introduced using the same RNP/ssODN strategy as described above. This allowed the selection of edited cells by exposure to 0.5 μM Ouabaïn for 48 h post-nucleofection. Finally, after one week of selection, the obtained edited populations were pooled, splitted and used for further characterization or freezing. Primers used for PCR amplification of the edited loci and their SANGER sequencing are listed below. Except for NGS and CGH, all experiments were performed using the obtained pools of edited cells.
sgRNA sequences (20nt)
| Targeted Locus | gRNA protospacer sequence |
|---|---|
| ATP1A1 – Intron 04 | 5′-GAGUUCUGUAAUUCAGCAUA-3′ |
| POLE – Exon 09 (Exo-null) | 5′-UGUCAAAUGCCAAAACCACA-3′ |
| POLE – Exon 11 (N363K) | 5′-CAUCAUGGUCACCUACAAGG-3′ |
| POLE – Exon 13 (L424V) | 5′-GGGCAGUCAUAAUCUCAAGG-3′ |
| Targeted Locus | gRNA protospacer sequence |
|---|---|
| ATP1A1 – Intron 04 | 5′-GAGUUCUGUAAUUCAGCAUA-3′ |
| POLE – Exon 09 (Exo-null) | 5′-UGUCAAAUGCCAAAACCACA-3′ |
| POLE – Exon 11 (N363K) | 5′-CAUCAUGGUCACCUACAAGG-3′ |
| POLE – Exon 13 (L424V) | 5′-GGGCAGUCAUAAUCUCAAGG-3′ |
sgRNA sequences (20nt)
| Targeted Locus | gRNA protospacer sequence |
|---|---|
| ATP1A1 – Intron 04 | 5′-GAGUUCUGUAAUUCAGCAUA-3′ |
| POLE – Exon 09 (Exo-null) | 5′-UGUCAAAUGCCAAAACCACA-3′ |
| POLE – Exon 11 (N363K) | 5′-CAUCAUGGUCACCUACAAGG-3′ |
| POLE – Exon 13 (L424V) | 5′-GGGCAGUCAUAAUCUCAAGG-3′ |
| Targeted Locus | gRNA protospacer sequence |
|---|---|
| ATP1A1 – Intron 04 | 5′-GAGUUCUGUAAUUCAGCAUA-3′ |
| POLE – Exon 09 (Exo-null) | 5′-UGUCAAAUGCCAAAACCACA-3′ |
| POLE – Exon 11 (N363K) | 5′-CAUCAUGGUCACCUACAAGG-3′ |
| POLE – Exon 13 (L424V) | 5′-GGGCAGUCAUAAUCUCAAGG-3′ |
ssODN sequences (120nt)
| Targeted locus | ssODN |
|---|---|
| ATP1A1 - Intron 04 | 5′-AAGACGGAGAAGATATCTGATGTGTACTACGAATTCATAT*GCTGAATTACAGAACTCACGTCATCGTTTTGAGGTTCCTCTTCTGTAGCAGCTCTGATGCTATAAGCCAAGAAACAAAGA-3′ |
| POLE - Exon 9 (Exo-null) | 5′-GTTTAGAGCTTGGCTTTATGCTTATTTTGTCCCCACAGGATCCTGT*GGTTTTGGCATTTGCCATTGCTACGACCAAACTGCCCCTCAAGTTTCCTGATGCTGAGACAGACCAGATTATGA-3′ |
| POLE - Exon 11 (N363K) | 5′-CACCACGCAACGCCCTCCCTCTCAAATGCTGCCCAGTTACTCATAGAGAAGACACAGACTCACCAGTCAAAAAAGTCGCCTT*TGTAGGTGACCATGATGGTGGGTTTGGTCTCCTGGAC-3′ |
| POLE - Exon 13 (L424V) | 5′-CTGCTCCGTGGCCATCCGGCACATGTCCTCCGGGTCTAGCTCCACGGGATCATAGCCTAGCTTGGCCTTGGCGGCAGCCT*TGACATTATGACTGCCCACAGGAAGGTAACTGTCCCTCTT-3′ |
| Targeted locus | ssODN |
|---|---|
| ATP1A1 - Intron 04 | 5′-AAGACGGAGAAGATATCTGATGTGTACTACGAATTCATAT*GCTGAATTACAGAACTCACGTCATCGTTTTGAGGTTCCTCTTCTGTAGCAGCTCTGATGCTATAAGCCAAGAAACAAAGA-3′ |
| POLE - Exon 9 (Exo-null) | 5′-GTTTAGAGCTTGGCTTTATGCTTATTTTGTCCCCACAGGATCCTGT*GGTTTTGGCATTTGCCATTGCTACGACCAAACTGCCCCTCAAGTTTCCTGATGCTGAGACAGACCAGATTATGA-3′ |
| POLE - Exon 11 (N363K) | 5′-CACCACGCAACGCCCTCCCTCTCAAATGCTGCCCAGTTACTCATAGAGAAGACACAGACTCACCAGTCAAAAAAGTCGCCTT*TGTAGGTGACCATGATGGTGGGTTTGGTCTCCTGGAC-3′ |
| POLE - Exon 13 (L424V) | 5′-CTGCTCCGTGGCCATCCGGCACATGTCCTCCGGGTCTAGCTCCACGGGATCATAGCCTAGCTTGGCCTTGGCGGCAGCCT*TGACATTATGACTGCCCACAGGAAGGTAACTGTCCCTCTT-3′ |
Underligned: PAM mutation, Bold: Knock In, *predicted site of DSB in the genome.
ssODN sequences (120nt)
| Targeted locus | ssODN |
|---|---|
| ATP1A1 - Intron 04 | 5′-AAGACGGAGAAGATATCTGATGTGTACTACGAATTCATAT*GCTGAATTACAGAACTCACGTCATCGTTTTGAGGTTCCTCTTCTGTAGCAGCTCTGATGCTATAAGCCAAGAAACAAAGA-3′ |
| POLE - Exon 9 (Exo-null) | 5′-GTTTAGAGCTTGGCTTTATGCTTATTTTGTCCCCACAGGATCCTGT*GGTTTTGGCATTTGCCATTGCTACGACCAAACTGCCCCTCAAGTTTCCTGATGCTGAGACAGACCAGATTATGA-3′ |
| POLE - Exon 11 (N363K) | 5′-CACCACGCAACGCCCTCCCTCTCAAATGCTGCCCAGTTACTCATAGAGAAGACACAGACTCACCAGTCAAAAAAGTCGCCTT*TGTAGGTGACCATGATGGTGGGTTTGGTCTCCTGGAC-3′ |
| POLE - Exon 13 (L424V) | 5′-CTGCTCCGTGGCCATCCGGCACATGTCCTCCGGGTCTAGCTCCACGGGATCATAGCCTAGCTTGGCCTTGGCGGCAGCCT*TGACATTATGACTGCCCACAGGAAGGTAACTGTCCCTCTT-3′ |
| Targeted locus | ssODN |
|---|---|
| ATP1A1 - Intron 04 | 5′-AAGACGGAGAAGATATCTGATGTGTACTACGAATTCATAT*GCTGAATTACAGAACTCACGTCATCGTTTTGAGGTTCCTCTTCTGTAGCAGCTCTGATGCTATAAGCCAAGAAACAAAGA-3′ |
| POLE - Exon 9 (Exo-null) | 5′-GTTTAGAGCTTGGCTTTATGCTTATTTTGTCCCCACAGGATCCTGT*GGTTTTGGCATTTGCCATTGCTACGACCAAACTGCCCCTCAAGTTTCCTGATGCTGAGACAGACCAGATTATGA-3′ |
| POLE - Exon 11 (N363K) | 5′-CACCACGCAACGCCCTCCCTCTCAAATGCTGCCCAGTTACTCATAGAGAAGACACAGACTCACCAGTCAAAAAAGTCGCCTT*TGTAGGTGACCATGATGGTGGGTTTGGTCTCCTGGAC-3′ |
| POLE - Exon 13 (L424V) | 5′-CTGCTCCGTGGCCATCCGGCACATGTCCTCCGGGTCTAGCTCCACGGGATCATAGCCTAGCTTGGCCTTGGCGGCAGCCT*TGACATTATGACTGCCCACAGGAAGGTAACTGTCCCTCTT-3′ |
Underligned: PAM mutation, Bold: Knock In, *predicted site of DSB in the genome.
Genomic PCR primers
| Targeted locus | PCR primer | Product |
|---|---|---|
| ATP1A1 – Intron 04 - Fwd | 5′-TTGCAACCGTCCAGCTACC-3′ | 598 bp |
| ATP1A1 – Intron 04 - Rev | 5′-GCCAACATGAATCGCTGTGC-3′ | – |
| POLE – Exon 9 (Exo-null) - Fwd | 5′-CCTGTGATCAGCTGCTCTCTTA-3′ | 626 bp |
| POLE – Exon 9 (Exo-null) - Rev | 5′-AAGCCAAGAAATGGCGTTCC-3′ | – |
| POLE – Exon 11 (N363K) - Fwd | 5′-TGTACCCTCCCCTTTCGAGT-3′ | 843 bp |
| POLE – Exon 11 (N363K) - Rev | 5′-CAGGTCAGGCTCTAATGCCC-3′ | – |
| POLE – Exon 13 (L424V) - Fwd | 5′-GTCGGAGAGGCCACAAACTC-3′ | 609 bp |
| POLE – Exon 13 (L424V) - Rev | 5′-CACAGTAAGGAGACCGGCAC-3′ | - |
| Targeted locus | PCR primer | Product |
|---|---|---|
| ATP1A1 – Intron 04 - Fwd | 5′-TTGCAACCGTCCAGCTACC-3′ | 598 bp |
| ATP1A1 – Intron 04 - Rev | 5′-GCCAACATGAATCGCTGTGC-3′ | – |
| POLE – Exon 9 (Exo-null) - Fwd | 5′-CCTGTGATCAGCTGCTCTCTTA-3′ | 626 bp |
| POLE – Exon 9 (Exo-null) - Rev | 5′-AAGCCAAGAAATGGCGTTCC-3′ | – |
| POLE – Exon 11 (N363K) - Fwd | 5′-TGTACCCTCCCCTTTCGAGT-3′ | 843 bp |
| POLE – Exon 11 (N363K) - Rev | 5′-CAGGTCAGGCTCTAATGCCC-3′ | – |
| POLE – Exon 13 (L424V) - Fwd | 5′-GTCGGAGAGGCCACAAACTC-3′ | 609 bp |
| POLE – Exon 13 (L424V) - Rev | 5′-CACAGTAAGGAGACCGGCAC-3′ | - |
Genomic PCR primers
| Targeted locus | PCR primer | Product |
|---|---|---|
| ATP1A1 – Intron 04 - Fwd | 5′-TTGCAACCGTCCAGCTACC-3′ | 598 bp |
| ATP1A1 – Intron 04 - Rev | 5′-GCCAACATGAATCGCTGTGC-3′ | – |
| POLE – Exon 9 (Exo-null) - Fwd | 5′-CCTGTGATCAGCTGCTCTCTTA-3′ | 626 bp |
| POLE – Exon 9 (Exo-null) - Rev | 5′-AAGCCAAGAAATGGCGTTCC-3′ | – |
| POLE – Exon 11 (N363K) - Fwd | 5′-TGTACCCTCCCCTTTCGAGT-3′ | 843 bp |
| POLE – Exon 11 (N363K) - Rev | 5′-CAGGTCAGGCTCTAATGCCC-3′ | – |
| POLE – Exon 13 (L424V) - Fwd | 5′-GTCGGAGAGGCCACAAACTC-3′ | 609 bp |
| POLE – Exon 13 (L424V) - Rev | 5′-CACAGTAAGGAGACCGGCAC-3′ | - |
| Targeted locus | PCR primer | Product |
|---|---|---|
| ATP1A1 – Intron 04 - Fwd | 5′-TTGCAACCGTCCAGCTACC-3′ | 598 bp |
| ATP1A1 – Intron 04 - Rev | 5′-GCCAACATGAATCGCTGTGC-3′ | – |
| POLE – Exon 9 (Exo-null) - Fwd | 5′-CCTGTGATCAGCTGCTCTCTTA-3′ | 626 bp |
| POLE – Exon 9 (Exo-null) - Rev | 5′-AAGCCAAGAAATGGCGTTCC-3′ | – |
| POLE – Exon 11 (N363K) - Fwd | 5′-TGTACCCTCCCCTTTCGAGT-3′ | 843 bp |
| POLE – Exon 11 (N363K) - Rev | 5′-CAGGTCAGGCTCTAATGCCC-3′ | – |
| POLE – Exon 13 (L424V) - Fwd | 5′-GTCGGAGAGGCCACAAACTC-3′ | 609 bp |
| POLE – Exon 13 (L424V) - Rev | 5′-CACAGTAAGGAGACCGGCAC-3′ | - |
NGS and CGH analysis
For both approaches, genomic DNA of two million cells for each condition was extracted with DNeasy Blood & Tissue Kit (69504, QIAGEN) and dosed with Qubit Fluorometer (Thermo Fisher) for accurate DNA quantification.
For whole genome sequencing (WGS), library preparation and sequencing, the short-insert paired-end libraries for the whole genome sequencing were prepared with PCR free protocol using KAPA HyperPrep kit (Roche) with some modifications. In short, 1.0 μg of genomic DNA was sheared on a Covaris™ LE220-Plus (Covaris). The fragmented DNA was size-selected with AMPure XP beads (Agencourt, Beckman Coulter), end-repaired, adenylated and Illumina platform compatible adaptors with unique dual indexes and unique molecular identifiers (Integrated DNA Technologies) were ligated. The libraries were quality controlled on an Agilent 2100 Bioanalyzer with the DNA 7500 assay (Agilent) for size and quantified by Kapa Library Quantification Kit for Illumina platforms (Roche).
The libraries were sequenced on NovaSeq6000 (Illumina) in paired-end mode with a read length of 2 × 151 bp following the manufacturer's protocol for dual indexing. Image analysis, base calling and quality scoring of the run were processed using the manufacturer's software Real Time Analysis (RTA 3.4.4) and followed by generation of FASTQ sequence files. After sequencing, reads were mapped to the human genome (hs37d5) using the BWA-mem with default parameters (27). Alignment files (BAM format) containing only properly paired, uniquely mapping reads were processed using picard tools version 1.110 (http://broadinstitute.github.io/picard/) to add read groups and remove duplicates. The Genome Analysis Tool Kit was used for base recalibration (28). Processed BAM files were submitted to variant calling for single nucleotide variants and small insertions and deletions using HaplotypeCaller (GATK v4.1.9.0). To allow for a comparable mutation analysis, a common set of callable bases for all the samples was computed. The program CallableLoci from GATK (v3.6) was used with default parameters to compute the bases with coverage higher of 10 reads and quality reads to reliable call genomic variants. The callable bases from all the samples were intersected to yield a total of 2733 callable mega bases. For the analysis, only the variants predicted in these genomic regions were used.
Hard filters to remove potential artefactual variants were applied following the best practices from GATK. Thus, variants with QualByDepth (QD) lower than 2, FisherStrand (FS) lower than 200, RMSMappingQuality (MQ) lower than 40, MappingQualityRankSumTest (MQRS) lower than −12.5, ReadPosRankSumTest (RPRS) lower than −8 or higher than 20, or BaseQRankSum (BQR) lower than -4 or higher than 4, were discarded. Variants with an allele frequency lower than 0.3 were also discarded to avoid contamination or sub-clonal variants. Sigprofiler with default parameters was used to compute the mutational signatures (29).
For comparative genomic hybridization (CGH), genomic DNA samples were processed with CytoScan™ HD Array Kit and Reagent Kit Bundle (901835, Applied Biosystems) following manufacturer's instructions. Microarrays were hybridized by using microfluidic station, scanned using Affymetrix GeneChip Scanner 3000 7G System (00-0213, Applied Biosystems) and analyzed with Chromosome Analysis Suite (ChAS) software. The WGS data are available via SRA under the BioProject number PRJNA896776.
TMB determination and MMR classification of human tumors
Tumor mutation burden (TMB) was evaluated using the TruSightTM Oncology 500 kit (Illumina) and NextSeq 550 sequencer. Briefly, the kit and pipeline consider non-synonymous and synonymous SNVs and indel mutations at an allelic frequency (AF) of 5% limit of detection, followed by a post-processing step to remove artefact, variants in low confidence regions, driver mutations, and germline variants. DNA extraction from FFPE tumor blocks was performed using the MAXWELL 16® FFPE Plus LEV DNA PURIF KIT (Promega). At least 5 ng of DNA was used for sequencing according to manufacturer's protocol. Variants detection was performed with a minimal profounder 150X TSO500 local app version 2.1.0.60.
MMR immunohistochemistry and MSI testing were performed according to standard procedures (30). To evaluate MSI, the Pentaplex-PCR comprising five mononucleotide markers (BAT-25, BAT26, NR-21, NR-22 and NR-24) from the NCI panel was used. For all tumors analyzed by PCR, at least 20% tumor-cellularity was obtained. Tumors were classified as MSI-H when at least two microsatellite markers showed instability, MSI-Low when only one marker showed instability and MSS when none of the markers showed instability. The expression of four mismatch repair (MMR) proteins MLH1, MSH2, MSH6 and PMS2 was evaluated by immunohistochemistry as described before (30).
Drug sensitivity assays
RPE-1 were seeded at 4000 cell/well in 96-well plate with 200 μl of complete medium. After 24 h, medium was replaced by fresh medium complemented with different drugs at varying doses. Cell viability, after indicated times of drug exposure, was quantified using CellTiter AQ one solution MTS reagent (G5421, Promega, Madison, WI) according to manufacturer's instructions and measured using a Mithras plate reader (Berthold).
Immunofluorescence
Cells grown on glass coverslips were pulse-labeled before fixing with 2 μM EdU (5-ethynyl-2′-deoxyuridine, C10340, Thermo Fisher Scientific) in complete medium for 30 min. Cells were then fixed and extracted with 4% paraformaldehyde/PBS containing 0,5% of Triton X-100 at RT for 15 min. After washing with PBS, 0.1% Tween20, cells were saturated with 5% BSA/PBS for 60 min and subsequently EdU detection was performed by click-chemistry according to manufacturer's instructions (C10340, Thermo Fisher Scientific). After washing in PBS/0.1% Tween20, cells were incubated with primary antibodies in PBS/0.1%Tween20 complemented with 1% BSA at 4°C overnight. Anti-ɣ-H2AX antibody (JBW301, Millipore) was used at a dilution of 1:5000 and anti-phospho-DNAPK antibody (AB18192, abcam) was used at a dilution of 1:1000. Cells were then washed 3 times for 10 min in PBS/0.1% Tween20 and incubated with fluorescently labeled secondary antibodies (Alexa 488-Goat anti-rabbit IgG, A-11008 and Alexa 555-Goat anti-mouse IgG, A-21422, Thermo Fisher Scientific) in PBS/0.1%Tween20 complemented with 1% BSA for 1 h. To label cell boundaries (actin cytoskeleton), Alexa555-labeled phalloidin was used according to manufacturer's instructions (A34055, Thermo Fisher Scientific). After three subsequent washes in PBS/0.1% Tween20 as before, DNA was labeled for 20 min with 5 μg/ml Hoechst 33342 (H3570, Thermo Fisher Scientific) in PBS. After a final wash in PBS for 5 min, coverslips were dipped in water and dried before mounting with ProLong Diamond Antifade (P35970, Thermo Fisher Scientific). Micrographs were taken on a Nikon Eclipse Ni microscope with a DS-Qi2 camera. Counting of nuclei of all cells and of EdU + cells and quantification of ɣ-H2AX and phosphorylated DNAPK in nuclei were performed using FIJI and Photoshop CS5.
Western blot
Cells were taken up in SDS sample buffer (2000 cells/ μl), (161–0747, BioRad), migrated on NuPAGE 4–12% Bis–Tris gels (NP0322, Thermo Fisher Scientific) and transferred onto nitrocellulose membranes (10600062, Amersham). Membranes were blocked in 5% milk for 40 min and then incubated with primary antibodies overnight, washed in PBS/Tween 0.1% and then incubated with a secondary HRP-labeled antibody for 1h, washed again and bands were finally visualized using the ECL plus assay (1863031, Thermo Fisher Scientific) and visualized on a Chemidoc imaging system (BioRad).
Fiber stretching
Dual sequential labeling of RPE-1 cells was done by pulse labeling for 25 min with 50 mM of the nucleotide analog IdU, washing twice with warm media, followed by a second pulse labeling with 100 mM CIdU for 25 min. Cells were then trypsinized and pelleted by centrifugation and resuspended in ice cold PBS. Cells were lysed with spreading buffer (200 mM Tris–HCl pH 7.5/50 mM EDTA/0.5% SDS), and the released DNA fibers were stretched on silanized microscopy slides and then fixed with methanol/acetic acid (3:1) for 15 min at 20°C. This was followed by denaturation at room temperature with 2.5 M HCl for 1 h, then blocking for 1 h with PBS/1% BSA/0.1% Tween20. ldU and CIdU were immunodetected with an anti-BrdU (347580, Becton Dickinson) and the rat anti-CIdU (C117-7513, Abcys) primary antibodies, and with the anti-mouse Alexa 488 (A11029) and anti-rat Alexa 555 (A21094) secondary antibodies (Invitrogen) respectively. The whole fibers were marked with a single-stranded DNA antibody (MAB3034, Merck Millipore) and the secondary Alexa Fluor 647 goat anti-mouse (A21241, Invitrogen). DNA fibers were analyzed on a Nikon Eclipse Ni microscope with a DS-Qi2 camera. Image analysis was performed by measuring the lengths of the CIdU tracks manually with FIJI software. As fibers were labeled with anti-DNA antibody, only intact fibers were scored.
Chromosome spreading
For each condition, 1 million cells were seeded in 100 mm dishes and treated 24 h later with 0.1μg/ml of colcemid solution KaryoMAX™ (15212012, Thermo Scientific) overnight to block cells in metaphase. Cells were then resuspended in 1 ml PBS and incubated 20 min at 37°C with an additional 10 ml of prewarmed 75 mM KCl solution. After incubation, 600 μl of fixation solution (methanol : acetic acid at 3:1 ratio cooled at −20°C) were added to the cells. The cells were then centrifuged for 10 min at 200g at 4°C to remove supernatant until 1ml final. To fix the cells, fixation solution was added drop by drop under gentle vortexing to 5ml then more rapidly until 10 ml final. After that, cells were washed by centrifugation/resuspension two times with 10ml of fixation solution. Drops of fixed cell solution were then spread on ethanol cleaned glass slides, dried, and imaged with inverted microscope (20× objective).
RESULTS
To compare the phenotypes of different POLE exonuclease mutants in a strictly isogenic human background, we chose to introduce the respective mutations as knock-ins into the endogenous POLE gene using CRISPR/Cas9-based techniques. We opted first for RPE-1 cells, which are well characterized non-transformed and stable diploid epithelial cells, immortalized with an exogenic copy of human telomerase. Furthermore, these cells have wildtype copies of p53 and are mismatch repair proficient (microsatellite stable, MSS) and thus resemble as far as possible normal human cells, without showing features of cancer cells.
To introduce specific POLE mutations by CRISPR homologous directed repair (HDR), RPE cells were cotransfected with two couples of sgRNA-Cas9 RNP complexes and their associated ssODN donor templates (scheme in Figure 1A). The first RNP/ssODN couple aimed at introducing the desired POLE mutation at its endogenous locus, while in parallel the second couple targeted the ATP1A1 gene, introducing two point mutations in this essential K+/Na+ pump, rendering successfully CRISPRed cells resistant to the drug Ouabain (26) (for details, see Materials and Methods). To suppress repair of the Cas9- introduced DNA double strand breaks (DSBs) by non-homologous end joining (NHEJ) and to enhance repair via error-free homologous recombination, we exposed the cells 4 h before and directly after transfection to the DNA-PK inhibitor Nu7441 (31). 48h after transfection cells were then exposed to Ouabain to select for edited cells. After one week, obtained clones were pooled and the POLE locus around the desired mutations sequenced. By adjusting the amounts and transfection conditions of donor ssODN and RNP complexes, we managed to obtain edited cell pools harboring either the (likewise edited) wild-type control allele or the mutant L424V, N363K and exo-null alleles (D275A/E277A, (32) to apparently 100% without detectable NHEJ repair by-products as verified by SANGER sequencing (Figure 1A, right panel).
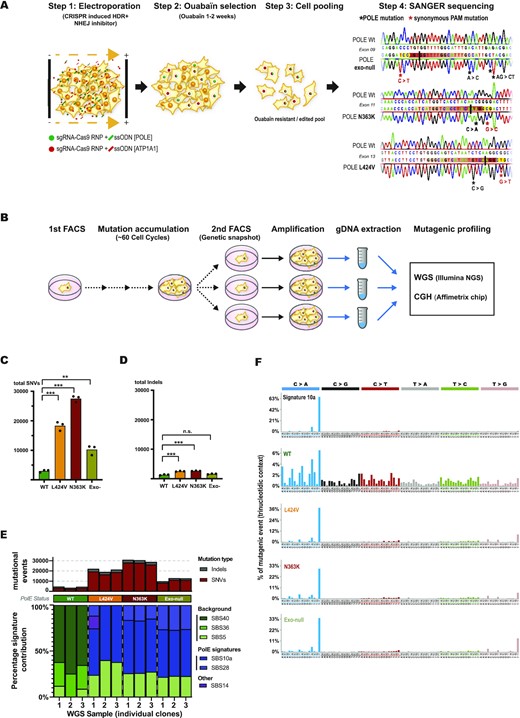
CRISPR-mediated knock-ins of POLE exonuclease mutations in RPE-1 cells show different levels of SNVs but recapitulate all the mutational signature of POLE-mutated tumors. (A) Scheme of the applied CRISPR-mediated gene editing strategy (for details see text) and verification of edited sequences by Sanger-sequencing. (B) Scheme of long-term clonal expansion, gDNA preparation and subsequent WGS analysis of edited cell clones. (C, D) Graphs showing the amount of accumulated SNVs per genome (C) and accumulated indels per genome (D) for each time three independent clones of edited wildtype (green), L424V (orange), N363K (red) and exo-null (asparagus) cells. SNV statistics: WT versus L: P< 0.0001, WT versus N: P< 0.0001, WT versus exo-null: P= 0.0018, N versus L: P= 0.0010, N versus exo-null: P< 0.0001, L versus exo-null: P= 0.0033. Indel statistics: WT versus L: P= 0.0005, WT versus N: P= 0.0002, WT versus exo-null: n.s., L versus N: n.s., L versus exo-null: P= 0.0008, N versus exo-null: P= 0.0003. (E) Quantification of mutational events for each edited wildtype-, L424V-, N363K- and exo-null RPE-1 cell clone (upper panel) with their corresponding trinucleotidic mutational signature contribution determined by Sigprofiler algorithm (lower panel). (F) Mutational signature profiles according to Alexandrov et al. (2020) of tumor signature 10a (upper graph) and below the WGS generated profiles of edited wildtype-, L424V-, N363K- and exo-null RPE-1 cell clones.
Even though in POLE mutated cancers the founding POLE mutation is generally present in a heterozygous state, two considerations convinced us to characterize the different mutants in a homozygous set-up. Firstly, we reasoned that before a developing cancer becomes symptomatic, a vast amount of cell cycles and S-phases must pass, in which additional mutations will accumulate. This number of S-phases cannot readily be simulated in cell culture, even by culturing the cells for months. Therefore, having both alleles mutated might accelerate the manifestation of a mutator phenotype and of its cell biological effects. Secondly, in heterozygous POLE mutated cells, an adaptive silencing of the mutant allele during long-term culture was recently described for several POLE exonuclease mutations (33), which we had to avoid by working with homozygous mutations.
To validate our CRISPR model, we first kept the edited RPE-1 cell pools on exponential growth for at least 3 weeks and then sequenced them a second time to rule out that minor initial contaminations of not edited cells might outgrow the mutant population during our experimental settings (Supplementary Figure S1A). We also verified that the cellular protein amount of the POLE mutants did not differ importantly from the wildtype situation (Supplementary Figure S1B).
To assess the mutational potential of the POLE mutants, we sorted single cells of all RPE-1 POLE edited pools and the equally edited POLE wildtype controls by FACS to establish several clonal populations that we let proliferate and expand for two months (around sixty cell cycles). We then single cell sorted again and expanded the subclones once more until obtaining enough cells for gDNA preparation suitable for WGS analyses. Every single cell of this second amplification step contained thus a genetic ‘screenshot’ of all the mutations acquired during the first two months of growth by its founder cell with an allele frequency of 50% (heterozygous) or 100% (homozygous), (scheme in Figure 1B). WGS analysis of three subclones for each mutant (and the edited wildtype control) revealed a remarkable increase in mutations accumulated in the POLE mutants. Whereas L424V cells had acquired around six times more SNVs than the wildtype, the N363K clones had accumulated around nine times more SNVs.
Since no POLE N363K mutated tumors were yet sequenced and to verify that our findings reflected in vivo situations, we determined tumor mutation burdens (TMBs) of POLE N363K mutated glioblastomas, colorectal- and duodenal cancers and verified also their giant-cell- and hypermutated state (Supplementary Table S1 and Supplementary Figure S1C). Finally, the exo-null mutant had acquired only three and a half times more SNVs than the wildtype (Figure 1C). In contrast and as expected for proofreading mutants, the amount of indels did not vary significantly between the N363K and L424V mutants and had increased only by a factor of two compared to wildtype cells (Figure 1D). Expressed in mut/Mb per single cell cycle, the wildtype clones accumulated 0.02 mut/Mb, the L424V clones 0.12 mut/Mb, the N363K clones 0.18 mut/Mb and the exo-null clones 0.07 mut/Mb per cell cycle. In this counting scheme, similar mutations found in more than one wildtype clone were not counted in any sample since these mutations were most likely already present in the parental RPE-1 cells before gene editing. To assure that this approach did not bias towards a higher mutagenicity of the mutants in respect to the wildtype, we added two different counting scenarios in which we either omitted all mutations present in more than one sample (wildtype and mutants) or counted all mutations, regardless of their frequency. Both alternative scenarios reproduced the highest mutagenicity of the N363K mutants, followed by the L424V mutants and a relative low mutagenicity of the exo-null mutants compared to wildtype clones (Supplementary Figure S1D). Furthermore, we verified using the WGS data that no clone had acquired additional mutations in the POLE gene either during CRISPR-editing or long-term culture that could have acted as suppressor-mutants.
In 2013, Alexandrov et al. established signatures of mutational processes in different cancers, taking into account the trinucleotide context of SNVs, and could assign a well-defined mutational profile to specifically colorectal- and uterine cancers containing POLE exonuclease domain mutants (Signature 10, (34)). This signature was defined by TCT→TAT and TCG→TTG mutations. In 2020 Alexandrov et al., refined this classification and split Signature 10 into Signature 10a (mainly TCT→TAT) and 10b (mainly TCG→TTG), (35) to appreciate the different amounts of TCT→TAT and TCG→TTG that different POLE exonuclease domain mutants contribute to the total mutational count (33,36). To further validate the POLE-edited mutants, we used the SigProfiler tools developed from Alexandrov et al. to generate for each mutant clone its mutational signatures. Whereas the mutational profile of the edited wildtype controls reproduced only unspecific background signatures, all the N363K, L424V and the exo-null clones recapitulated the POLE exonuclease-associated Signature 10a and its associated signature 28 (Figure 1E). Figure 1F emphasizes the contribution of the highly specific trinucleotide-mutational contribution of signature 10a in the mutant clones compared to the wildtype clones whereas Supplementary Figure S1E depicts the contribution of all detected signatures for all individual samples. Interestingly, Signature 10b was absent in the mutant clones, as it was absent also for e.g. the POLE exonuclease mutant S459F, using a similar approach as ours by Hodel et al. (33). Furthermore, Hodel and coworkers demonstrated that Signature 10b is linked to an additional defect in mismatch repair. However, our RPE-1 cell lines used here are mismatch repair proficient. These data all together show first that in only around 60 cell cycles, the knocked-in POLE exonuclease mutants increased massively the amount of acquired mutations. Second, they demonstrate that the glioblastoma-predisposing mutant N363K is more mutagenic than the L424V mutant and that on the other hand the blunt exonuclease-null mutant has a significant lower mutagenic potential than both cancer-associated PPAP mutants N363K and L424V. Finally, they confirm that the generated knock-in mutants recapitulate the mutational signatures of cancers with POLE exonuclease mutations.
To follow up with a cell biological characterization of the mutants, we next assessed their proliferation. We realized that the classical PPAP mutant L424V cell pool proliferated similarly to the wildtype control, but that in contrast the PPAP N363K mutant pool showed a marked growth defect. Interestingly, also exo-null mutant cells depicted a growth defect compared to wildtype cells (Figure 2A). One reason for slowing down or even blocking cellular proliferation is DNA damage. A sole defect in proofreading should however not lead to DNA damage, as also MMR deficient cells (which are likewise hypermutated by single nucleotide substitutions) and the cancer associated, extremely mutagenic POLE exonuclease mutant P286R do not show increased DNA damage (12). We quantified the phosphorylation of the histone variant H2AX (γH2AX), a major indicator of DNA double strand breaks (DSBs) and replication stress, in POLE wildtype and POLE mutant cells (Figure 2B). As expected for a mutant affecting only proofreading, L424V cells had despite a six times higher mutational rate (Figure 1C) only slightly elevated γH2AX levels compared to wildtype cells. In contrast, N363K cells showed a much stronger increase in γH2AX signal. Likewise, also the exo-null mutant depicted higher DNA damage (even though its mutational impact is below that of L424V), suggesting that both the N363K mutation and the exo-null mutations (located in the active site of the exonuclease domain) did in fact not only affect exonuclease activity, but somehow caused DNA damage. We also labeled cells with EdU, a nucleotide analogue that is incorporated into DNA during replication, which can subsequently be fluorescently labeled by click-chemistry. This allowed us to quantify DNA damage exclusively in replicating cells, which actively employ mutant Polϵ at the time of analysis. As also shown in Figure 2B, DNA damage was as expected higher in cells in S-phase. Thus, the growth defects of the N363K and exo-null mutants were proportional to their increased levels of DNA damage, but not to their mutational impact (Figure 1C).
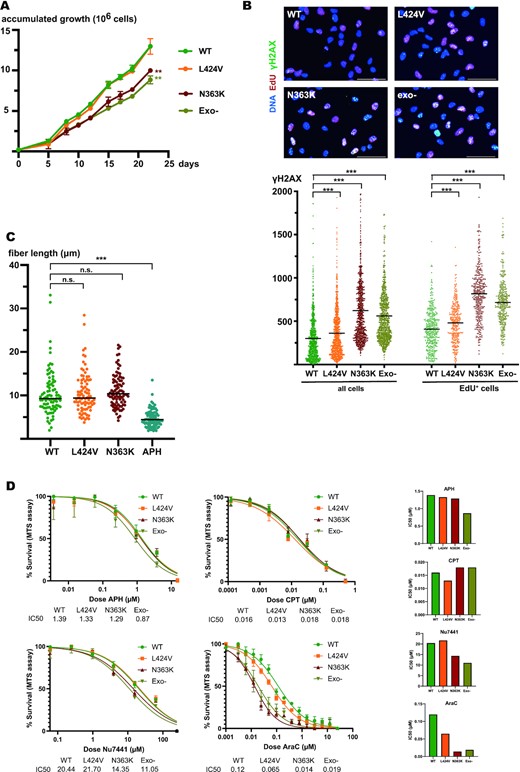
In contrast to the non-glioblastoma predisposing mutant L424V, the N363K- and exo-null mutants depict a growth defect, accumulate DNA damage, and show a higher sensibility to the chain-terminating drug cytarabine. (A) Graph of accumulated growth of edited wildtype (green), L424V (orange), N363K (red), and exo-null (asparagus) RPE-1 cells. WT versus L: n.s., WT versus N: ** P≤0.01, WT versus exo-null: ** P≤0.01. (B) Upper panel: Immunofluorescence images of edited wildtype-, L424V-, N363K- and exo-null RPE-1 cells. Shown is a merge of signals for DNA (blue), EdU (red) and γH2AX (green). Lower panel: Scatter plots showing the quantification of γH2AX in these cells (wildtype in green, L424V in orange, N363K in red and exo-null in asparagus) for all cells (left) and exclusively S-phase cells (EdU+), (right). All differences of γH2AX levels between edited wildtype- and mutant cells are statistically significant (P<0.0001). (C) Scatter plots of CldU tracks of DNA from replicating edited wildtype (green), L424V (orange), N363K (red) and APH-treated edited wildtype (blue) RPE-1 cells. For each condition, 90 fibers were counted. *** P<0.0001. (D) Plots of dose–response survival curves (MTS assay) to determine IC50 (μM) of edited RPE-1 cells treated with aphidicolin (APH), camptothecin (CPT), Nu7441 and cytarabin (AraC). On the right, histograms representing calculated IC50 for each compounds/cell lines.
To be able to compare and to integrate the damage caused by the N363K mutation to classical DNA damage treatments, we exposed in parallel wildtype cells to 1 μM camptothecin (CPT) for 1 h. CPT is an irreversible inhibitor of topoisomerase I and causes DSBs particularly during replication. As expected, DNA damage of CPT-treated cells was much higher in S-phase, showing an approximately seven-fold higher DNA damage as in non-treated cells. In direct comparison, (not treated) N363K cells depicted approximately a two-fold increase of the DNA damage level of replicating wildtype cells (Supplementary Figure S2). Taken these results together, our data showed that particularly the glioblastoma-predisposing N363K mutant (and the generated exo-null mutant) accumulated a substantial amount of DNA damage during S-phase compared to both wildtype and the L424V mutant, which does not predispose to glioblastoma. Noteworthy, compared to a harsh CPT treatment, which increased DNA damage approximately seven fold, the chronic DNA damage level in the N363K mutant was around two times higher as in the wildtype. Considering the long-term viability of the mutant cells and the normal development of germline mutated individuals, this rather modest increase (compared to CPT treatment) seemed reasonable.
Next, we wanted to test if the POLE mutants, and particularly the N363K mutant, somehow compromised the primary polymerase activity of POLE (e.g. through unanticipated larger structural distortions of the enzyme), which could eventually explain the increased DNA damage. Therefore, we quantified the overall speed of replication forks in the different mutants using two-color labeling of replicating DNA followed by DNA fiber stretching. As a control, we added to wildtype cells the reversible polymerase inhibitor aphidicolin (APH) that blocks or slows down replication forks. Whereas in APH treated cells fork speed was drastically reduced as expected, we did not find significantly different fork speeds for all the POLE mutants compared to wildtype controls, suggesting that the primary polymerase activity of POLE was not affected by the different exonuclease-domain mutations (Figure 2C).
We next wanted to assess if the replisomes of the mutant cells were less stable or more prone to disassembly or inactivation during fork blocking or DNA damaging conditions. Therefore, we performed several drug tolerance assays in which either replication forks were slowed down (aphidicolin) or blocked by DNA damaging agents (CPT) or DNA repair was impaired independently of replication (using NU7441, a DNA-PK inhibitor). We did not find any differences in drug tolerance to either aphidicolin or CPT between wildtype and mutants and thus concluded that mutant replisomes did not differ in their stability or capacity of fork restart and DNA repair (Figure 2D, upper panels). We detected a slightly increased sensibility to inhibition of DNA repair via NHEJ particularly in the N363K and the exo-null mutants, likely due to their increased DNA damage baseline as reported above (Figure 2B). However, in contrast and as a confirmation of their specifically impaired exonuclease function, all mutants showed a marked sensibility to cytarabine, a nucleotide analogue that, once incorporated, terminates a nascent DNA strand and whose removal in order to continue replication is dependent on POLE exonuclease activity (37,38). Paralleling their growth defect and increased DNA damage, the N363K and the exo-null mutants displayed an even higher sensibility to cytarabine than the L424V mutant (Figure 2D).
As pointed out above, several lines of evidence suggested that it was not the number of acquired SNVs, which determined if a mutant accumulated DNA damage (e.g the exo-null mutant had a lower mutational impact as the L424V mutation but showed increased DNA damage compared to the wildtype, which the L424V mutant did not). To corroborate this supposition further, we silenced the mismatch repair system through knocking down Mlh1 in wildtype-, N363K- and L424V-cells during several days of culture and quantified then DNA damage (scheme in Figure 3A). As shown in Figure 3B (left panel), we managed to reduce Mlh1 protein levels to undetectable levels for at least six days by two repeated transfections with an siRNA targeted against Mlh1, without detecting any growth difference compared to control-transfected cells. Cells were finally fixed and γH2AX was quantified by immunofluorescence. As expected, silencing mismatch repair and resulting increased SNV levels (4) did not substantially increase DNA damage (Figure 3B, right panel).
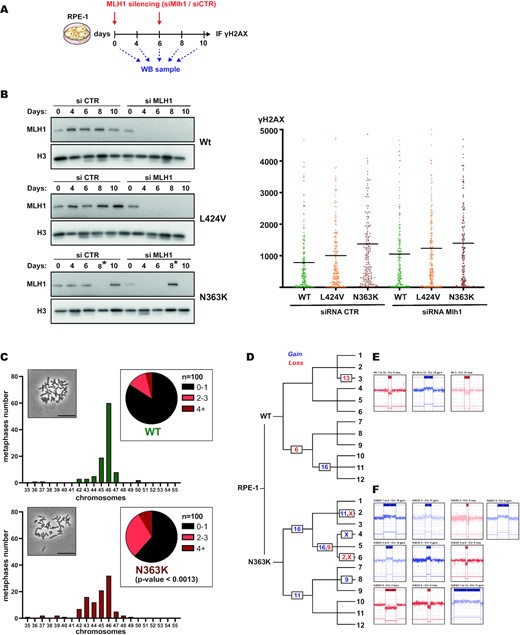
The N363K mutant accumulates DNA damage independently of functional mismatch repair and shows numerical and structural chromosomal aberrations. (A) Scheme of siRNA transfection and cell sampling for long-term silencing of Mlh1. (B) Left panel: Western blot analysis of edited wildtype, L424V and N363K RPE-1 cells during transfection with either control siRNA or siRNA directed against Mlh1. Asterisks mark switched lanes of siCTR and siMLH1 for the 8 days time point during gel loading. Right panel: Scatter plots showing the quantification of γH2AX by immunostaining in these cells (wildtype in green, L424V in orange and N363K in red). (C) Plots and pie charts showing the distribution of chromosome numbers in analyzed metaphases of edited wildtype and N363K RPE-1 cells. An equal number of 100 metaphases per sample were counted. Statistical difference between these distributions was calculated using the Chi-squared test. Inserts: Micrographs of metaphases, scale bar 50 μm. (D–F) Diagram depicting the lineage of subclones generated from wildtype and N363K edited RPE-1 cells and indicating the chromosomal number of detected aberrations by CGH arrays. Gains are indicated in blue, losses in red. Representative sketches of the genomic regions (generated by ChAS, Thermo Scientific) for each aberration are depicted in (E) for wildtype and (F) for N363K clones.
Taken together, our results so far showed that whereas all POLE mutants were impaired in their proofreading activity (high SNV-specific mutagenic potential and entailing a specific sensitivity to cytarabine), their primary polymerase activity was not affected (similar overall fork speed and no increased sensibility to fork slowing- or blocking agents). However, we demonstrated that particularly the N363K- and the exo-null mutant cells proliferate slower than the wildtype and the L424V mutant, and that they accumulate DNA damage during replication.
To find an explanation for the increased DNA damage in the N363K and exo-null mutant, we wondered if particularly these mutants would lead to fork block and subsequent DNA damage only after engaging in a proofreading cycle. Due to the mutation, such an engagement could (with a certain probability) block the enzyme from resuming DNA polymerization. This defect would not be visible by fiber stretching since a proofreading cycle is statistically engaged around only once for every replicated megabase (6), which is an event too rare to be statistically detectable by fiber stretching. While a formal proof of our idea of mutant POLE enzymes blocked in abortive proofreading cycles is experimentally difficult to achieve, we reasoned that the resulting replication stress and DNA damage would thus not be high enough to block the cell cycle through the activation of the replication checkpoint (statistically one engaged proofreading cycle every Mb per cell cycle, see above). Several recent studies showed that cells with a relative low amount of replication stress and DNA damage eventually continue with damage and stretches of underreplicated DNA into mitosis and do not necessarily fail completely mitosis, but accumulate structurally and numerically chromosomal aberrations, leading to aneuploidy and chromosome translocations (39,40), for review (41).
We prepared metaphase spreads of RPE-1 wildtype and N363K cells and quantified numerical chromosomal aberrations. As shown in Figure 3C, N363K cells displayed an obvious increase in aneuploid metaphases compared to wildtype cells (P < 0.0013). As expected for an outcome of mitotic division with stretches of underreplicated DNA, most metaphases had <46 chromosomes and were missing chromosomes. Next, we subjected wildtype and N363K cells to CGH analyses. We started with single cells being either wildtype or carrying the N363K mutation and created after identical times of cultivation subclones, which we let amplify again. These subclones were split and amplified two more times. A final group of 12 independent clones of each wildtype and N363K were finally subjected to CGH analysis (Figure 3D). During this experiment (as described for the setup of the WGS experiments reported above), both wildtype and N363K cells were cultivated in parallel, under identical conditions and for the same span of time to assure a comparable amount of cell cycles during which chromosomal aberration could have had occurred (since the N363K mutation showed a growth defect compared to wildtype cells as shown above, N363K cells might have passed even fewer cycles during the same given time). Whereas in wildtype clones only three independent chromosomal aberrations detectable by CGH were observed (Figure 3E), we detected ten independent events in N363K (Figure 3F). We deep-sequenced in all these clones the POLE locus to confirm that no clone had acquired additional mutations in the POLE gene itself (Supplementary Table S2). These results demonstrated that indeed the N363K POLE mutation causes chromosomal instability on a numerical as well as on a structural level.
As we reported earlier, particularly the N363K mutation predisposes in vivo to giant cell glioblastoma characterized by cells with bizarrely enlarged, deformed and often multiple nuclei (20). These nuclear defects also suggest severe problems of this mutant in maintaining a normal ploidy. However, we could not detect macroscopic nuclear aberrations in the N363K edited, not transformed RPE-1 cells. Therefore, we next analyzed how the N363K mutation would affect already transformed cells. We chose the human colon cancer cell line HCT-116 since colon cancer predisposition is shared among all POLE exonuclease mutations. We managed to introduce the N363K mutation in HCT116 cells using the same CRISPR/Cas9 approach as described above and managed to obtain homozygous N363K mutated HCT116 cells (unfortunately, we technically failed in introducing the L424V mutant into HCT116 cells). We realized promptly that the growth defect of the N363K HCT116 cells compared to control edited wildtype HCT116 cells was strikingly high (Figure 4A).
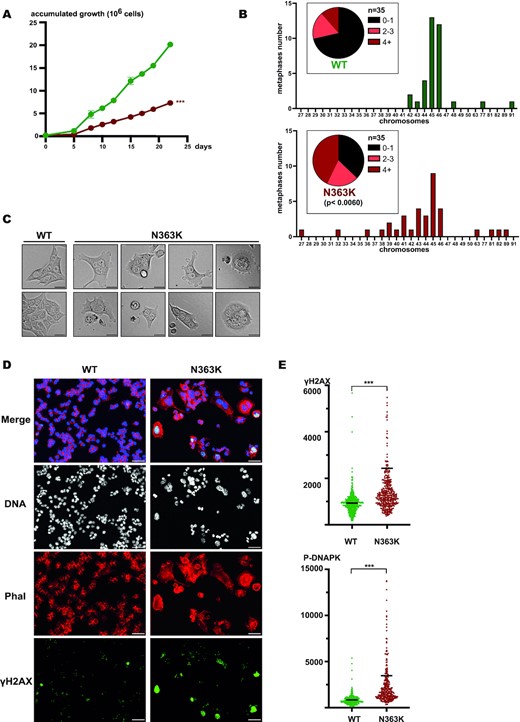
The single point mutation N363K in the POLE gene causes a strong growth defect, severe aneuploidy, deformed and enlarged nuclei and multinucleated cells in HCT116 colon cancer cells. (A) Plot of accumulated growth of edited wildtype (green) and N363K (red) HCT116 cells. *** P≤0.0001. (B) Plots showing the distribution of chromosome numbers in analyzed metaphase spreads of edited wildtype and N363K HCT116 cells. An equal number of 35 metaphases was analyzed. (C) Representative brightfield images of edited wildtype (left) and N363K (right) HCT116 cells. Scale bar 50 μm. (D) Immunofluorescence images of edited wildtype- and N363K HCT116 cells. Shown is a merge of signals for DNA, actin (phalloidin) and γH2AX as well as the separate images for DNA (white), actin (red) and γH2AX (green). Scale bar 50 μm. (E) Scatter plots showing the quantification of γH2AX (upper panel) and phosphorylated DNAPK (lower panel) in edited wildtype- (green) and N363K (red) HCT116 cells. *** P≤0.0001. Bars indicate average of values, to visualize more clearly γH2AX- and phosphorylated DNAPK levels, ordinate range was chosen to spread bulk values and to omit very high scoring N363K cells.
We then prepared metaphase spreads of wildtype and N363K edited HCT116 cells and revealed an even stronger aneuploidy of the N363K mutant as what we had observed in edited RPE-1 cells. Again, chromosomes losses were predominant (Figure 4B).
We analyzed N363K HCT116 cells by phase contrast microscopy and found many dead or dying cells, but most importantly that many cells contained either multiple or heavily enlarged nuclei or even undefinable nuclear masses (Figure 4C (gallery) and Supplementary Figure S3 for an overview micrograph). Of note, we accordingly observed many metaphase spreads with apparently two or even more chromosome sets but could not include these into our quantification shown in Figure 4B since it is impossible to rule pro forma out that these spreads originated from two individual, adjacent mitotic cells.
We next performed immunofluorescence of N363K HCT116 cells to visualize chromatin and to quantify DNA damage. To be able to unequivocally identify enlarged and malformed individual cells and their nuclei, we counterstained in addition the cellular cytoskeleton with fluorescently labeled phalloidin. Whereas wildtype edited HCT116 cells showed equally sized and regular nuclei, which were individually separated by the phalloidin signal, many N363K HCT116 cells were massively enlarged and contained giant- or multiple nuclei. These abnormal nuclei often displayed extremely elevated γH2AX signals (Figure 4D), phenocopying the extremely strong DNA damage present particularly in cells with abnormal and enlarged giant nuclei in sections of giant cell glioblastomas (25). A quantification of γH2AX in the whole cell population likewise revealed a strong increase of DNA damage in HCT116 N363K cells compared to wildtype edited HCT116 cells (Figure 4E, upper panel). Likewise, phosphorylated DNAPK, a marker for DNA double strand breaks was also augmented (Figure 4E, lower panel). These data together showed that solely the single point mutation N363K in POLE is sufficient to phenocopy in transformed cultured cells the abnormal cellular shape and the deformed, massively enlarged nuclei of giant cell glioblastomas.
DISCUSSION
Somatic POLE exonuclease mutations are found in sporadic colon- and endometrial cancers. Germline POLE exonuclease mutations are responsible for the PPAP syndrome (together with POLD1 exonuclease mutations, see above) showing a similar tumor spectrum, complemented by rare upper gastrointestinal-, ovarian-, breast- and brain cancer, depending on the particular mutation (42). All these tumors are characterized by an extreme high burden of SNVs. Intriguingly, several of these POLE cancer-associated mutations are far more mutagenic as artificially designed complete exonuclease-deficient (exo-null) mutants. Recently, several important reports elucidated some of the mechanistic rationales behind this apparent paradox (12,13,43). Common to these cancer-associated mutations is besides a disabled exonuclease activity the creation of a polymerase-activity, which is less selective and less controlled. As expected, these circumstances lead to massively increased rates of single nucleotide substitutions, but in contrast, no other replication-associated defects like less stable replication forks, problems in fork restart or DNA breaks were so far reported for these mutants. The discovery and first clinical description of the hereditary cancer-associated exonuclease N363K mutation by Rohlin et al., suggested that this mutation predisposes not only to the classical POLE exonuclease tumor spectrum of colon- and endometrial cancers, but curiously in addition to a rare cancer characterized by extreme high DNA-damage, the giant cell glioblastoma. Our discovery and clinical description of a second family carrying the same mutation validated the responsibility of the N363K mutation for the predisposition to giant cell glioblastoma in addition to the classical PPAP tumor spectrum (see above and (20)). In addition, with a much lower frequency, glioblastomas (but not giant cell glioblastomas) were also found in carriers of the L424V mutation (two cases at 47 and 31 years old (44,45)). Here, we validated the hypermutated nature of N363K mutated giant cell glioblastomas, colorectal- and duodenal cancers (Supplementary Table S1, Supplementary Figure S1C) and present data showing that this POLE N363K mutation stands out from all the other so far characterized cancer-associated POLE exonuclease mutations by increasing DNA damage, chromosomal aberrations, aneuploidy and by its potential to disrupt on its own severely nuclear and cellular organization, minute hallmarks of giant cell glioblastoma.
We used CRISPR/Cas techniques to precisely generate the herein characterized POLE mutants in strict isogenic backgrounds and to select edited cells likewise via CRISPR/Cas conferred ouabain-resistance. We complemented this strategy by blocking imprecise NHEJ-mediated repair of CRISPR/Cas-generated double strand breaks (DSBs), which promoted error-free homologous recombination-mediated repair. This strategy allowed us to generate both homogeneous cell pools and clones, prerequisite to assess and precisely compare the impact of the different mutations. Using WGS, we then confirmed that the mutants indeed increase massively SNVs during proliferation compared to control edited wildtype cells. We further showed that both the L424V and the N363K mutants exceed mutational rates of the (not cancer-associated) engineered exonuclease-deficient (exo-null) mutant (32). We thus confirmed that POLE mutants found in human cancers are usually more mutagenic than artificial, complete exonuclease-deficient mutants.
In a next step, we verified that the mutational signature of the generated mutants reproduced the typical signature of POLE exonuclease-mutated cancers. Whereas wild-type edited control cells accumulated (few) SNVs independent of sequence context, the L424V, N363K and the exo-null mutants highly preferentially mutated the trinucleotide sequence TCT to TAT, characteristic for POLE exonuclease proofreading mutants (35). Interestingly, the N363K mutation revealed a significantly higher mutagenicity than both the L424V and the exo-null mutation. Compared to the study of Albertson et al. (10) that estimated the mutation rate of homozygous exo-null mouse fibroblasts to 0.1–10 mut/Mb per cell cycle, we found mutation rates that were somewhat lower (roughly 0.07 mut/Mb per cell cycle for the exo-null and 0.18 mut/Mb per cycle for the N363K mutation). However, Albertson et al. used mouse-, not human cells and employed exclusively genetic screens, but not WGS, to determine mutational rates. Li et al. (11) estimated using WGS a mutation rate of 1.6 mut/Mb per cycle for the heterozygous, but much higher mutagenic P286R allele, also using mouse fibroblasts. Finally, Hodel et al. (33), introduced via CRISPR/Cas9 editing in a heterozygous manner the POLE S459F mutation in human HCT116 cancer cells and followed a similar approach as ours to determine mutational frequencies. Clones of this mutant acquired around 500 mutations per exome during around 54 cycles, which equals roughly 0.15 Mut/Mb per cell cycle, around what we find in our homozygous model. Different experimental approaches, different cells (primary versus repair-deficient cancer cells) and different alleles make it thus difficult to compare mutational potential of mutants in the existing literature. Finally, here reported counts are also influenced by our quite stringent mutation calling during WGS (see Materials and Methods).
Importantly, we then found that the mutational impact of the mutants did not correlate with their growth properties. Both the mutant with the highest (N363K) and lowest (exo-null) mutational impact showed a significant growth defect, whereas the intermediate mutagenic L424V mutant proliferated as the wildtype. We found an explanation for this behavior by our discovery that both growth-retarded mutants suffer from DNA damage. Up to now, no POLE exonuclease mutant was reported to increase DNA damage and the most mutagenic mutant known, P286R, being more than a thousand times more mutagenic than the wildtype, was explicitly shown not to increase DNA damage (11). Thus, the N363K mutation constitutes a new group of cancer associated POLE exonuclease mutants that do not only increase SNV frequencies but cause chronic DNA damage.
Our first assumption that the N363K mutation might not only affect proofreading activity but somehow also restricts normal polymerase activity was proven incorrect since our experiments quantifying overall replication fork speed demonstrated that forks with mutated POLE were not slower, neither more sensitive to fork block. However, the elevated sensitivity of all in this work analyzed POLE mutants to cytarabine proved once more their specific defect in exonuclease activity (37,38). We also gained additional evidence that the amount of SNVs in the mutants was not responsible for the observed DNA damage since silencing mismatch repair and thus increasing SNV frequencies in the L424V or N363K mutant even further did not lead to more DNA damage.
We then considered the possibility that only after engaging in a proofreading cycle (likely abortive or unsuccessful), particularly the DNA-damage causing mutants were stuck in a way that resumption of replication was not possible. The exchanged amino acids might have caused a distinct distortion or electrostatic change at or near the exonuclease active site that would keep the nascent DNA strand bound too tight for an efficient hand over back to the polymerase domain. In this scenario, resumption of replication would not be possible, provoking prolonged fork block, as proposed by Xing et al. in a study using S. cerevisiae (12). As pointed out before, such events would not be frequent enough to be detectable or even statistically significant by DNA fiber analysis. However, they would leave their marks in DSBs (46) or stretches of underreplicated DNA at the end of S-phase. Whereas important shortfalls during replication are normally detected by the S-phase checkpoint, resulting in slowing down or halt of the cell cycle, minor defaults can pass undetected and be carried over into mitosis (39,47), (for review, (41)). If subsequently the attempt of cell division does not end fatal, daughter cells will lack parts of their DNA and show signs of unrepaired DNA damage such as missing DNA stretches or entire chromosomes. This is precisely what we observed for the N363K mutant. These cells showed a significant increase in aneuploid metaphases compared to wildtype cells, while having nearly exclusively lost chromosomes. Since our WGS analysis was not designed to detect larger chromosomal aberrations, we additionally performed CGH arrays with N363K and wildtype clones after cultivation over approximately sixty cell cycles. We showed that N363K cells had accumulated more than three times the number of chromosomal aberrations found in wildtype cells. We thus conclude that the N363K mutation indeed entails chromosomal aberrations, however, in a subtle way so that cells pass obviously without readily detectable activation of the S-phase- or DNA damage checkpoint.
Importantly, Dahl and coworkers published very recently a study introducing the human N363K mutation in yeast (pol2-N378K) (43). The authors could verify that this pol2-N378K mutation has a higher mutagenicity as the yeast exonuclease-null mutation and has a strongly reduced proofreading activity. Based on structural studies from homologous bacterial and viral enzymes, the authors reason that passing the mismatched nascent DNA into the exonuclease site is hampered and thus polymerization beyond a mismatch is more likely. Furthermore, modeling the N363K mutation on the yeast enzyme, Hamzaoui et al., localized the mutation at the surface of the enzyme, hindering passage of the DNA strand into the exonuclease site (48). These considerations coincide with our model that once the mismatched DNA managed to enter the exonuclease site, its way back to the polymerase domain is similarly blocked or hampered, resulting in a blocked fork. Finally, to exclude that downstream-acquired suppressor-mutations in the edited POLE-gene might have caused unknown structural changes and falsified our analyses of the mutants, we also deep-sequenced the POLE exons of our mutant clones but did not find any other mutations as the ones specifically introduced by our CRISPR-approach.
As the name precises, giant cell glioblastomas contain cells with a drastically altered shape. Cells are very heterogenic, often increased in size containing one massively enlarged nucleus, several nuclei or even difficult to characterize chromatin masses (25). Whereas in our study the simply immortalized, but untransformed N363K RPE-1 cells maintained albeit their DNA damage and chromosomal aberrations a normal cellular and nuclear shape, the situation was entirely different for HCT116 cancer cells carrying the N363K mutation. These cells showed an even more pronounced growth defect, depicted an even stronger aneuploidy, and had lost their normal cellular and nuclear organization. By phase contrast microscopy we observed many dying cells but most strikingly giant cells containing two or more nuclei or undefinable nuclear masses. We further counterstained the cytoskeleton with fluorescent phalloidin to assure that multiple nuclei or gross nuclear masses were indeed contained in one single cell (49). Such ‘giant’ cells often depicted an extreme level of DNA damage (γH2AX), as it is the case in giant cell glioblastoma. Thus, our data argue that the POLE N363K mutation is sufficient to induce the ‘giant cell’ phenotype in at least already transformed cells. Even though HCT116 cells are MMR deficient, RPE-1 cells with silenced MMR did not reproduce the giant cell phenotype, making it unlikely that the MMR status is involved in developing the giant cell phenotype. Moreover, POLE N363K-mutated giant cell glioblastomas can be both MMR proficient or -deficient (Supplementary Table S1). However, we do not know if in untransformed cells (in this study RPE-1 cells) likewise ‘giant cells’ are produced but are readily eliminated by still active control mechanisms during or after mitosis, which often are disabled in transformed cells. Live microscopy following cells over several cycles could approach this question.
Giant, (pseudo-) polyploid cells are known indicators of worse prognosis, treatment failure and metastatic potential in cancers (for review, (49)). Still a subject of intense research, one described key mechanism for pseudo-polyploidization is a (partially) failed mitosis, caused by DNA replication stress, DNA damage and checkpoint defects (39,47) (for review, (41)). Importantly, in yeast low levels of DNA polymerases were directly shown to cause replication stress, chromosome aberrations and aneuploidy (50,51). Furthermore, aneuploidy was also shown to be linked to abnormal nuclear shape (52,53). It is however noteworthy that in POLE N363K patients, only glioblastomas, but not colon- and endometrial cancers were reported to contain giant cells. Why in POLE N363K patients only cancerous glia cells show the ‘giant cell’ phenotype, whereas in our study a colon cancer cell line (HCT116) was readily transformed into ‘giant cells’, is not clear, maybe the fact that we worked in this study with cells carrying the N363K mutation in a homozygous manner, whereas patients always keep one POLE wildtype allele, might explain this difference. However, to the best of our knowledge, no single point mutation of a gene is known that changes as profoundly nuclear-cellular organization, even more strikingly as paradigm lamin-mutations in e.g. Hutchinson-Gilford Progeria do (54).
To conclude, our work provides a molecular and cell biological explanation for the increased tumor spectrum of the POLE N363K exonuclease mutation. We demonstrated that particularly this mutation causes besides massive SNV accumulation also DNA damage, chromosome aberrations, aneuploidy, and multinucleated cells. POLE exonuclease-mutated cancers are a priori classified as prime targets for immunotherapy due to their extreme high mutagenicity and richness of neoantigens (14–16). However, aneuploidy was reported to be a counterindication for immunotherapy (55). Furthermore, well established, intrinsically elevated DNA damage sensitizes cancer cells to chemotherapy. In the future, it will thus be important to (re-)assess known cancer-associated POLE exonuclease mutants for DNA damage, structural chromosomal defects, and ploidy to possibly re-evaluate therapeutic strategies.
DATA AVAILABILITY
The WGS data areavailable via SRA using the BioProject number PRJNA896776. All sequences of sgRNA used in CRISPR-Cas mediated gene editing are detailed in Materials and Methods.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
ACKNOWLEDGEMENTS
We want to express our deep gratitude to Jérôme Moreaux at the Institute of Human Genetics, IGH, in Montpellier for having generously provided laboratory space, equipment and reagents indispensable for the completion of this study.
FUNDING
Ligue Regionale Contre le Cancer Haute Garonne; AAP 2018 Translational Research @IUCT-O.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments