





ABSTRACT
N-heterocycles are suspected to play an important role in the chemical origin of life. Despite their detection in meteorites and in Titan’s atmosphere, their extra-terrestrial chemical formation networks remain elusive. Furthermore N-heterocyclics are undetected in the interstellar medium. This paper assesses the photostability of protonated N-hetero(poly)acenes after ultraviolet (UV) and vacuum ultraviolet (VUV) excitation. It provides information on their ability to retain the N atom into the cycle to generate larger N-containing species or functionalized N-heterocyles. Protonated N-hetero(poly)acenes were generated using electrospray ionization and injected into a linear ion trap where they were irradiated by radiation of 4.5 to 10 eV using the DESIRS beamline at the synchrotron SOLEIL. The photodissociation action spectra of protonated pyridine, quinoline, isoquinoline, and acridine were measured by recording the photofragment yields as a function of photon energy. The four systems exhibit dissociation channels associated with H2 and HCN/HNC loss but with different branching ratios. The results indicate that increasing the size of the N-hetero(poly)acenes increases the chance of retaining the N atom in the larger fragment ion after photodissociation but it remains that all the protonated N-hetero(poly)acenes studied lose their N atom at part of a small neutral photofragment, with high propensity. Therefore, protonated N-hetero(poly)acenes in interstellar space are unlikely precursors to form larger N-containing species. However, protonated pyridine, quinoline, isoquinoline, and acridine are most likely to retain their N atoms in planetary atmospheres where UV radiation at the planet’s surface is typically restricted to wavelengths greater than 200 nm – suggesting such environments are possible substrates for prebiotic chemistry.
1 INTRODUCTION
Nitrogen-containing, cyclic organic molecules (N-heterocycles) are the building blocks for many biomolecules including nucleobases, which are essential components of the genetic material in all terrestrial living organisms. This implicates N-heterocycles in the chemical origin of life (Peeters et al. 2003, 2005; Sandford et al. 2020). While photochemical routes to produce these molecules have been proposed for the prebiotic Earth (Xu, Chmela & Green 2020), these were restricted to wavelengths of light longer than 200 nm (6.2 eV) at the surface (Ranjan & Sasselov 2016). It is not known how and where these compounds are formed beyond Earth where the ultraviolet (UV) radiation fields are harsher and incorporate significantly shorter wavelengths. This is true even for prototypical N-heterocycles such as pyridine (C5H5N), quinoline (C9H7N), and acridine (C13H9N). Yet simple N-heterocycles and nucleobases have been detected from meteorites (Hayatsu 1964; Folsome et al. 1971; Stoks & Schwartz 1981; Martins et al. 2008; Callahan et al. 2011; Pearce & Pudritz 2015; Martins 2018) and in extraterrestrial atmospheres (Ali et al. 2015). One proposed mechanism for pyridine formation is from acetylene polymerization, where one C2H2 unit is substituted with an HCN molecule instead, leading to direct incorporation of a nitrogen atom into a growing aromatic ring (Ricca, Bauschlicher & Bakes 2001). The viability of this formation path depends primarily on the HCN/C2H2 ratio, which is typically low in circumstellar envelopes and thus does not favour this mechanism but can be fairly large in carbon-rich stars such as the AGB star IRC+10216 (Peeters et al. 2005). However, Tielens (2013) has reviewed how UV light drives chemical processes in the interstellar medium (ISM) and Peeters et al. (2005) have exposed that neutral N-heterocycles are readily destroyed by UV radiation and that their stability decreases further with the number of nitrogen atoms incorporated in the ring – note that the nucleobases are derived from purine and pyrimidine containing 4 and 2 nitrogen atoms in the backbones, respectively.
The propensity of N-heterocycles to be destroyed in photodominated regions (PDRs), including circumstellar envelopes and diffuse interstellar clouds, implies a low probability of detecting them and instead points the search towards dense molecular clouds and in particular regions with a high HCN/C2H2 ratio. Attempts to detect pyridine, quinoline, isoquinoline, and nucleobase derivatives using radioastronomy in circumstellar envelopes of carbon-rich stars and dark molecular clouds have been yet unsuccessful (Simon & Simon 1973; Kuan et al. 2004; Charnley et al. 2005; Brünken et al. 2006). The explanation given to rationalize the observation of N-heterocycles in meteorites is that after their formation in circumstellar envelopes, they are ejected and incorporated into icy interstellar grains in cold molecular clouds, thus shielding them from harsh UV radiation and where they can be functionalized by photoprocessing to form nucleobase-type structures (Peeters et al. 2005).
In space, proton transfer reactions are the most likely routes to form protonated molecules. The major source of protons in the ISM are H3+ molecules, which are strong proton donors (Larsson, Geppert & Nyman 2012; Etim et al. 2017) and found in high concentrations in dense molecular clouds (Geballe & Oka 1996; McCall et al. 1999) and in PDRs (McCall et al. 1998). The high proton affinity of the N-heterocycles (as Lewis bases) suggest they are likely to become protonated in such regions of space. Gas-phase electronic spectra of protonated pyridine (monocyclic), quinoline/isoquinoline (bicyclic), and phenanthridine (tricyclic) reveal absorption features only in the UV (Hansen, Blanksby & Trevitt 2015a; Hansen et al. 2015b; Noble, Dedonder & Jouvet 2015; Féraud et al. 2017), preventing their detection in the ISM based on comparison with known diffuse interstellar bands (DIBs). However, tricyclic protonated acridine does exhibit absorption in the visible spectral region but no match was found with known DIBs (Noble et al. 2015). Note that these laboratory studies on the mono- and bicyclic species found that photodissociation, at wavelengths accessible within a planetary atmosphere (above 200 nm), was dominated by the loss of H2 in the case of pyridine (Hansen et al. 2015b), leading to a reactive pyridyne species with its ring intact and still incorporating its nitrogen atom, while negligible single-photon photodissociation was observed for quinoline/isoquinoline (Hansen et al. 2015a). If this photochemistry – conservation of the N atom in the ring of the ionic photofragments – is reproduced at higher excitation energies, then protonated N-heterocycles would be good candidates as intermediates in the growth of complex N-heterocycles in space. They could evolve into either larger PANHs such as N-containing rubicenes, tetrabenzopentacenes, and corannulenes or functionalized N-heterocycles such as nicotinic acid detected in meteorites (Smith et al. 2014) and thereafter synthesized using simulated conditions of astrophysical ice analogues (Smith, Gerakines & Callahan 2015).
All N-heterocycles formed in space and containing only one N atom in the ring are unlikely to play a direct role in the formation of nucleobases, even those derived from pyrimidine with two N atoms in the ring. Indeed, no viable gas-phase chemical route in ISM conditions to insert an additional N atom to an existing pyridine-type structure is known. However, it has been postulated that quinoline and isoquinoline could potentially be precursors of nucleobases through ice reactions (Parker et al. 2015). Nevertheless, mono nitrogen containing heterocycles could be alkylated (Naraoka et al. 2017) and/or functionalized (Rodriguez et al. 2019) potentially leading to good precursors for coenzymes such as nicotinamide adenine dinucleotide (Smith et al. 2014).
In this paper, the photodissociation of protonated N-hetero(poly)acenes is investigated following excitation with UV and VUV light in the 4.5–10 eV photon energy range. The photodissociation action spectra of protonated pyridine (PH+), quinoline (QH+), isoquinoline (iQH+), and acridine (AcH+) were recorded using a linear ion trap coupled to the DESIRS beamline at the SOLEIL synchrotron. These spectra do not allow to retrieve the absolute photodissociation quantum yield of the protonated species nor to elucidate the structural arrangements of the ionic photofragments produced. However, they provide relative fragmentation branching ratio, and inform on the chemical composition of the surviving ionic photofragments so that the propensity of retaining the N atom into the ionic fragments as a function of photoexcitation energies and size of the parent ions can be discussed.
2 METHODS
The photodissociation of protonated N-hetero(poly)acenes following excitation with UV and VUV light was studied using action spectroscopy with an LTQ linear ion trap coupled (Milosavljević et al. 2012) to the DESIRS undulator-based VUV beamline (Nahon et al. 2012) at the SOLEIL synchrotron facility in Saint-Aubin (France). Protonated species were generated from their corresponding neutral precursor (pyridine, quinoline, isoquinoline, and acridine) purchased from Sigma-Aldrich by infusing a 10 μM solution in methanol into the ion trap mass spectrometer via its electrospray ionization source. The pressure in the ion trap was maintained at ≈ 10−3 mbar using He as a buffer gas to provide efficient trapping conditions. The protonated species of interest (precursor ions), PH+ (m/z 80), QH+ (m/z 130), AcH+ (m/z 180), and iQH+ (m/z 130) were isolated through mass selection prior to being photoexcited by the synchrotron radiation in the UV range from 4.5 to 7.5 eV in steps of 0.1 eV and in the VUV range from 7.0 to 10.0 eV in steps of 0.2 eV. A gas filter filled with Kr gas was used to prevent pollution from higher harmonics of the undulator synchrotron radiation for the 7–10 eV range. For the low-lying energy region (4.5 to 7.5 eV), an additional Suprasil window was introduced into the synchrotron radiation path. The experiments were carried out with a typical photon flux of 1012 to 1013 photons s−1 and a photon resolution of ≈10 meV at 10 eV (monochromator exit slit width set to 200 μm). The photofragmentation yields were kept below 10 per cent by setting the irradiation time to 5 s to prevent multiple sequential one-photon absorption processes leading to spurious peaks in the MS2 spectra. The data processing of the recorded mass spectra was done using the MassJ package developed by Giuliani (2021) written in the julia language (Bezanson et al. 2012). The error bars given as colour shaded zones in Figs 1, 2, and 3 result from the statistical standard error from the averaging process (Giordano 2016) and the error propagation as it was done by Wenzel et al. (2020) and Jacovella et al. (2022). Photodissociation action spectra were corrected by the incident photon flux as measured by a calibrated VUV photodiode (IRD AXUV100). As aforementioned, the photodissociation absolute quantum yield cannot be extracted as the number of ions interrogated per trapping cycle by the synchrotron light is unknown.
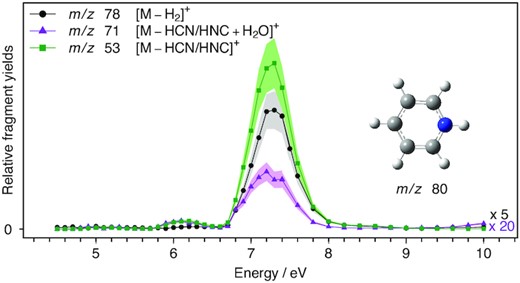
UV/VUV photodissociation action spectra of PH+ over the 4.5–10 eV range. The black trace is obtained by monitoring H2 loss and the green one by monitoring HCN and/or HNC loss. The purple trace represents the channel of phenylium C6H5+ reacting with water molecules present in the background gas in the ion trap.
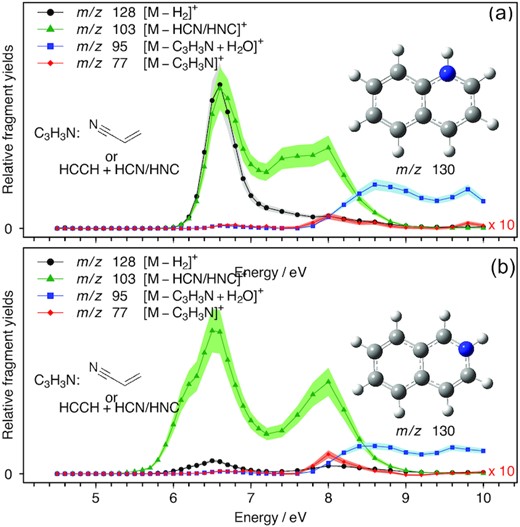
UV/VUV photodissociation action spectra of QH+ (a) and iQH+ (b) over the 4.5–10 eV range. Black traces are obtained by monitoring H2 loss. Green and red traces by monitoring HCN or HNC loss and C3H3N loss, respectively. The blue traces represent the channel of phenylium C6H5+ reacting with water molecules present in the background gas in the ion trap.
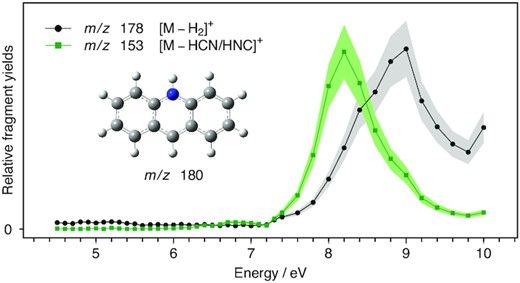
UV/VUV photodissociation action spectra of AcH+ over the 4.5–10 eV range. Black trace is obtained by monitoring H2 loss and the green one by monitoring HCN/HNC loss.
Thermodynamical dissociation thresholds were calculated using the CBS-QB3 compound method (Wood et al. 2006) implemented in the Gaussian 16 package (Frisch et al. 2016).
3 RESULTS AND DISCUSSION
3.1 Results
The VUV photodissociation action spectra of PH+, QH+, iQH+, and AcH+ over the 4.5–10.0 eV range are shown in Figs 1, 2(a and b), and 3, respectively. The black curves represent the spectra obtained by monitoring the ion signal of the photofragments corresponding to the loss of H2 from the precursor ions. The green curves depict the intensity of signal from HCN and/or HNC loss and red curves correspond to the loss of 53 amu from the protonated precursors. The purple and blue curves result from the reaction of background water with photofragments obtained upon HCN/HNC and 53 amu loss, respectively.
3.1.1 Protonated pyridine (PH+)
The UV/VUV photodissociation action spectra of PH+ displayed in Fig. 1 exhibit two dissociation channels. The major channel corresponds to the HCN/HNC loss (green and purple curves) and the minor one (≈ 6 times weaker at 7.3 eV) to H2 loss depicted in black. The spectra of both channels consist of one intense band centred at 7.3 eV and a weak one at 6.1 eV. We note that the band observed by Hansen et al. (2015b) at 5.1 eV while monitoring the H2 dissociation channel is not visible at this scale. The photofragment formed upon photoexcitation followed by H2 elimination has been unambiguously elucidated by studies of its ion–molecule reaction kinetics (Hansen et al. 2015b). Fragment m/z 78 corresponds to the 2-pyridinylium ion obtained by 1,2-elimination of H2. The major dissociation channel yields to the formation of C4H5+ ionic fragment upon the loss HCN or HNC. The assignment of this m/z 53 ion cannot be made based on thermodynamical dissociation thresholds alone as the formation of the five most stable C4H5+ isomers (Muller et al. 2020) requires < 5 eV of excitation energy. At least one of the C4H5+ isomers formed appears to be reactive with background water (purple trace in Fig. 1a) present in the ion trap forming an ion at m/z 71. Reactive monitoring studies with isomer isolation using different precursors or ion mobility techniques would likely provide insight into the structure of the isomers formed upon HCN/HNC loss.
3.1.2 Protonated quinoline (QH+) and isoquinoline (iQH+)
The UV/VUV photodissociation action spectra of QH+ and iQH+ are shown in Figs 2(a) and (b), respectively. Three dissociation channels are observed, corresponding to loss of H2 (black), HCN/HNC (green), and C3H3N (red). In both systems, the dominant dissociation channel is the loss of HCN/HNC at photolysis energies below 8.5 eV, and C3H3N loss at energies above 8.5 eV. Additionally, H2 loss is observed with a band maximum at 6.6 eV. For this band, the relative intensities of HCN/HNC loss and H2 loss are equivalent for QH+, but for iQH+ the HCN/HNC loss is 8 times more intense than the H2 loss channel. The bands observed by Hansen et al. (2015a) at 5.2 and 5.3 eV in the H2 loss channel for QH+ and iQH+, respectively, are not observed in our spectra. These bands were explained by a resonance-enhanced two-photon photodissociation process in the pulse-laser study of Hansen et al. (2015a) and are not observed in this study where the experimental conditions were configured to minimize the likelihood of such multiphoton processes. The spectra consist of 4 bands with their maxima located at 6.6, 8.0, 8.6, and 9.8 eV. The structure of the m/z 103 photoproduct cannot be confirmed based on the spectra obtained. The m/z 77 channel is weak, and almost all the initially formed m/z 77 photoproducts react with background water present in the ion trap to give m/z 95, as seen with the same photoproduct ion from protonated benzonitrile (Jacovella et al. 2022), leading us to infer that the structure corresponding to m/z 77 is most likely the phenylium ion (c −C6H|$_5^+)$|.
3.1.3 Protonated acridine (AcH+)
The UV/VUV photodissociation action spectra of AcH+ represented in Fig. 3 show two dissociation channels of similar fragmentation yield. The black trace corresponds to H2 loss and the green one to HCN/HNC loss. The band maxima are located at 8.2 and 9.0 eV for HCN/HNC and H2 loss, respectively. The shift to higher energy of the band maximum of the H2 loss action photodissociation spectrum compared to the HCN/HNC loss spectrum cannot be explained the opening of a new thermodynamical route around 8.5 eV since the threshold for H2 elimination is far lower in energy, predicted at 4.77 eV by our calculations. This change in the fragmentation yields between the two channels may reflect a change in the nature of the excited state populated, leading to different dissociation mechanisms and outcomes.
3.2 Discussion
The UV/VUV photodissociation action spectra of PH+ and AcH+ exhibit similarities between the H2 and HCN/HNC loss channels. However, the intensities of these two loss channels are identical, whereas the loss of an HCN/HNC dominates (6 times stronger than H2 loss at 7.3 eV) for PH+. Furthermore, the photoproducts obtained upon HCN/HNC elimination show a drastic change in reactivity with water molecules. The m/z 53 formed from PH+ has a weak but non-negligible reactivity with water, while the m/z 153 created from AcH+ presents no reactivity at all with water in the limit of detection of the machine.
The UV/VUV photodissociation action spectra of QH+ and iQH+ present more complex spectroscopic signatures and larger complexity of fragmentation routes than those of PH+ and AcH+. The inclusion of the N atom in the naphtalene-type structure in position 1 (QH+) or 2 (iQH+) does not significantly affect the energy level diagram and selection rules since the band maxima in both spectra are identical. Yet, the dissociation dynamics appear to be influenced by the position of the N atom for the fragmentation route leading to m/z 128 + H2.
The photochemistry of both the monocyclic and bicyclic protonated N-heterocycles (PH+/QH+/iQH+) is dominated by the loss of HCN/HNC when excited in the 4.5–10 eV photon energy range. These processes disrupt the ring systems and eliminate the nitrogen atom, making these compounds unlikely participants in the growth and functionalization of N-heterocycles in interstellar space as free gas-phase species. In regions of space where the UV radiation field destroys protonated N-heterocycles, it is also likely to dissociate HCN/HNC leading to a hydrogen atom and a cyanide diatom (Chenel et al. 2016), thereby triggering organo-nitrogen chemistry. Although the nucleobases, and many N-heterocyclic biomolecules, are mono- or bicyclic, for completeness, we also studied the tricyclic protonated acridine cation in case it might form some smaller, reactive cyclic species susceptible to functionalization. Yet it was observed that, while important, HCN/HNC loss does not dominate its photochemistry. Instead, an H2 loss channel emerges at high-photon energies from which the structure of the corresponding ionic fragment remains elusive. It is not conceivable how this species could be a realistic progenitor for any prebiotic compounds.
4 CONCLUSIONS
In this paper, we present UV/VUV photodissociation action spectra of PH+, QH+, iQH+, and AcH+ spanning a wide UV-VUV photon energy range 4.5–10 eV. The photodissociation spectra reveal that photolysis of protonated pyridine and protonated quinoline/isoquinoline is dominated by the loss of HCN/HNC (–27 amu) across these photon energies, making these precursor ions unlikely agents in the formation of more complex N-heterocycles in interstellar space. While the tricyclic species, acridine, is able to survive as an N-heterocyclic species by eliminating H2 at the highest photolysis energies used, it does not form the basis for any reasonable bottom-up synthesis of a prebiotic precursor. Therefore, we conclude that if gas-phase protonated N-heterocycles are involved in growth mechanisms, these should take place within environments shielded from short wavelengths of UV radiation, such as dense planetary atmospheres or very dense interstellar molecular clouds.
ACKNOWLEDGEMENTS
The authors thank the staff from SOLEIL for smoothly running the facility and providing beamtime under project 20191313. This work was partially supported by the Agence Nationale de la Recherche Scientifique, France, under Project Number ‘ANR-08-BLAN-0065’.
DATA AVAILABILITY
The data underlying this article will be shared on reasonable request to the corresponding author.

{kind=link}
{kind=link}
{kind=link}