





Abstract
Solid-state astrochemical reaction pathways have the potential to link the formation of small nitrogen-bearing species, like NH3 and HNCO, and prebiotic molecules, specifically amino acids. To date, the chemical origin of such small nitrogen-containing species is still not well understood, despite the fact that ammonia is an abundant constituent of interstellar ices towards young stellar objects and quiescent molecular clouds. This is mainly because of the lack of dedicated laboratory studies. The aim of this work is to experimentally investigate the formation routes of NH3 and HNCO through non-energetic surface reactions in interstellar ice analogues under fully controlled laboratory conditions and at astrochemically relevant temperatures. This study focuses on the formation of NH3 and HNCO in CO-rich (non-polar) interstellar ices that simulate the CO freeze-out stage in dark interstellar cloud regions, well before thermal and energetic processing start to become relevant. We demonstrate and discuss the surface formation of solid HNCO through the interaction of CO molecules with NH radicals – one of the intermediates in the formation of solid NH3 upon sequential hydrogenation of N atoms. The importance of HNCO for astrobiology is discussed.
1 INTRODUCTION
The detection of glycine, the simplest amino acid, in cometary samples recently returned to Earth by the STARDUST mission has boosted detailed investigations of the origin and fate of (pre-) biotic molecules in the interstellar medium (ISM; Elsila, Glavin & Dworkin 2009; Garrod 2013). However, although an increasing number of laboratory and theoretical studies show that complex species form in the solid phase, on the surface of icy grains, we still lack understanding of the complete surface formation pathways at play. The nitrogen chemistry of the ISM is particularly important within this context, because of its potential to reveal the formation routes of the simplest amino acids or their possible precursors. From the ∼180 species unambiguously identified in the ISM, about one-third contains nitrogen atoms, but only NH3, XCN, and possibly NH4+ are identified as constituents of interstellar ices. Solid NH3 is generally found with a typical abundance of 5 per cent with respect to water ice towards low- and high-mass young stellar objects (YSOs; Gibb et al. 2004; Bottinelli et al. 2010; Öberg et al. 2011). Solid isocyanic acid (HNCO) has not been identified in the solid phase yet, but its direct derivative, the cyanate ion (OCN−), has been found in interstellar ices with abundances between 0.3 and 0.6 per cent with respect to water ice. The assignment of solid OCN− is often attributed to either the entire, so-called, XCN band or to a single component (2165 cm−1) of the full band (van Broekhuizen et al. 2005). More recently, Öberg et al. (2011) found a correlation between CO and the XCN band that supports the identification of the latter as OCN−. Another possible N-bearing component of interstellar ices is NH4+. Although the unambiguous assignment of NH4+ is still under debate (Gálvez et al. 2010), it can potentially be one of the carriers of the 5–8 μm bands, and its presence in interstellar ices is consistent with previously obtained laboratory results (Boogert et al. 2008). The existence of interstellar solid NH4+ is indeed constrained by the hypothesis that NH4+ helps to maintain charge balance between positive and negative ions within interstellar ices. Öberg et al. (2011) assigned NH4+ abundances of 2.3 and 4.3 per cent with respect to water ice towards low- and high-mass protostars, respectively. The formation of OCN− and NH4+ is commonly associated with a later stage of molecular cloud evolution, when thermal processing of the ice by a newly formed protostar becomes important. NH3 and HNCO are commonly considered the precursors of NH4+ and OCN− (Demyk et al. 1998; Lowenthal, Khanna & Moore Marla 2002), and therefore are expected to be formed in an earlier evolutionary stage of dark clouds, when temperatures are as low as 10–20 K and the formation routes through non-energetic atom and radical addition surface reactions dominate.
To date, laboratory experiments on the non-energetic surface formation routes of nitrogen-containing species have mainly focused on the formation of ammonia (NH3), hydroxylamine (NH2OH), and various nitrogen oxides (NO, NO2, N2O) (Hiraoka et al. 1995; Hidaka et al. 2011; Congiu et al. 2012a, 2012b; Fedoseev et al. 2012; Ioppolo et al. 2014; Minissale et al. 2014). This work extends previous studies on the solid-state formation of ammonia to non-polar (CO-rich) ices, and, at the same time, discusses the link between the surface formation of HNCO. In the accompanying paper (Fedoseev, Ioppolo & Linnartz 2014), we investigate the deuterium enrichment of all the ammonia isotopologues as produced through the competition between hydrogenation and deuteration of nitrogen atoms. These results are not further discussed in this paper.
Reaction (4) is exothermic. The reactivity of CO with NH has been investigated in a combined experimental and quantum chemical study by Himmel, Junker & Schnöckel (2002) via photo-induced dissociation of HN3 in a 12 K Ar matrix. In their work, matrix experiments indicated that NH(3Σ) reacts with CO under laboratory conditions to form HNCO(1A’). An activation barrier of ∼4200 K was derived by means of CCSD(T), CASSCF, and MP2 calculations carried out to evaluate geometries and energies at the transition state for this spin-forbidden reaction. Although the value of this barrier could be considered quite high (e.g. the activation barrier for CO + H ∼500 K; Fuchs et al. 2009), experiments and simulations yield consistent data when taking into account experimental and computational inaccuracies. Reaction (4) has often been proposed in spectroscopic studies to explain the formation of HNCO in mixed interstellar ice analogues processed by proton or UV radiation (e.g. Raunier et al. 2003; van Broekhuizen et al. 2005 and references therein). However, to the best of our knowledge, there are no studies available from the literature on the investigation of reaction (4) with non-energetic input. Reaction (5) is endothermic (4800 K) and therefore is unlikely to occur under cold dense molecular cloud conditions (Nguyen et al. 1996).
2 EXPERIMENTAL PROCEDURE
2.1 Experimental setup
All experiments (summarized in Table 1) are performed in an ultrahigh vacuum (UHV) setup (SURFRESIDE2), constructed to investigate solid-state atom addition reactions at cryogenic temperatures. The system has been extensively described in Ioppolo et al. (2013), and therefore only a brief description is given here. SURFRESIDE2 consists of three UHV chambers with a room-temperature base-pressure in the range of 10−9–10−10 mbar. A rotatable gold-coated copper substrate is placed in the centre of the main chamber, where gasses are introduced and deposited with monolayer precision onto the substrate surface through two metal deposition lines. A monolayer (ML) corresponds to about 1015 molecules cm−2. The substrate temperature is varied between 13 and 300 K using a He closed-cycle cryostat with an absolute temperature accuracy better than ∼2 K. Both of the two other UHV chambers contain an atom beam line and are connected to the main chamber with angles of 45° and 135° with respect to the substrate (see Figs 1, 3, and 4 in Ioppolo et al. 2013). In one atom line, a commercially available thermal cracking source (Dr. Eberl MBE-Komponenten GmbH, see Tschersich 2000) is used to generate H/D atoms. In the other atom line, a microwave plasma atom source (Oxford Scientific Ltd, see Anton et al. 2000) can be used to generate H/D/N/O atoms or radicals, such as OH. A custom made nose-shape quartz pipe is placed in between each atom source and the substrate. These pipes are designed in a way that products formed upon thermal cracking (e.g. H from H2) or plasma dissociation (e.g. N from N2) experience at least four collisions with the pipe walls before reaching the substrate. This is done to quench electronically or ro-vibrationally excited states before impacting on the ice. A considerable fraction of non-dissociated molecules (e.g. H2/D2 and N2) are present in the beam. The method to derive atom flux values is described in Ioppolo et al. (2013). We want to stress that the N-atom flux is an effective flux, estimated by measuring the amount of products of a series of barrierless reactions involving N atoms in the solid phase. The H-atom flux used here is an absolute flux. In this case, the amount of H atoms present in the beam is directly measured by the quadrupole mass spectrometer (QMS) in the gas phase. The latter measurement neglects that not every H atom will stick to the surface of the substrate and therefore will be unavailable for further reactions, and it also does not consider H-atom recombination on the ice surface. The absolute H-atom flux, therefore, is an upper limit for the effective H-atom flux.
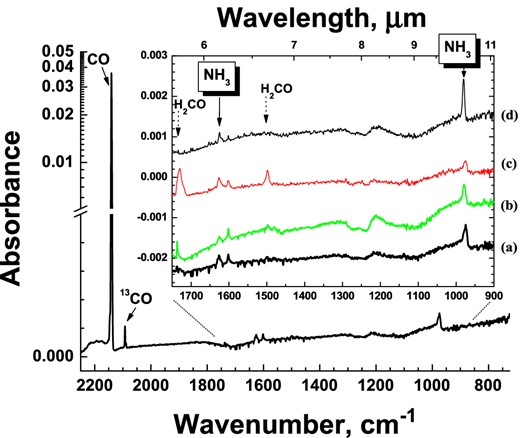
A RAIR difference spectrum from a co-deposition of N:H:N2:CO = 1:20:100:100 at 13 K with a total N-atom fluence of 3×1014 (±40 per cent) atoms cm−2 (experiment 1.1) is shown in the large panel. In the inset, four spectra from different co-deposition experiments are shown in a narrower spectral range: (a) is a zoom-in of the aforementioned spectrum; (b) is for a co-deposition of N:H:N2:CO = 1:20:100:500 (experiment 1.3); (c) is for a co-deposition of N:H:N2:CO = 1:100:100:100 (experiment 1.4); and (d) is for the deposition of NH3:CO = 1:500 with a total deposited NH3 amount of 0.3 ML, corresponding to the N-atom total fluence of the experiments (a)–(c). All spectra are for 13 K and plotted with offsets for clarity.
Metal shutters separate the atom beam lines from the main chamber. The atom beam sources as well as the molecular dosing lines in the main chamber can be operated independently. This versatile design allows for the sequential (pre-deposition) or simultaneous (co-deposition) exposure of selected interstellar ice analogues to different atoms (e.g. H/D/O/N). In the present study, co-deposition experiments are largely used. The ice composition is monitored in situ by means of reflection absorption infrared spectroscopy (RAIRS) in the range between 4000–700 cm−1 and with a spectral resolution of 1 cm−1 using a Fourier transform infrared (FTIR) spectrometer. The main chamber gas-phase composition is monitored by a QMS, which is placed behind the rotatable substrate, and is mainly used during TPD experiments. Here, RAIRS is used as the main diagnostic tool, complemented with TPD data to constrain the experimental results. Although QMS provides us with a better sensitivity, preference is given to the RAIRS due to the in situ nature of the method.
2.2 Performed experiments
The formation of solid NH3 and HNCO is studied for a selected set of well-defined experimental conditions. First, all the used gases (CO, H2/D2, and N2) are prepared in distinct pre-pumped (< 10−5 mbar) dosing lines. Pure H2/D2 gas (Praxair 5.0/Praxair 2.8) is introduced into the tungsten capillary pipe of the thermal cracking source. Pure N2 gas (Praxair 5.0) is dissociated in the plasma chamber of the microwave plasma source. A simultaneous co-deposition of H/D and N atoms with CO gas (Linde 2.0) is performed on the surface of the bare gold substrate, typically at 13 K. RAIR difference spectra are acquired every 5 min with respect to the spectrum of the bare gold substrate. For the crucial experiments, once the co-deposition is completed, a new spectrum is taken and used as background reference. Two additional control experiments are then performed on top of the previously grown ice. The first one is a co-deposition of CO molecules with H/D (i.e. without N) atoms and the second one is a co-deposition of CO molecules with N (i.e. without H/D) atoms. These two experiments are performed under exactly the same experimental conditions used for the very first co-deposition experiment in order to allow for a direct comparison. This procedure guarantees that the production of NH3 and HNCO is the cumulative outcome of a low-temperature co-deposition of H/D, N, and CO, ruling out other possible formation pathways due to contaminations in the atom lines or in the main chamber. Co-deposition experiments of H/D + N + CO are repeated a second time and a TPD experiment is performed right afterwards to monitor desorption of the formed species by means of the QMS.
A complementary set of control experiments is used to further verify the HNCO formation under astronomically relevant conditions. In this case, pure NH3 vapour is introduced into the microwave plasma source, and the plasma dissociation products (i.e. NH and NH2 radicals together with NH3, H, N, H2, and N2) are co-deposited with CO molecules. During this co-deposition experiment, RAIR difference spectra are acquired every 5 min with respect to a spectrum of the bare gold substrate. After completion of the co-deposition, a TPD experiment is performed and desorbing species are monitored by means of the QMS. The presence of NH and NH2 radicals in the beam is verified by performing a co-deposition of NH3 plasma dissociation products with D atoms and observing the N–D stretching mode in the mid-IR. For clarity, in Table 1 only the relevant experiments performed in this study are listed.
List of the performed experiments.
| Ref. N | Experiment | Ratio | Tsample | Rdep | Atom-fluxTL | Atom-fluxPL | t | TPDa | Detection of | Detection of |
|---|---|---|---|---|---|---|---|---|---|---|
| (K) | (ML min−1) | (1015 cm−2 min−1) | (1015 cm−2 min−1) | (min) | NH3b | HNCOb | ||||
| Verification of NH3 formation | ||||||||||
| CO | H (from H2) | N (from N2) | ||||||||
| 1.1 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | Y | N |
| 1.2 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 180 | – | Y | N |
| 1.3 | N:H:N2:CO | 1:20:100:500 | 13 | 2.5 | 0.1 | 0.005 | 60 | – | Y | N |
| 1.4 | N:H:N2:CO | 1:100:100:100 | 13 | 0.5 | 0.5 | 0.005 | 60 | – | Y | N |
| 1.5 | N:H:N2:CO | 1:100:100:100 | 25 | 0.5 | 0.5 | 0.005 | 60 | – | N | N |
| CO | D (from D2) | N (from N2) | ||||||||
| 1.6 | N:D:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | Y | N |
| H2O | H (from H2) | N (from N2) | ||||||||
| 2.1 | N:H:N2:H2O | 1:20:100:500 | 15 | 2.5 | 0.1 | 0.005 | 90 | – | – | – |
| 2.2 | N:H:N2:H2O | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | – | – |
| Verification of HNCO formation through hydrogenation of N atoms in CO-rich ice analogues | ||||||||||
| CO | H (from H2) | N (from N2) | ||||||||
| 3.1 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 90 | QMS2K/5K | Y | N |
| 3.2 | N:H:N2:CO | 1:6:100:100 | 13 | 0.5 | 0.03 | 0.005 | 90 | QMS2K/5K | Y | Y/N |
| 3.3 | N:H:N2:CO | 1:6:100:100 | 13 | 0.5 | 0.03 | 0.005 | 90 | RAIRSc | Y | Y/N |
| 3.4 | N:H:N2:CO | 1:4:100:100 | 13 | 0.5 | 0.02 | 0.005 | 90 | QMS2K/5K | Y | Y |
| 3.5 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 3.6 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS0.4K/5K | Y/N | Y |
| 3.7 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 180 | QMS2K/5K | Y/N | Y |
| 3.8 | N:H:N2:CO | 1:1.5:100:100 | 13 | 0.5 | 0.0075 | 0.005 | 90 | QMS2K/5K | N | Y |
| 3.9 | N:H:N2:CO | 1:1:100:100 | 13 | 0.5 | 0.005 | 0.005 | 90 | QMS2K/5K | N | Y |
| 3.10 | N:H:N2:CO | 1:2:100:100 | 25 | 0.5 | 0.01 | 0.005 | 90 | QMS1K/5K | N | N |
| 3.11 | N:H:N2:CO | 1:1.5:100:100 | 25 | 0.5 | 0.0075 | 0.005 | 90 | QMS2K/5K | N | N |
| Isotope shift experiments confirming the formation of HN13CO, H15N13CO and D15NCO | ||||||||||
| 13CO | H (from H2) | N (from N2) | ||||||||
| 4.1 | N:H:N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 4.2 | N:H:N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 360 | RAIRSc | Y | Y |
| 13CO | H (from H2) | 15N (from 15N2) | ||||||||
| 4.3 | 15N:H:15N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 13CO | D (from D2) | 15N (from 15N2) | ||||||||
| 4.4 | 15N:D:15N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y/N |
| Formation of HNCO further constrained via interaction of CO with NH3 plasma dissociation products | ||||||||||
| CO | NH3(dissociated) | |||||||||
| 5.1 | CO:NH3(dissociated) | nn | 13 | 0.5 | – | n | 90 | QMS2K/5K | – | Y |
| 5.2 | CO:NH3 | nn | 13 | 0.5 | – | – | 90 | QMS2K/5K | – | N |
| 5.3 | CO:NH3(dissociated) | nn | 70 | 0.5 | – | n | 90 | QMS5K | – | N |
| 13CO | NH3(dissociated) | |||||||||
| 5.4 | 13CO:NH3(dissociated) | nn | 13 | 0.5 | – | n | 90 | QMS2K/5K | – | Y |
| Confirmation of the presence of NH3 plasma dissociation products in the beam | ||||||||||
| D (from D2) | NH3(dissociated) | |||||||||
| 6.1 | D:NH3(dissociated) | nn | 13 | – | 0.05 | n | 60 | – | – | – |
| 6.2 | D:NH3 | nn | 13 | – | 0.05 | – | 60 | – | – | – |
| 6.3 | NH3(dissociated) | nn | 13 | – | – | n | 60 | – | – | – |
| Ref. N | Experiment | Ratio | Tsample | Rdep | Atom-fluxTL | Atom-fluxPL | t | TPDa | Detection of | Detection of |
|---|---|---|---|---|---|---|---|---|---|---|
| (K) | (ML min−1) | (1015 cm−2 min−1) | (1015 cm−2 min−1) | (min) | NH3b | HNCOb | ||||
| Verification of NH3 formation | ||||||||||
| CO | H (from H2) | N (from N2) | ||||||||
| 1.1 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | Y | N |
| 1.2 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 180 | – | Y | N |
| 1.3 | N:H:N2:CO | 1:20:100:500 | 13 | 2.5 | 0.1 | 0.005 | 60 | – | Y | N |
| 1.4 | N:H:N2:CO | 1:100:100:100 | 13 | 0.5 | 0.5 | 0.005 | 60 | – | Y | N |
| 1.5 | N:H:N2:CO | 1:100:100:100 | 25 | 0.5 | 0.5 | 0.005 | 60 | – | N | N |
| CO | D (from D2) | N (from N2) | ||||||||
| 1.6 | N:D:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | Y | N |
| H2O | H (from H2) | N (from N2) | ||||||||
| 2.1 | N:H:N2:H2O | 1:20:100:500 | 15 | 2.5 | 0.1 | 0.005 | 90 | – | – | – |
| 2.2 | N:H:N2:H2O | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | – | – |
| Verification of HNCO formation through hydrogenation of N atoms in CO-rich ice analogues | ||||||||||
| CO | H (from H2) | N (from N2) | ||||||||
| 3.1 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 90 | QMS2K/5K | Y | N |
| 3.2 | N:H:N2:CO | 1:6:100:100 | 13 | 0.5 | 0.03 | 0.005 | 90 | QMS2K/5K | Y | Y/N |
| 3.3 | N:H:N2:CO | 1:6:100:100 | 13 | 0.5 | 0.03 | 0.005 | 90 | RAIRSc | Y | Y/N |
| 3.4 | N:H:N2:CO | 1:4:100:100 | 13 | 0.5 | 0.02 | 0.005 | 90 | QMS2K/5K | Y | Y |
| 3.5 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 3.6 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS0.4K/5K | Y/N | Y |
| 3.7 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 180 | QMS2K/5K | Y/N | Y |
| 3.8 | N:H:N2:CO | 1:1.5:100:100 | 13 | 0.5 | 0.0075 | 0.005 | 90 | QMS2K/5K | N | Y |
| 3.9 | N:H:N2:CO | 1:1:100:100 | 13 | 0.5 | 0.005 | 0.005 | 90 | QMS2K/5K | N | Y |
| 3.10 | N:H:N2:CO | 1:2:100:100 | 25 | 0.5 | 0.01 | 0.005 | 90 | QMS1K/5K | N | N |
| 3.11 | N:H:N2:CO | 1:1.5:100:100 | 25 | 0.5 | 0.0075 | 0.005 | 90 | QMS2K/5K | N | N |
| Isotope shift experiments confirming the formation of HN13CO, H15N13CO and D15NCO | ||||||||||
| 13CO | H (from H2) | N (from N2) | ||||||||
| 4.1 | N:H:N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 4.2 | N:H:N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 360 | RAIRSc | Y | Y |
| 13CO | H (from H2) | 15N (from 15N2) | ||||||||
| 4.3 | 15N:H:15N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 13CO | D (from D2) | 15N (from 15N2) | ||||||||
| 4.4 | 15N:D:15N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y/N |
| Formation of HNCO further constrained via interaction of CO with NH3 plasma dissociation products | ||||||||||
| CO | NH3(dissociated) | |||||||||
| 5.1 | CO:NH3(dissociated) | nn | 13 | 0.5 | – | n | 90 | QMS2K/5K | – | Y |
| 5.2 | CO:NH3 | nn | 13 | 0.5 | – | – | 90 | QMS2K/5K | – | N |
| 5.3 | CO:NH3(dissociated) | nn | 70 | 0.5 | – | n | 90 | QMS5K | – | N |
| 13CO | NH3(dissociated) | |||||||||
| 5.4 | 13CO:NH3(dissociated) | nn | 13 | 0.5 | – | n | 90 | QMS2K/5K | – | Y |
| Confirmation of the presence of NH3 plasma dissociation products in the beam | ||||||||||
| D (from D2) | NH3(dissociated) | |||||||||
| 6.1 | D:NH3(dissociated) | nn | 13 | – | 0.05 | n | 60 | – | – | – |
| 6.2 | D:NH3 | nn | 13 | – | 0.05 | – | 60 | – | – | – |
| 6.3 | NH3(dissociated) | nn | 13 | – | – | n | 60 | – | – | – |
Experiments are performed in co-deposition under different laboratory conditions; different co-deposition ratios are given; Ref. N is the reference number; Tsample is the substrate temperature during co-deposition; Rdep is the deposition rate of a selected molecule expressed in ML min−1 under the assumption that 1 L (Langmuir) exposure leads to the surface coverage of 1 ML; Atom-fluxTL is the thermal cracking source atom flux; Atom-fluxPL is the MW plasma source atom flux; absolute uncertainties of H/D- and N- fluxes are 50 and 40 per cent, respectively; t is the time of co-deposition; TPD is the temperature programmed desorption experiment performed afterwards with the TPD rate indicated; Detection of NH3 is the detection of ammonia either by RAIRS or QMS at the end of co-deposition; Detection of HNCO is the detection of isocyanic acid at the end of co-deposition; n – the exact NH3 plasma beam composition is not determined, nn – since the exact NH3 plasma beam composition is unknown the co-deposition ratio is not listed.
aTwo numbers are given for the TPD rate: the first number is the TPD rate that is used below 50 K to gently remove the bulk of CO/N2 ice, and the second number is the TPD rate above 50 K. A higher TPD rate above 50 K is used in order to have a higher peak-to-noise ratio in the QMS. Routinely, 1.5 or 2 K min−1 are used as TPD rates below 50 K. Since no difference is found in the results between the two rates, 2 K min−1 is indicated everywhere.
bY/N means that the detection is uncertain.
cgradual warm-up followed by the acquiring of RAIR spectra is used instead of QMS.
List of the performed experiments.
| Ref. N | Experiment | Ratio | Tsample | Rdep | Atom-fluxTL | Atom-fluxPL | t | TPDa | Detection of | Detection of |
|---|---|---|---|---|---|---|---|---|---|---|
| (K) | (ML min−1) | (1015 cm−2 min−1) | (1015 cm−2 min−1) | (min) | NH3b | HNCOb | ||||
| Verification of NH3 formation | ||||||||||
| CO | H (from H2) | N (from N2) | ||||||||
| 1.1 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | Y | N |
| 1.2 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 180 | – | Y | N |
| 1.3 | N:H:N2:CO | 1:20:100:500 | 13 | 2.5 | 0.1 | 0.005 | 60 | – | Y | N |
| 1.4 | N:H:N2:CO | 1:100:100:100 | 13 | 0.5 | 0.5 | 0.005 | 60 | – | Y | N |
| 1.5 | N:H:N2:CO | 1:100:100:100 | 25 | 0.5 | 0.5 | 0.005 | 60 | – | N | N |
| CO | D (from D2) | N (from N2) | ||||||||
| 1.6 | N:D:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | Y | N |
| H2O | H (from H2) | N (from N2) | ||||||||
| 2.1 | N:H:N2:H2O | 1:20:100:500 | 15 | 2.5 | 0.1 | 0.005 | 90 | – | – | – |
| 2.2 | N:H:N2:H2O | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | – | – |
| Verification of HNCO formation through hydrogenation of N atoms in CO-rich ice analogues | ||||||||||
| CO | H (from H2) | N (from N2) | ||||||||
| 3.1 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 90 | QMS2K/5K | Y | N |
| 3.2 | N:H:N2:CO | 1:6:100:100 | 13 | 0.5 | 0.03 | 0.005 | 90 | QMS2K/5K | Y | Y/N |
| 3.3 | N:H:N2:CO | 1:6:100:100 | 13 | 0.5 | 0.03 | 0.005 | 90 | RAIRSc | Y | Y/N |
| 3.4 | N:H:N2:CO | 1:4:100:100 | 13 | 0.5 | 0.02 | 0.005 | 90 | QMS2K/5K | Y | Y |
| 3.5 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 3.6 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS0.4K/5K | Y/N | Y |
| 3.7 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 180 | QMS2K/5K | Y/N | Y |
| 3.8 | N:H:N2:CO | 1:1.5:100:100 | 13 | 0.5 | 0.0075 | 0.005 | 90 | QMS2K/5K | N | Y |
| 3.9 | N:H:N2:CO | 1:1:100:100 | 13 | 0.5 | 0.005 | 0.005 | 90 | QMS2K/5K | N | Y |
| 3.10 | N:H:N2:CO | 1:2:100:100 | 25 | 0.5 | 0.01 | 0.005 | 90 | QMS1K/5K | N | N |
| 3.11 | N:H:N2:CO | 1:1.5:100:100 | 25 | 0.5 | 0.0075 | 0.005 | 90 | QMS2K/5K | N | N |
| Isotope shift experiments confirming the formation of HN13CO, H15N13CO and D15NCO | ||||||||||
| 13CO | H (from H2) | N (from N2) | ||||||||
| 4.1 | N:H:N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 4.2 | N:H:N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 360 | RAIRSc | Y | Y |
| 13CO | H (from H2) | 15N (from 15N2) | ||||||||
| 4.3 | 15N:H:15N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 13CO | D (from D2) | 15N (from 15N2) | ||||||||
| 4.4 | 15N:D:15N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y/N |
| Formation of HNCO further constrained via interaction of CO with NH3 plasma dissociation products | ||||||||||
| CO | NH3(dissociated) | |||||||||
| 5.1 | CO:NH3(dissociated) | nn | 13 | 0.5 | – | n | 90 | QMS2K/5K | – | Y |
| 5.2 | CO:NH3 | nn | 13 | 0.5 | – | – | 90 | QMS2K/5K | – | N |
| 5.3 | CO:NH3(dissociated) | nn | 70 | 0.5 | – | n | 90 | QMS5K | – | N |
| 13CO | NH3(dissociated) | |||||||||
| 5.4 | 13CO:NH3(dissociated) | nn | 13 | 0.5 | – | n | 90 | QMS2K/5K | – | Y |
| Confirmation of the presence of NH3 plasma dissociation products in the beam | ||||||||||
| D (from D2) | NH3(dissociated) | |||||||||
| 6.1 | D:NH3(dissociated) | nn | 13 | – | 0.05 | n | 60 | – | – | – |
| 6.2 | D:NH3 | nn | 13 | – | 0.05 | – | 60 | – | – | – |
| 6.3 | NH3(dissociated) | nn | 13 | – | – | n | 60 | – | – | – |
| Ref. N | Experiment | Ratio | Tsample | Rdep | Atom-fluxTL | Atom-fluxPL | t | TPDa | Detection of | Detection of |
|---|---|---|---|---|---|---|---|---|---|---|
| (K) | (ML min−1) | (1015 cm−2 min−1) | (1015 cm−2 min−1) | (min) | NH3b | HNCOb | ||||
| Verification of NH3 formation | ||||||||||
| CO | H (from H2) | N (from N2) | ||||||||
| 1.1 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | Y | N |
| 1.2 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 180 | – | Y | N |
| 1.3 | N:H:N2:CO | 1:20:100:500 | 13 | 2.5 | 0.1 | 0.005 | 60 | – | Y | N |
| 1.4 | N:H:N2:CO | 1:100:100:100 | 13 | 0.5 | 0.5 | 0.005 | 60 | – | Y | N |
| 1.5 | N:H:N2:CO | 1:100:100:100 | 25 | 0.5 | 0.5 | 0.005 | 60 | – | N | N |
| CO | D (from D2) | N (from N2) | ||||||||
| 1.6 | N:D:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | Y | N |
| H2O | H (from H2) | N (from N2) | ||||||||
| 2.1 | N:H:N2:H2O | 1:20:100:500 | 15 | 2.5 | 0.1 | 0.005 | 90 | – | – | – |
| 2.2 | N:H:N2:H2O | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 60 | – | – | – |
| Verification of HNCO formation through hydrogenation of N atoms in CO-rich ice analogues | ||||||||||
| CO | H (from H2) | N (from N2) | ||||||||
| 3.1 | N:H:N2:CO | 1:20:100:100 | 13 | 0.5 | 0.1 | 0.005 | 90 | QMS2K/5K | Y | N |
| 3.2 | N:H:N2:CO | 1:6:100:100 | 13 | 0.5 | 0.03 | 0.005 | 90 | QMS2K/5K | Y | Y/N |
| 3.3 | N:H:N2:CO | 1:6:100:100 | 13 | 0.5 | 0.03 | 0.005 | 90 | RAIRSc | Y | Y/N |
| 3.4 | N:H:N2:CO | 1:4:100:100 | 13 | 0.5 | 0.02 | 0.005 | 90 | QMS2K/5K | Y | Y |
| 3.5 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 3.6 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS0.4K/5K | Y/N | Y |
| 3.7 | N:H:N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 180 | QMS2K/5K | Y/N | Y |
| 3.8 | N:H:N2:CO | 1:1.5:100:100 | 13 | 0.5 | 0.0075 | 0.005 | 90 | QMS2K/5K | N | Y |
| 3.9 | N:H:N2:CO | 1:1:100:100 | 13 | 0.5 | 0.005 | 0.005 | 90 | QMS2K/5K | N | Y |
| 3.10 | N:H:N2:CO | 1:2:100:100 | 25 | 0.5 | 0.01 | 0.005 | 90 | QMS1K/5K | N | N |
| 3.11 | N:H:N2:CO | 1:1.5:100:100 | 25 | 0.5 | 0.0075 | 0.005 | 90 | QMS2K/5K | N | N |
| Isotope shift experiments confirming the formation of HN13CO, H15N13CO and D15NCO | ||||||||||
| 13CO | H (from H2) | N (from N2) | ||||||||
| 4.1 | N:H:N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 4.2 | N:H:N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 360 | RAIRSc | Y | Y |
| 13CO | H (from H2) | 15N (from 15N2) | ||||||||
| 4.3 | 15N:H:15N2:13CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y |
| 13CO | D (from D2) | 15N (from 15N2) | ||||||||
| 4.4 | 15N:D:15N2:CO | 1:2:100:100 | 13 | 0.5 | 0.01 | 0.005 | 90 | QMS2K/5K | Y/N | Y/N |
| Formation of HNCO further constrained via interaction of CO with NH3 plasma dissociation products | ||||||||||
| CO | NH3(dissociated) | |||||||||
| 5.1 | CO:NH3(dissociated) | nn | 13 | 0.5 | – | n | 90 | QMS2K/5K | – | Y |
| 5.2 | CO:NH3 | nn | 13 | 0.5 | – | – | 90 | QMS2K/5K | – | N |
| 5.3 | CO:NH3(dissociated) | nn | 70 | 0.5 | – | n | 90 | QMS5K | – | N |
| 13CO | NH3(dissociated) | |||||||||
| 5.4 | 13CO:NH3(dissociated) | nn | 13 | 0.5 | – | n | 90 | QMS2K/5K | – | Y |
| Confirmation of the presence of NH3 plasma dissociation products in the beam | ||||||||||
| D (from D2) | NH3(dissociated) | |||||||||
| 6.1 | D:NH3(dissociated) | nn | 13 | – | 0.05 | n | 60 | – | – | – |
| 6.2 | D:NH3 | nn | 13 | – | 0.05 | – | 60 | – | – | – |
| 6.3 | NH3(dissociated) | nn | 13 | – | – | n | 60 | – | – | – |
Experiments are performed in co-deposition under different laboratory conditions; different co-deposition ratios are given; Ref. N is the reference number; Tsample is the substrate temperature during co-deposition; Rdep is the deposition rate of a selected molecule expressed in ML min−1 under the assumption that 1 L (Langmuir) exposure leads to the surface coverage of 1 ML; Atom-fluxTL is the thermal cracking source atom flux; Atom-fluxPL is the MW plasma source atom flux; absolute uncertainties of H/D- and N- fluxes are 50 and 40 per cent, respectively; t is the time of co-deposition; TPD is the temperature programmed desorption experiment performed afterwards with the TPD rate indicated; Detection of NH3 is the detection of ammonia either by RAIRS or QMS at the end of co-deposition; Detection of HNCO is the detection of isocyanic acid at the end of co-deposition; n – the exact NH3 plasma beam composition is not determined, nn – since the exact NH3 plasma beam composition is unknown the co-deposition ratio is not listed.
aTwo numbers are given for the TPD rate: the first number is the TPD rate that is used below 50 K to gently remove the bulk of CO/N2 ice, and the second number is the TPD rate above 50 K. A higher TPD rate above 50 K is used in order to have a higher peak-to-noise ratio in the QMS. Routinely, 1.5 or 2 K min−1 are used as TPD rates below 50 K. Since no difference is found in the results between the two rates, 2 K min−1 is indicated everywhere.
bY/N means that the detection is uncertain.
cgradual warm-up followed by the acquiring of RAIR spectra is used instead of QMS.
3 RESULTS AND DISCUSSION
3.1 Formation of NH3
A series of co-deposition experiments (see experiments 1.1–1.6 in Table 1) is performed to simulate the formation of NH3 under dense cold interstellar cloud conditions, i.e. when gas-phase CO has accreted onto the grains and the UV field is still negligible. A RAIR difference spectrum from a co-deposition of N:H:N2:CO = 1:20:100:100 at 13 K with a total N-atom fluence of 3×1014 atoms cm−2 (±40 per cent) is shown in the large panel of Fig. 1. 12CO (2140 cm−1) and 13CO (2092 cm−1) are both visible. The inset in Fig. 1 shows this experiment in a smaller spectral range (Fig. 1a), as well as two more N:H:CO co-deposition experiments at 13 K for different mixing ratios (Figs 1b and c), and a control experiment with only NH3 and CO molecules co-deposited at 13 K (Fig. 1d). The same N-atom effective flux (8×1010 atoms s−1 cm−2) within a 40 per cent accuracy and total N-atom total fluence are used in the first three experiments. It should be noted that the amount of N2 in the final mixed ice cannot be disregarded, since the N-atom beam comprises a considerable amount of non-dissociated N2 molecules that, unlike H2, can freeze out at 13 K and form a solid layer of ice (Cuppen & Herbst 2007).
The formation of NH3 is confirmed in Fig. 1 [and inset Fig. 1(a)] by the appearance of two absorption features at ν2 = 975 cm−1 and ν4 = 1625 cm−1 (Abouaf-Marguin, Jacox & Milligan 1977; Koops, Visser & Smit 1983; Nelander 1984). In addition, a third feature at ν3 = 3430 cm−1 is observed in the region of the N–H and O–H stretching modes (not shown in the figure). Furthermore, solid H2CO (ν2 = 1728 cm−1 and ν3 = 1499 cm−1) shows up as a result of CO hydrogenation. Formation of H2CO by H-atom addition to CO has been previously studied (e.g. Hiraoka et al. 1994; Watanabe & Kouchi 2002; Zhitnikov & Dmitirev 2002; Fuchs et al. 2009). Ongoing hydrogenation can form solid CH3OH that is below its detection limit here. A small feature around 1600 cm−1 can be assigned to either H2O impurity or to the aggregate of NH3. The latter assignment is supported by a negligible admixture of O2 or H2O in the N2 bottle used in the experiments. Small negative peaks in the range between 1350 and 1750 cm−1 are water vapour absorptions along the path of the FTIR beam outside the UHV chamber. Unfortunately, these absorptions are still visible in some of the spectra despite the use of a dry air purged system.
Fig. 1(b) shows the co-deposition of N:H:N2:CO = 1:20:100:500 at 13 K. In this case, the deposition rate of CO is five times higher than in the experiment plotted in Fig. 1(a). Figs 1(a) and (b) show the same NH3 final amount, but the H2CO peaks are more prominent in the spectrum with higher CO abundance. This is expected, since the effective CO surface coverage for the experiment with N2:CO = 100:500 is about 1.7 times higher than for N2:CO = 100:100 (i.e. 50 per cent CO surface coverage for N2:CO = 100:100 versus 87 per cent CO surface coverage for N2:CO = 100:500). Clearly, CH3OH abundances are still below the detection limit.
Fig. 1(c) shows a co-deposition spectrum with an absolute H-atom flux five times higher (N:H:N2:CO = 1:100:100:100) than the one in Fig. 1(a). This results in a further increase of the formed H2CO, consistent with previous work (e.g. Fuchs et al. 2009). In contrast with the H2CO final yield, ammonia absorption features and, therefore, the corresponding formation yield does not increase: the ν4 total absorbance shows the same value, while the ν2 total absorption is even 35–40 per cent less intense than the one in Fig. 1(a). This apparent inconsistency can be explained by the ν2 mode (symmetrical deformation) being more sensitive to the ice mixture composition than the ν4 mode (degenerate deformation), particularly with H2CO around which can form hydrogen bonds with NH3. The fact that the ν2 mode is significantly more sensitive to environmental changes than the ν4 mode was also found by Abouaf-Marguin et al. (1977). The latter work shows that when the hydrogen bonds are formed, the position of the ν2 band is shifted as much as 70–80 cm−1 with respect to the position of the monomeric NH3, while this difference is only 10–30 cm−1 for the ν4 mode. Fig. 1 in Hagen & Tielens (1982) further illustrates this for a 10 K CO matrix. In addition, more H2 is expected to be trapped in the growing matrix in the higher H-atom flux experiment, and this will further affect the environment in which NH3 is isolated.
Finally in Fig. 1(d), a RAIR spectrum of NH3 co-deposited with CO molecules is shown. The total amount of deposited NH3 is 0.3 ML. This number is about the same as for the N-atom total fluence in each of the three aforementioned experiments. A ratio NH3:CO = 1:500 is chosen to reproduce the ratio used in Fig. 1(b). The total absorbance of deposited NH3 molecules in Fig. 1(d) is about 10 and 40 per cent higher for the ν4 and ν2 modes, respectively, compared to the abundances of NH3 formed by N-atom hydrogenation in the experiments depicted in Figs 1(a) and (b). Although these differences are within the flux uncertainties, the larger difference in the ν2 mode is likely due to the higher sensitivity of the ν2 mode towards its environment.
In general, Fig. 1 shows that under our experimental conditions the final NH3 yield is determined by the total amount of available nitrogen atoms at the ice surface, as the integrated area of the ν4 mode stays near constant in all four plots, while for the ν2 mode this varies significantly in plot (c). The experiments indicate that the hydrogenation of the deposited N atoms is a faster and more efficient process than the hydrogenation of CO ice. Very low activation barriers are therefore expected for reactions (1)–(3). This is consistent with N-atom hydrogenation experiments in a solid N2 matrix by Hiraoka et al. (1994) and Hidaka et al. (2011). The Hidaka et al. (2011) experiments are tested under our experimental conditions, i.e. a co-deposition of N:H:N2 = 1:20:100 is performed at 15 K, and NH3 formation is also observed in this experiment.
Finally, we performed a few co-deposition (control) experiments of H- and N-atom beams with H2O instead of CO (see experiments 2.1 and 2.2 in Table 1). A strong broadening of the NH3 absorbance features due to hydrogen bonds and a considerable overlap of H2O and NH3 absorption features do not allow for an unambiguous assignment of NH3 peaks in these experiments, TPD using the QMS does not help to overcome the problem since co-desorbing H2O gives similar m/z numbers to NH3 complicating the assignments. Therefore, these experiments will not be further discussed in this section.
3.2 Temperature dependence
A co-deposition experiment using the same deposition rates discussed before (N:H:N2:CO = 1:100:100:100) is repeated for different substrate temperatures (13 and 25 K) to study the temperature effect on the N-atom hydrogenation in CO-rich ices. The temperatures chosen are below the desorption values of N2, N, and CO molecules (Acharyya et al. 2007). The goal of these experiments is to determine which mechanism is responsible for the formation of NH3 in a CO-rich environment, i.e. a Langmuir–Hinshelwood (L-H), Eley–Rideal (E-R), or ‘hot-atom’ mechanism. Since both E-R and ‘hot-atom’ mechanisms exhibit very limited sample temperature dependence over the short range of temperatures, one expects to find similar NH3 final yields in both experiments. This kind of dependence is found for example for NO + N co-deposition experiments (Ioppolo et al. 2013). In the case that L-H is responsible for the formation of ammonia, then the resulting NH3 formation rate is a rather complex combination of many individual processes that are temperature dependent (i.e. lifetime of H atoms on the surface, hopping rate of H and N atoms, and H-atom recombination rate). In this case, the NH3 yield should drop significantly at 25 K due to the shorter residence time of H atoms on the ice surface. For instance, the lifetime of H atoms on a water ice surface at 25 K is more than 1000 times shorter than at 13 K (Cuppen & Herbst 2007). A decrease of H2CO and CH3OH formation yields with increasing temperature was already observed by Watanabe et al. (2006) and Fuchs et al. (2009) in CO hydrogenation experiments, and this observation was explained assuming a L-H formation mechanism. Fig. 4 in the accompanying paper (Fedoseev et al. 2014) shows for similar experimental conditions a substantial drop in the amount of ammonia formation between 15 and 17 K further constraining the proposed L-H mechanism.
The two spectra in Fig. 2 show that neither H2CO nor NH3 is detected at 25 K by means of RAIRS. This is fully consistent with surface processes following an L-H mechanism, as suggested in the previous work that focused on the hydrogenation of N atoms trapped in an N2 matrix (Hidaka et al. 2011).
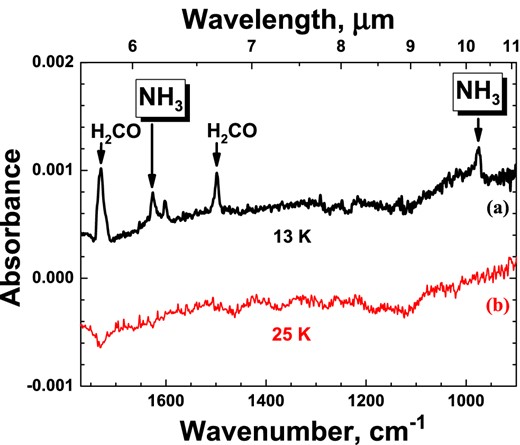
Two RAIRs difference spectra of the same co-deposition experiment N:H:N2:CO = 1:100:100:100 with the same total N-atom fluence of 3×1014 (±40 per cent) atoms cm−2 at 13 (a) and 25 K (b) (see experiments 1.4 and 1.5). Spectra are plotted with offsets.
3.3 Formation of HNCO
The surface formation pathway of ammonia through the sequential hydrogenation of N atoms leads to the formation of NH and NH2 intermediates that can also react with other species in the ice to form different molecules. One obvious candidate in a CO surrounding is HNCO formed through surface reactions (4) and (5). In the experiments shown in Figs 1 and 2, each NH and NH2 intermediate will face at least one neighbouring CO molecule as with the chosen co-deposition ratios for deposited N atoms there are 100 CO and 100 N2 molecules. However, in our RAIR spectra, solid HNCO and its hydrogenation products (e.g. NH2CO and NH2CHO) cannot be detected. Even sensitive mass spectrometry does not show any clear evidence for the formation of these species (masses 42–45 m/z). Thus, reactions (4) and (5) most likely experience an activation barrier and consequently are overtaken by reactions leading to NH3 formation. This is consistent with Himmel et al. (2002), who indeed suggested the presence of an activation barrier even though some reactivity of NH towards CO at 10 K was found.
Here, a new set of experiments is presented to study the surface formation of HNCO through the reaction of CO molecules with NH and NH2. The specific goal of these experiments is to prohibit the (fast) formation of ammonia, and to simultaneously increase the probability for NH and NH2 intermediates to react with CO molecules, overcoming any activation barriers. Such a set of experiments, in fact, is more representative for the actual processes taking place on interstellar grains, where H- and N-atom accretion rates are so low that once NH and NH2 radicals are formed, these experience a relatively long time to react with other ice molecules (∼ several days) before another impacting H atom contributes to the formation of ammonia. Thus, to reproduce this scenario, N atoms are co-deposited with CO molecules with the same rates as described in Section 3.1, while the H-atom co-deposition rate is substantially decreased (20 times less) to prevent full hydrogenation of N atoms, offering a pathway to the formed NH and NH2 to react with CO (see experiments 3.1–3.11 in Table 1). TPD experiments combined with QMS data are used to study the expected low HNCO final yield.
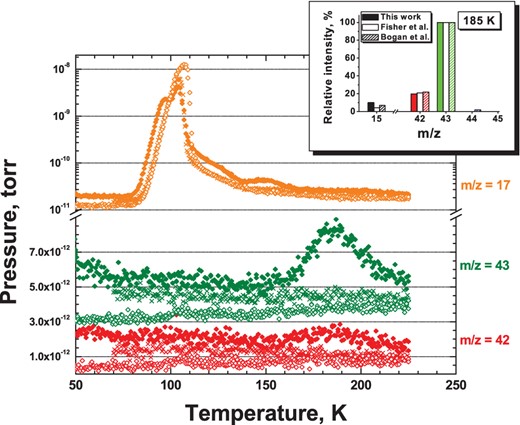
Three selected N + H + CO co-deposition experiments are presented in Fig. 3. After co-deposition of CO molecules with H and N atoms with a given ratio at 13 K, the ice is gently and linearly warmed up to 50 K with a rate of 2 K min−1 to remove the bulk of the CO ice. A rate of 5 K min−1 is used during the second part of the TPD (up to 225 K) in order to have a higher peak-to-noise ratio of the selected masses in the mass spectrometer. The correlation between the NH3 and HNCO final yields for different H-atom co-deposition ratios is investigated by integrating the corresponding area of the selected species from their QMS mass signal over time (i.e. m/z = 17 for NH3 and 42, 43 for HNCO). Fig. 3 shows the decrease of the NH3 formation yield (peak centred at 120 K), and the corresponding gradual increase of the HNCO formation yield (peak centred at 185 K) that follows the decrease of H-atom co-deposition ratio from N:H:N2:CO = 1:20:100:100 to N:H:N2:CO = 1:2:100:100. In the first experiment, only traces of HNCO are detected by the QMS, while the NH3 signal is maximum. In the third experiment, with a 10 times smaller H flux, the HNCO final yield is maximum while only traces of NH3 are present. The intermediate case, corresponding to a co-deposition ratio of N:H:N2:CO = 1:6:100:100, results in the presence of both NH3 and HNCO molecules.
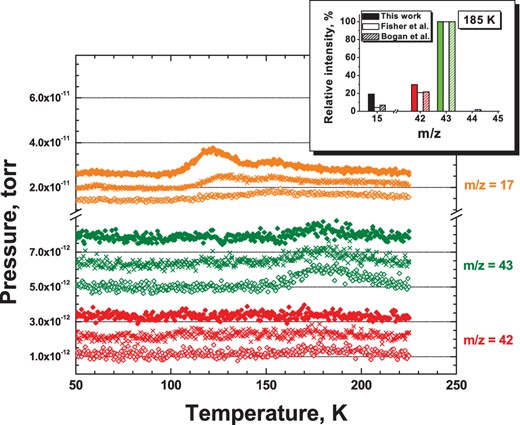
The TPD spectra for three different experiments: co-deposition of N:H:N2:CO = 1:20:100:100 at 13 K (filled diamonds); co-deposition of N:H:N2:CO = 1:6:100:100 at 13 K (crosses); and co-deposition of N:H:N2:CO = 1:2:100:100 at 13 K (open diamonds), see experiments 3.1, 3.2, and 3.5, respectively. The total N-atom fluence in each of the three experiments is 4.5×1014 (± 40 per cent) atom cm−2. Peaks at m/z = 17 are due to NH3 (centred at 120 K) and background H2O (centred at 155 K), m/z = 42 and 43 (middle and lower panel) are the two most intense signals from HNCO. Plots are shown with offsets. In the top-right corner, an inset is shown with the relative intensities for m/z = 15, 42, and 43 (HNCO), as derived in this study and compared to the available literature.
HNCO can be assigned in the TPD experiments to the desorbing species peaked at 185 K by looking at the electron ionization fragmentation pattern (see Bogan & Hand 1971; Fischer et al. 2002). The inset in Fig. 3 compares the ratio between m/z = 43, 42, and 15 (i.e. HNCO+, NCO+, HN+) in our experiment and literature values.
To further constrain this assignment, similar experiments are performed with H atoms co-deposited with 14/15N atoms and 13CO molecules (see experiments 4.1–4.3 in Table 1). In both cases (14N and 15N), a consistent isotopic shift of both peaks at m/z = 42 and 43 is observed, while the ratio between these two peaks stays constant.
Pure HNCO is known to desorb slightly above 120 K (Theule et al. 2011). However, as shown in Fig. 3, a much higher desorption temperature is found in our experiments. Upon desorption of the bulk of CO ice, HNCO may form ammonium isocyanate (NH4+OCN−) or hydronium isocyanate (H3O+OCN−) in presence of NH3 or H2O, respectively (reactions 6 and 7). This indeed shifts the desorption temperature of HNCO to higher values. Reactions (6) and (7) can take place during the thermal processing of mixed NH3:HNCO and H2O:HNCO ices and have been extensively studied by Raunier et al. (2003) and Theule et al. (2011), respectively. Under our experimental conditions, both NH3 and H2O are present in the ice sample during the TPD: i.e. NH3 is a product of N-atom hydrogenation, and H2O originates from background deposition (see the second peak around 155 K for m/z = 17 (OH+) in Fig. 3). In addition, the low final yield of HNCO (<1 ML) indicates that this molecule likely occupies the surface spots with the highest binding energy. This further shifts the desorption temperature to higher values. Unfortunately, the presence of background water in the main UHV chamber gives both m/z = 18 (H2O+) and 17 (OH+). CO that is present in the main chamber after co-deposition gives m/z = 16 signal (O+). As a consequence, it is not possible to make unambiguous assignments for the base counter parting HNCO acid. Finally, it should be noted that both NH2CHO (formamide) and (NH2)2CO (urea), two possible chemical derivatives of HNCO and NH4+NCO−, respectively, cannot be observed under our experimental conditions and must be under the detection limit of both QMS and RAIRS.
3.3.1 Control experiments
We performed several control experiments to constrain the formation of HNCO and OCN− at low temperatures by using RAIRS and QMS techniques during TPD experiments (see experiments 3.3 and 4.2 in Table 1). RAIR spectra can only be used to identify new species formed in the ice when their final yield is >0.1 ML. To enhance the RAIR signal for species, like HNCO and OCN−, we performed co-deposition experiments two times longer (experiment 3.3) and four times longer (experiment 4.2) than the corresponding experiments shown in Fig. 3. Unfortunately, the main infrared absorption band of HNCO at 2260 cm−1 (Teles et al. 1989) lies too close to the adsorption features of atmospheric CO2 that is present along the path of our IR beam, outside the UHV chamber. In addition, CO infrared features overlap with the strongest OCN− band (van Broekhuizen et al. 2005), making OCN− detectable only after desorption of the bulk of CO ice. Therefore, a co-deposition experiment with 13CO (experiment 4.2) is shown in Fig. 4. In this figure, some infrared spectra acquired at different temperatures during TPD are presented in two selected spectral regions. The left-hand panel covers the spectral range where HN13CO should be observed (Teles et al. 1989), while the right-hand panel shows the range where O13CN− is expected. Solid HN13CO can only be observed at 13 K, while O13CN− is clearly present in the spectra taken at 35 and 50 K, well before desorption at 185 K. Thus, the present results support the hypothesis that HN13CO is formed already at 13 K, during co-deposition, and as soon as 13CO desorbs, HN13CO reacts with NH3 or H2O to yield NH4+O13CN− and H3O+O13CN−, respectively. Formation of OCN− is also observed upon desorption of the bulk of the ice in experiment 3.3 using regular 12CO isomers.
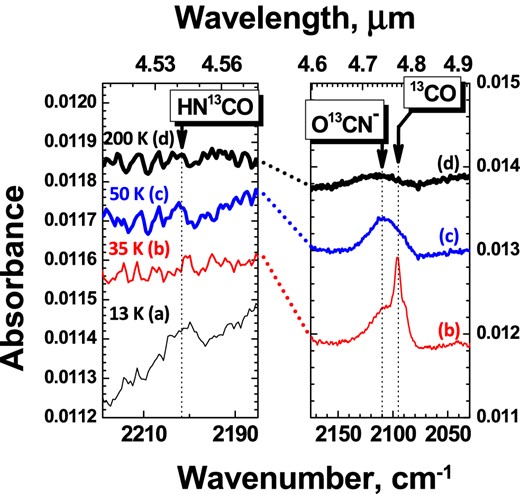
Four RAIR difference spectra obtained after co-deposition of N:H:N2:13CO = 1:2:100:100 at 13 K followed by a TPD of the ice (experiment 4.2): (a) 13 (b) 35 (c) 50 and (d) 200 K. The left-hand panel shows the strongest absorption feature of HN13CO in the mid-IR; the right-hand panel shows the strongest absorption feature of O13CN−. The total N-atom fluence is 1.8×1015 atoms cm−2 (± 40 per cent). In the right-hand panel, the 13 K plot is not shown because of the very high absorption of bulk 13CO. Some non-desorbed 13CO is still present in the plot at 35 K.
Below, additional arguments are discussed that are in favour of the HNCO formation through the interaction of NH/NH2 with CO molecules: (i) the co-deposition experiment that yields HNCO at 13 K is repeated for 25 K where it does not result in a QMS detection of HNCO or NH3 (experiments 3.10 and 3.11). This indicates that the involved formation mechanism for both species depends on the H-atom lifetime on the ice surface that is known to decrease substantially for increasing co-deposition temperature; (ii) a two times longer co-deposition time is applied for identical settings and leads to a two times larger HNCO area on the QMS TPD spectra (experiment 3.7); (iii) neither HNNH nor H2NNH2 are observed (within our detection limits) but both are expected to show up during TPD in the case that non-reacted NH and NH2 become mobile after the bulk of the ice has desorbed.
To further verify the reactivity of NH/NH2 radicals with CO molecules, we performed another set of RAIRS experiments (see experiments 5.1–5.4): a co-deposition of CO molecules with fragmentation products formed by discharging NH3 in the microwave plasma for different experimental conditions. The microwave discharge of ammonia results in the beam containing various plasma dissociation products (i.e. along with the non-dissociated NH3 molecules the beam may contain NH and NH2 radicals, H and N atoms as well as H2 and N2 molecules). The presence of NH and NH2 radicals is confirmed by co-depositing the products of NH3 plasma dissociation with an overabundance of D atoms under the same experimental conditions as in experiments 5.1–5.4. In this case, two broad features are observed in the N–D stretching vibrational mode region (2438 and 2508 cm−1, respectively) together with the absorption features due to non-dissociated NH3. This indicates that at least part of the NH3 is decomposed into NH and NH2 radicals and N atoms that then react with D atoms to form the observed N–D bonds.
The QMS TPD spectra obtained after co-deposition of CO molecules with NH3 plasma dissociation products at 13 K results again in a mass peak at m/z = 43 for 185 K that can be assigned to HNCO according to the electron ionization fragmentation pattern (see the filled diamonds in the main panel and the inset in Fig. 5). Additionally, the open diamonds in Fig. 5 represent a TPD spectrum after co-deposition of unprocessed NH3 and CO that proves that the formed HNCO is not the result of thermal processing of mixed NH3:CO ice. Cross symbols in Fig. 5 represent data from the experiment where CO molecules are co-deposited with NH3 plasma dissociation products at a substrate temperature of 70 K, well above the CO desorption temperature (peaked at 29 K, Acharyya et al. 2007). This experiment confirms that the formed HNCO does not originate from gas-phase reactions or contaminations in the microwave plasma source and that the presence of CO on the surface is a prerequisite to form HNCO. All the aforementioned experiments are performed by using the same procedure: first the ice is warmed up with a 2 K min−1 rate from 13 to 50 K to remove the bulk of CO ice; then a rate of 5 K min−1 is used from 50 to 225 K. A control experiment is performed with a different isotope (experiment 5.4); when NH3 plasma dissociation products are co-deposited with 13CO, m/z values of 43 and 44 present a clear feature at 185 K, the ratio 44/43 is 0.27, while m/z = 42 is not found.
The TPD spectra of three distinct experiments: co-deposition of NH3 plasma fragments with CO at 13 K (filled diamonds); co-deposition of NH3 plasma fragments with CO at 70 K (crosses); and co-deposition of NH3 and CO at 13 K when the microwave discharge is turned off (open diamonds), see experiments 5.1, 5.3, and 5.2, respectively. Three m/z values are selected: masses 42 and 43 are the two most intense signals from HNCO, while 17 comes from NH3. Plots are shown with offsets for clarity. In the top-right corner, the inset compares the relative intensities of the masses assigned in this study to HNCO and the available literature values.
3.3.2 HNCO formation pathway
As mentioned in Section 3.3, NH2CHO (the product of sequential HNCO hydrogenation) is not observed in any of our experiments. This is not surprising since H-atom addition to HNCO involves an activation barrier. Nguyen et al. (1996) used ab initio calculations to study the reaction of H atoms with isocyanic acid, and an activation barrier of 1390 K was found for H-atom addition to the nitrogen atom of HNCO. This makes the formation of NH2CHO unlikely under our experimental conditions, since it would imply a second consequent reaction involving an activation barrier, while the lack of H atoms is used in the experiments resulting in HNCO formation.
4 ASTROCHEMICAL IMPLICATIONS
This laboratory work is motivated by several of the main conclusions of the Spitzer c2d Legacy ice survey (Öberg et al. 2011). The evolutionary steps of interstellar ice formation can be divided into three main stages: an early phase, driven by fast H-atom addition reactions in cold molecular clouds, before cloud core formation; a later CO freeze-out stage, when chemistry in the ices is driven by accreting CO molecules to a large extent; and the protostellar phase, where thermal and UV processing shape the ice content.
During the first stage, a H2O-rich (polar) ice is formed. In this phase, the relative abundances of CO2 (in H2O), CH4, and NH3 correlate with H2O ice suggesting their co-formation. This indicates that most of the solid NH3 is formed during an early evolutionary stage. Our laboratory experiments are designed to study the non-energetic surface formation of NH3 through the hydrogenation of N atoms under cold dense cloud conditions. These conditions approximately resemble the first stage of interstellar ice formation. We therefore performed some experiments co-depositing H and N atoms with water, but the spectral confusion due to the overlap of features from H2O and NH3, as well as the strong broadening of the NH3 absorption bands in a polar ice made an unambiguous assignment of NH3 ice formation far from trivial. Therefore, this work mostly focuses on the investigation of the NH3 formation in non-polar mixtures containing CO ice. This way, newly formed NH3 can be easily identified, because ammonia features are sharper and do not overlap with features of other species in the ice. Although CO-rich ices better represent the second phase of interstellar ices, when CO molecules freeze out onto the grains, some of our conclusions on the surface formation of ammonia in CO-rich ice can also be extended – within limits – to the formation of NH3 in H2O-rich ice. For instance, we find that the formation of NH3 by hydrogenation of N atoms proceeds barrierless or through a very small activation barrier at 13 K. Moreover, in agreement with previous works, we confirm an L-H mechanism as the main channel for the formation of solid NH3 (Hiraoka et al. 1995 and Hidaka et al. 2011). Our experimental results further constrain the findings described in Charnley, Rodgers & Ehrenfreund (2001), where the accretion of gas-phase ammonia in their model results in a solid NH3/H2O ratio of only ∼2 per cent, which is less than the observed values of 5 per cent (Gibb et al. 2004; Bottinelli et al. 2010; Öberg et al. 2011). However, the amount of NH3 ice on the grains could be higher assuming that N atoms also accrete onto grains and undergo hydrogenation. If this surface formation route of ammonia is included in the Charnley et al. (2001) model, the solid NH3/H2O ratio becomes ∼10 per cent, which is even above the observed value. Moreover, recent models that account for the formation of NH3 in the solid phase through hydrogenation of N atoms indicate a NH3/H2O ratio as high as 25 per cent (Garrod & Pauly 2011 and Vasyunin & Herbst 2013).
Our experimental results can explain why recent astrochemical models overestimate the surface production of NH3. We observe an efficient formation of HNCO ice in H+N+CO experiments. Solid HNCO is a product of the interaction between CO molecules and intermediates involved in the surface formation of NH3. In this scenario, NH3 ice is formed efficiently in a polar ice together with water during the first phase of interstellar ices. However, as soon as densities are high enough for CO to freeze out onto the grains, the formation of NH3 competes with the formation of HNCO in a non-polar ice. Our experiments reveal that the formation of NH3 in CO-rich ices is only efficient when the H-atom deposition rate is high enough to quickly hydrogenate all the N atoms to NH3 which can only occur on rather fast laboratory time-scales. If a slower H-atom deposition rate is used to simulate the slow accretion rate observed in the ISM as much as possible, the formation of NH3 is suppressed in favour of the formation of HNCO. In this case, formed NH or NH2 radicals have significantly more time to overcome the activation barrier of the reaction with the surrounding CO molecules before the next H atom arrives and eventually converts it to NH3. In space, the extremely low accretion rate of H atoms on the surface of the icy grains (unfortunately, not reproducible in the laboratory) gives days to each of the intermediates to overcome activation barriers and to react with the surrounding molecules (i.e. CO ice), before the next hydrogenation event occurs. This potentially explains the observed low ammonia abundances in non-polar ices and, at the same time, shows that solid HNCO is formed in molecular clouds.
Interstellar HNCO was first detected in the Sgr B2 molecular cloud complex by Snyder & Buhl (1972). Since its discovery, HNCO has been detected in different environments, as diverse as dark molecular clouds and hot cores in massive star-forming regions. Li et al. (2013) studied the spatial distribution of HNCO in massive YSOs that is consistent with a ‘hot’ gas-phase formation route. In cold dark molecular clouds, however, HNCO is expected to be efficiently formed in the solid phase. Quan et al. (2010) were able to reproduce the observed gas-phase abundances of HNCO and its isomers in cold and warm environments using gas–grain simulations, which include both gas-phase and grain-surface routes. Our work shows that HNCO is efficiently formed under dense cold cloud conditions, i.e. in non-polar ices with a reaction pathway that is linked to the formation of ammonia ice and does not require any energetic input, such as UV light or cosmic ray irradiation. Although our work indicates that HNCO should be detectable in CO-rich ices through its antisymmetric stretch mode at ∼2250 cm−1, this is not the case; the non-detection of solid HNCO in the ISM can be explained by efficient destruction pathways that include the hydrogenation of HNCO as well as thermal reactions with solid ammonia at temperatures slightly higher than its formation temperature. The latter mechanism is supported by a combined laboratory and modelling study that derives a low (48 K) activation energy barrier for reaction (6) to occur (Mispelaer et al. 2012).
Öberg et al. (2011) reported a close correlation between CO, CO2 (in CO), CO (in H2O), and the XCN band in support of their co-formation during the CO freeze-out stage as well as the identification of OCN− as a main carrier of the interstellar XCN band. Our TPD experiments shown in Fig. 4 simulate the heating of ice mantles by a newly formed protostar (i.e. the third phase of interstellar ices). This process leads to the formation of O13CN− detectable from its infrared absorption feature centred at 2202 cm−1 (in our experiments we used 13CO instead of the regular 12CO). While NH3 and HN13CO are formed through non-energetic surface reactions at low temperature, the formation of O13CN− occurs through the interaction of HN13CO with NH3 or H2O molecules at higher temperatures, when highly volatile species leave the ice. Thus, our results further constrain the assignment of either the entire or a single component (2165 cm−1) of the XCN band observed towards numerous YSOs to solid OCN− and show that OCN− is successfully formed in the interstellar ices without any UV or cosmic ray processing involved (see Gibb et al. 2004; Bottinelli et al. 2010; Öberg et al. 2011).
The presence of HNCO and OCN− in interstellar ices during the protostellar phase may be very important for astrobiology. The recent detection of amino acids in comets has boosted efforts to investigate the astrochemical origin of species such as glycine and alanine (Elsila et al. 2009). Since gas-phase routes to form these complex species seem to be inefficient, solid-phase formation pathways offer a strong alternative (Barrientos et al. 2012). During protostar formation, interstellar grains are exposed to thermal, UV, electron, and ion processing that can drastically modify the composition of the icy mantles. Especially in the case of cosmic ray irradiation, the energy released by the ions passing through a material causes the dissociation of hundreds of molecules along their path. These fragments can then recombine forming new and more complex species. Eventually, a complex polymeric refractory residue can be formed. As shown in Fig. 6, HNCO molecules are included as a peptide bond [–(H)N–C(O)–] between any two single amino acid. Moreover, even the simplest peptide, polyglycine, contains nothing but HNCO and CH2 components. Therefore, energetic processing (e.g. UV photolysis and cosmic ray irradiation) of HNCO:CH4 and HNCO:CH4:CH3OH-rich ices can be a possible pathway to form amino acids or peptide fragments. If OCN− is used in the aforementioned mixtures instead of HNCO, amino acid anions and their fragments can be formed as well. Such experiments will be the focus of a future laboratory study aimed to investigate the formation of the simplest amino acids and peptide fragments, but for now, it is important to conclude that convincing solid-state pathways are found that explain the effective formation of the elementary precursor species.
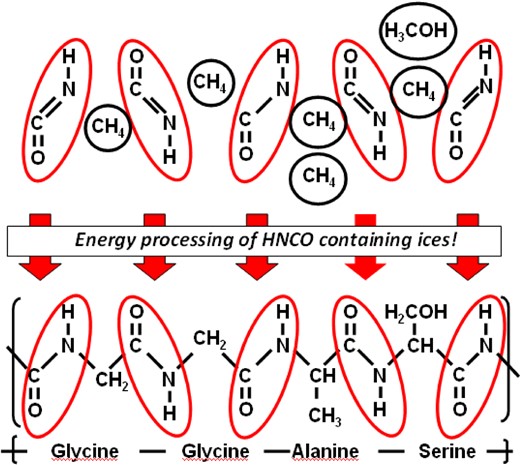
A schematic that illustrates the potential importance of HNCO as a simple bearer of peptide bonds for the production of amino acids in interstellar ices.
We thank Dr Herma Cuppen for useful discussions. This work is financially supported by the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 238258 (LASSIE), the Netherlands Organization for Scientific Research (NWO) through a VICI grant, and NOVA, the Netherlands Research School for Astronomy. Support for SI from the Niels Stensen Fellowship and the Marie Curie Fellowship (FP7-PEOPLE-2011-IOF-300957) is gratefully acknowledged.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}