






ABSTRACT
In recent years, the interplay of epigenetics and infection moved into the limelight. Epigenetic regulation describes modifications in gene expression without alterations of the DNA sequence. In eukaryotes, this mechanism is central for fundamental cellular processes such as cell development and differentiation, but it is also involved in more specific tasks such as the response to infection by a pathogen. One of the most common types of epigenetic changes is the modification of histones. Histones, the small protein building blocks that are wrapped with DNA are the fundamental packaging unit of chromatin. Histones can be modified by linking different moieties to them—one of the most abundant ones is acetylation. Histone acetylation is regulated by two main classes of enzymes, histone acetyl transferases (HAT) and their counterparts, histone deacetylases (HDAC). Given the high abundance and importance in regulating gene expression, histone acetylation is an excellent target for pathogens to manipulate the host cell to their advantage. Targeting HDACs gained particular interest in recent years, due to the increased use of HDAC inhibitors in clinical practice. Recently, the possibility to fight an infection with HDAC inhibitors was suggested as an alternative to overcome the ever-growing problem of antibiotic resistance. In this review, we focus on the regulation of HDACs and their involvement in immune cell function. We then highlight different mechanisms employed by pathogens to manipulate histone deacetylases and we discuss the possibility of HDAC inhibitors as therapeutics to fight infections.
INTRODUCTION
Epigenetics and the histone code
The study of epigenetic regulation, defined as ‘structural adaptation of chromosomal regions to register, signal or perpetuate altered activity states’, has become an emerging topic in life sciences over the past decades (Bird 2007). Such expression changes can be substantial in a cell and are crucial for its function. Three main mechanisms causing these changes in gene expression are known: DNA methylation, regulation by non-coding RNAs and histone modifications (Goldberg, Allis and Bernstein 2007).
DNA methylation, especially when found at promotors, is a repressive modification of DNA transcription caused by enzymes called DNA methyltransferases (DNMTs). In mammals, cytosine residues in CpG dinucleotides are the main target of this modification. Areas of the chromatin where these modifications accumulate are known as CpG islands, which are connected to transcriptional repression (Goll and Bestor 2005). This repressive effect is mostly connected to promotor methylation (Jones 2012), while methylation of gene body regions has been connected to active gene transcription, highlighting a core principle of epigenetic regulation, which is context-dependency (Maunakea et al. 2010; Arechederra et al. 2018). Moreover, methylated cytosines can be oxidized by TET (ten-eleven translocation) proteins, giving rise to several new modification types such as hydroxymethyl-, formyl- and carboxylcytosine, each of which has distinct effects. For detailed descriptions of these modifications and their effects on transcription, we refer the reader to one of the following reviews (Richa and Sinha 2014; Wu and Zhang 2017; Hardwick, Lane and Brown 2018; Yingqian Zhang and Zhou 2019).
The role of non-coding RNAs in epigenetics is not completely understood yet (Knowling and Morris 2011). However, more and more experimental evidence pushes some types of non-coding RNAs, namely long non-coding RNAs (lncRNA), in the realm of epigenetics. The functions of lncRNAs are manifold as they can repress gene expression, interfere with RNA polymerases, impede mRNA processing and change the cellular localization of proteins by binding to them (Jiao Chen, Ao and Yang 2019). Furthermore, lncRNAs and miRNAs seem to impact the epigenetic machinery at two levels: (i) their expression can be regulated by epigenetic mechanisms and (ii) they have been shown to repress key enzymes that drive epigenetic remodeling (epi-miRNAs) (Moutinho and Esteller 2017). In addition, miRNAs can be involved in establishing DNA methylation (Bao, Lye and Barton 2004).
The third mechanism is the post-translational modification (PTM) of histones. Histones are small highly basic proteins that are crucial for chromatin formation and DNA packaging. The DNA is wrapped around a core octamer complex consisting of two copies of histones H2A, H2B, H3 and H4. H1, the linker histone, sits on top of this complex like a clamp, to maintain the structural integrity. This complex of DNA and histone proteins is called the nucleosome, which then forms the chromatin fiber (Kornberg 1974). Each histone protein can be the target of numerous post-translational modifications, especially the histone tails, the N- or C-terminal parts of the core histone that stick out from the complex, are prone to modifications which include phosphorylation, ubiquitination, SUMOylation, methylation and acetylation (du Preez and Patterton 2013). Recently, novel histone modifications have been identified, for instance propionylation, crotonylation, succinylation and benzoylation (Barnes, English and Cowley 2019). Specific modifications on certain amino acids can drastically influence the transcription of genes associated with the respective histones (Berger 2007; Lawrence, Daujat and Schneider 2016). First, the modifications can directly interfere with the interaction between the different histones, as well as neighboring nucleosomes. Second, proteins and protein complexes—such as nucleosome remodelers—recruited to these modifications can further modify the chromatin structure or influence the activity of transcription factors by condensing the chromatin. However, the effects of a single modification cannot be seen in isolation, but rather in synergy between different modifications within the histone tails (Kouzarides 2007).
A key concept of epigenetics is the highly regulated balance between the addition and the removal of modifications by specific enzymes, so-called writers and erasers, as well as recognition proteins, the so-called readers. The writers, enzymes that add for example methyl or acetyl groups, include histone methyltransferases, histone acetyltransferases, and many others. The readers recognize these modifications and mediate their downstream effects. Numerous protein domains have evolved to detect these modifications, such as chromodomains and ankyrin domains for histone methylation, or bromodomains and double PHD finger domains for histone acetylation. The third group of enzymes, the erasers, are responsible for removing histone modifications, thus reversing their effect on gene expression. The most common examples of erasers are histone demethylases and histone deacetylases. Additionally, it should be noted that many proteins involved in epigenetic regulation can comprise more than one of these properties. The protein p300/CBP, for example, encodes a histone acetyltransferase domain (writer) as well as a bromodomain (reader) (Breen and Mapp 2018). The balanced interplay between writers, readers, and erasers is paramount to a variety of cellular processes (Biswas and Rao 2018).
One of the most abundant histone modifications is the reversible acetylation of lysine residues on histone tails. Histone acetylation is mostly connected to a transcriptional activation (Hebbes, Thorne and Crane-Robinson 1988; Barnes et al. 2019). The writers and erasers controlling the acetylation status of histones are histone acetyltransferases (HAT) and histone deacetylases (HDAC), respectively. In recent years, the role of HDACs in the immune response and their manipulation by certain pathogens became evident. In addition, the therapeutic use of HDAC inhibitors might open a new window to bypass the prominent problem of antibiotic resistance. These topics will be the main focus of this review.
HDACs and their regulation
Five classes of HDAC proteins that are split up in two groups are known to date. The first group, formed by Zn2+-dependent histone deacetylases, comprises class I (HDAC1, HDAC2, HDAC3, and HDAC8), class IIa (HDAC4, HDAC5, HDAC7 and HDAC9), class IIb (HDAC6 and HDAC10) and class IV (HDAC11). The second group, NAD+-dependent histone deacetylases, contains class III, the so-called Sirtuins (SIRT1-7). This classification is based on the homology of the enzymes to specific yeast proteins: Rpd3 for class I, Hda1 for class II and Sir2 for class III. Class IV shares sequence similarity with both class I and class II (H. P. Chen, Zhao and Zhao 2015).
However, despite their name, HDACs can also deacetylate non-histone proteins. The tumor suppressor p53 and the adaptor protein MyD88 are two of the most prominent non-histone targets of HDAC enzymes (AIto et al. 2001; Akihiro Ito et al. 2002; Menden et al. 2019). MyD88 is a crucial component of toll-like-receptor (TLR) signaling, leading to the activation of NF-κB, consequently promoting the production of pro-inflammatory cytokines (New et al. 2016). MyD88 is deacetylated by HDAC6, thereby decreasing its activity (Menden et al. 2019). Thus, acetylation of MyD88 is paramount in the TLR signaling cascade because it supports the interaction of MyD88 with downstream effectors such as TNF receptor associated factor-6 (TRAF6) (Kawai et al. 2004).
The regulation of HDAC activity is a complex and multilayered process with four main regulation mechanisms: gene expression, subcellular localization, protein complex formation and post-translational modifications (PTMs). The expression of histone deacetylase-coding genes has been studied in detail, but exceeds the scope of this review (Sengupta and Seto 2004). However, an interesting characteristic of HDACs is that some can regulate their own expression by interacting with their own promoter regions. For example in mice, HDAC1 autoregulates its own expression by deacetylating the promoter region and, thereby, repressing its own gene expression (Schuettengruber et al. 2003). In addition, HDAC1 seems to be involved in regulating the gene expression of HDAC2 and HDAC3 (Lagger et al. 2002).
The regulation dependent on the subcellular localization is seen mostly with class IIa HDACs since these proteins shuttle between the nucleus and the cytoplasm. A striking example for the regulation by subcellular localization is the interaction of HDAC4 and HDAC5 with the 14–3-3 family adaptor proteins. In mammals, the seven different isoforms of 14–3-3 proteins are highly conserved and they assist in various processes such as protein-protein interactions, protein folding, and protein localization (Stevers et al. 2018). HDAC4 and HDAC5 enzymes directly interact with 14–3-3 proteins, leading to an accumulation of these HDACs in the cytoplasm. This cytoplasmic retention of HDAC4 and HDAC5 dampens the transcriptional repression of a subset of genes (Grozinger and Schreiber 2000). For HDAC7, a similar mechanism of regulation is known (Kao et al. 2001). A different regulation mechanism, which is best studied in class I HDACs, is the control of HDAC activity through protein complex formation. HDAC1 and HDAC2 are crucial components of at least five different protein complexes: Mi-2/NuRD (nuclear remodeling deacetylase), Sin3 (switch intensive 3), CoREST (corepressor of REST), MiDAC (mitotic deacetylase), and the recently discovered BAHD1 (Bromo adjacent homology domain protein 1) (Kelly and Cowley 2013; Lakisic et al. 2016; Millard et al. 2017). As part of these complexes, the HDACs become maximally activated and are targeted to specific regions of chromatin. Moreover, through scaffolding proteins, HDACs are connected with other important epigenetic regulators (Lakisic et al. 2016; Millard et al. 2017). The NuRD complex is an excellent example to illustrate such a combinatorial assembly. The core MTA scaffolding proteins (MTA1, MTA2, MTA3) bridge HDAC1 and HDAC2 with subunits involved in nucleosome remodeling (CHD3, CHD4), histone demethylation (LSD1), binding to other subunits and histones (RBBP4, RBBP7, GATAD2A, GATAD2B), and binding to methylated DNA (MBD2). The combinatorial assembly of these subunits determines the function of NuRD in genomic targeting and association with specific transcription factors, in the mediation of cell type- specific transcriptional regulations, such as the repression of tumor suppressor genes (Lai and Wade 2011).
The recruitment of these complexes to specific chromatin regions is mediated by their interaction with different transcription factors or directly by histone-recognition motifs present in subunits of these complexes (Kelly and Cowley 2013; Millard et al. 2017; Adams, Chandru and Cowley 2018). Another example is the interaction of HDAC3 with NCoR (nuclear receptor corepressor) and SMRT (silencing mediator for retinoid and thyroid receptor) (J. Li et al. 2000). These HDAC3-containing complexes act as ligand-dependent transcription factors and are involved in the transcriptional repression of a specific subset of genes (J. Li et al. 2000). Interestingly, this complex is not only crucial for the activity of HDAC3, but also for the activity of class IIa HDACs, like HDAC4 and HDAC5 (Fischle et al. 2002). In addition, the NCoR/SMRT complex seems to be in competition with 14–3-3 proteins in the binding of HDAC3. 14–3-3 proteins promote the cytoplasmic localization of HDAC3, hence interfering with its repressive activity, demonstrating an elaborate interplay between the different types of HDAC regulation (Rajendran et al. 2011).
Another major regulatory mechanism of HDAC activity are post-translational modifications (PTM). There are numerous PTMs that regulate HDAC function, such as acetylation, SUMOylation, ubiquitination, and phosphorylation (Eom and Kook 2014). Furthermore, the regulation via PTMs is tightly connected to other regulatory mechanisms such as subcellular localization and protein complex formation. A well described example is the phosphorylation of specific residues in HDAC4 that enables the binding of 14–3-3 proteins. Without this interaction, HDAC4 accumulates in the nucleus, leading to a decrease in gene expression (Grozinger and Schreiber 2000). PTMs can, in addition, influence HDAC protein complex formation. Casein kinase 2 (CK2) phosphorylates two serine residues of HDAC1 that mediate its interaction with Sin3, Mi-2/NuRD, and CoREST (Pflum et al. 2001). As mentioned before, the interplay of HDAC1 with these complexes is essential for its histone deacetylase activity (Kelly and Cowley 2013).
These examples highlight the multiple levels of regulation that ensure the correct function of histone deacetylases, important for fundamental cellular processes such as cell division, metabolism, and others, but also complex intercellular processes like regulating the immune response.
HDACs and the immune response
The function of histone deacetylases in regulating immune cells is manifold (Busslinger and Tarakhovsky 2014). Here we focus on the involvement of HDACs in the development of different cell types of the innate and adaptive immune system as well as the regulation of cytokine signaling.
Innate immunity and cytokine signaling
The regulation of immune cells by HDACs starts during myeloid development. During this process hematopoietic stem cells differentiate into either myeloid cells—which are precursor cells of the innate immune system, such as macrophages and neutrophils—or into lymphoid cells, which later differentiate into B cells and T cells (Weiskopf et al. 2016).
HDAC5 and HDAC9, both members of class II, are involved in the differentiation of progenitor cells into macrophages. Especially HDAC5 seems to be a negative regulator of differentiation, since its expression is upregulated in non-differentiated cells (Baek et al. 2009). Also, HDAC3 plays a role in the differentiation process as it is recruited to promoter regions that are normally occupied by the transcription factor PU.1–a key player in the differentiation process of hematopoietic cells (Oikawa et al. 1999)—where it impedes its activity (Ueki, Zhang and Haymann 2008).
The function of another subgroup of macrophages, microglia cells, is also regulated by HDACs. The treatment of mouse microglia cells with HDAC inhibitors reduced the production of cytokines, such as IL-6, TNF-α, and IL-10. Furthermore, this treatment impedes the expression of markers associated with anti-inflammatory M2 macrophages (Kannan et al. 2013). Sirtuins are also implicated in macrophage function: a deficiency of SIRT2, which is highly expressed in myeloid cells, promotes phagocytosis in macrophages, probably through metabolic changes, without interfering with their development or their cytokine production (Ciarlo et al. 2017). In neutrophils, HDAC11 seems to be a key regulator of their activity, as neutrophils lacking HDAC11 show a stronger migratory phenotype and higher phagocytic capacity (Sahakian et al. 2017).
These examples illustrate that the regulation of immune cells is closely tied to the repression or activation of cytokine production and other immune signals. Indeed, broad range HDAC inhibitors can impair the expression of TLR-dependent pro-inflammatory cytokines such as IL-6 and TNF-α in primary mouse macrophages (Roger et al. 2011). Similar results can be seen in dendritic cells, where HDAC inhibition interferes with the expression and secretion of IL-12p40, a potent chemoattractant for macrophages and dendritic cells (Cooper and Khader 2007), and interferon-β (Bode et al. 2007), a cytokine central to the activity of dendritic cells (Barchet, Cella and Colonna 2005). A more specific example for the involvement of HDACs in pro-inflammatory signaling is HDAC4. A knock-out of HDAC4 reduces the expression of interferon stimulated genes (ISG), a phenotype rescued by reintroducing HDAC4 but not by any other HDAC (Lu et al. 2019). Sirtuins have also been recently described to act in the process of ISG expression. SIRT2 is activated in a type-I IFN-dependent manner, leading to a downstream activation of the transcription factor STAT1, promoting the expression of ISG (Kosciuczuk et al. 2019).
Interestingly, HDACs not only promote inflammatory responses, but can also inhibit them. In endothelial cells, HDAC6 is a key player in TLR-signaling. HDAC6 deacetylates MyD88, thereby impeding its interaction with TNF receptor associated factor-6 (TRAF6) and the subsequent expression of pro-inflammatory genes (Menden et al. 2019). However, HDACs also contribute to dampen the immune response as in activated macrophages HDAC1 is recruited to the IL-6 promoter by death domain-associated protein-6 (Daxx), leading to histone deacetylation and prevention of IL-6 overproduction (Yao et al. 2014). Furthermore, in macrophages, the retinoblastoma protein (Rb) can recruit HDAC1 and HDAC8 to the promoter region of Interferon-β and selectively inhibit its expression (Meng et al. 2016).
The role of HDACs in adaptive immunity
HDACs also regulate differentiation and function of adaptive immune cells. In T cells, HDAC3 plays an important role in the CD4-CD8 lineage commitment in the thymus. Indeed, Philips and colleagues have shown that HDAC3 expression maintains bipotency of thymocytes and that a knockout of HDAC3 redirects the cells towards CD8 lineage commitment (Philips et al. 2019). In T-regulatory (Treg) cells, a deficiency of HDAC5 reduces protein levels of Foxp3, a characteristic transcription factor of these cells. Furthermore, in CD8 cytotoxic T cells, a lack of HDAC5 impairs the production of Interferon-γ, a central pro-inflammatory cytokine for CD8 cytotoxic activity (Xiao et al. 2016). In addition, the production of the pro-inflammatory cytokine IL-17 is regulated by HDAC6 in CD4 helper T cells (Yan et al. 2017). The process of T cell differentiation in the thymus ends by generating terminally differentiated cells. This terminal differentiation goes hand in hand with specific metabolic changes within the cell. Recently, it was shown that these changes are dependent on the expression of FoxO1, a transcription factor regulated by SIRT1 (Jeng et al. 2018).
In B cells, a SIRT1 knockout shows a decrease in viability and IgM production (Han et al. 2019). Another histone deacetylase, HDAC3, is involved in regulating B cell maturation and function. The loss of HDAC3 impairs B cell maturation and changes the expression of numerous B cell genes. In addition, the absence of HDAC3 reduces productive VDJ rearrangement, a process essential for generating suitable B cell antigen receptors (Stengel et al. 2017).
These examples summarize a small fraction of what is known about the involvement of HDACs in the activation and regulation of immune cells. However, they underline the abundance of HDACs in these processes. For a more detailed discussion on this specific topic we refer the readers to excellent reviews about this topic (Shakespear et al. 2011; Busslinger and Tarakhovsky 2014). In the following sections of this review, we will focus on examples of how HDACs may be manipulated by pathogens and discuss the possibility to use HDAC inhibitors as treatment options against infectious diseases.
HDACs as targets during infection
Given the major role of histone deacetylases in numerous cell types and signaling pathways, changes in histone acetylation in the context of infection have been reported (Aung et al. 2006; Hamon et al. 2007; Hamon and Cossart 2008).
Bacterial compounds, such as LPS, change the histone acetylation pattern by inducing an immune response, consequently leading to changes in the expression of specific genes. More specifically, cells challenged with LPS modify their histone H3 acetylation profile, which is mediated by the activation of TLR-4, subsequently promoting the production of IL-8 (Angrisano et al. 2010). But, as mentioned before, histone deacetylases also represent excellent targets for pathogens to actively manipulate the host. First, we discuss three different mechanisms how pathogens can exploit histone deacetylases (Table 1): (i) alter HDAC activity, (ii) change the intracellular localization (often leading to HDAC degradation), or (iii) modify their expression at the gene level. Second, we discuss the potential of pathogens to directly target the acetylation of histones by encoding enzymes mimicking host HDACs.
Pathogens known to target different eukaryotic histone deacetylases.
| Pathogen | Effector | Target HDAC (complex) | Reference |
|---|---|---|---|
| Changes in HDAC activity | |||
| Anaplasma phagocytophilium | AnkA | HDAC1 | (Rennoll-Bankert et al. 2015) |
| Helicobacter pylori | Unknown | Unknown | (Ding et al. 2010) |
| Mycobacterium avium/tuberculosis | Unknown | HDAC1/2 (Sin3) | (Wang et al. 2005) |
| Listeria monocytogenes | LntA | HDAC1/2 (BAHD1) | (Lebreton et al. 2011) |
| Beta-herpesvirus | UL38 and UL29/28 | HDAC1/2 (Mi-2/NuRD) | (Terhune et al. 2010) |
| Gamma-herpesvirus | orf36 | HDAC1/2 | (Mounce et al. 2013) |
| Herpes simplex virus 1 | US3 | HDAC2 | (Walters et al. 2010) |
| Toxoplasma gondii | TgIST | HDAC1/2 (Mi-2/NuRD) | (Olias et al. 2016) |
| TgNSM | HDAC3 (NCoR/SMRT) | (Rosenberg and Sibley 2021) | |
| ROP18 | HDAC3 | (An et al. 2018) | |
| Changes in HDAC localization | |||
| Salmonella typhimurium | Unknown | SIRT1 | (Ganesan et al. 2017) |
| Listeria monocytogenes | InlB | SIRT2 | (Eskandarian et al. 2013) |
| Human immunodeficiency virus 1 | Vpr | HDAC1/3 | (Romani et al. 2016) |
| Vaccinia virus | C6 | HDAC4 | (Lu et al. 2019) |
| Changes in HDAC expression | |||
| Mycobacterium tuberculosis | Unknown | HDAC6/11 | (Wang et al. 2018) |
| Unknown | SIRT1 | (Cheng et al. 2017) | |
| Hepatitis C virus | Unknown | HDAC9 | (Chen et al. 2015) |
| Influenza A virus | PA-X | HDAC4 | (Galvin and Husain 2019) |
| Hepatitis B virus | HBx | HDAC1 | (Fu et al. 2018) |
| Pathogen | Effector | Target HDAC (complex) | Reference |
|---|---|---|---|
| Changes in HDAC activity | |||
| Anaplasma phagocytophilium | AnkA | HDAC1 | (Rennoll-Bankert et al. 2015) |
| Helicobacter pylori | Unknown | Unknown | (Ding et al. 2010) |
| Mycobacterium avium/tuberculosis | Unknown | HDAC1/2 (Sin3) | (Wang et al. 2005) |
| Listeria monocytogenes | LntA | HDAC1/2 (BAHD1) | (Lebreton et al. 2011) |
| Beta-herpesvirus | UL38 and UL29/28 | HDAC1/2 (Mi-2/NuRD) | (Terhune et al. 2010) |
| Gamma-herpesvirus | orf36 | HDAC1/2 | (Mounce et al. 2013) |
| Herpes simplex virus 1 | US3 | HDAC2 | (Walters et al. 2010) |
| Toxoplasma gondii | TgIST | HDAC1/2 (Mi-2/NuRD) | (Olias et al. 2016) |
| TgNSM | HDAC3 (NCoR/SMRT) | (Rosenberg and Sibley 2021) | |
| ROP18 | HDAC3 | (An et al. 2018) | |
| Changes in HDAC localization | |||
| Salmonella typhimurium | Unknown | SIRT1 | (Ganesan et al. 2017) |
| Listeria monocytogenes | InlB | SIRT2 | (Eskandarian et al. 2013) |
| Human immunodeficiency virus 1 | Vpr | HDAC1/3 | (Romani et al. 2016) |
| Vaccinia virus | C6 | HDAC4 | (Lu et al. 2019) |
| Changes in HDAC expression | |||
| Mycobacterium tuberculosis | Unknown | HDAC6/11 | (Wang et al. 2018) |
| Unknown | SIRT1 | (Cheng et al. 2017) | |
| Hepatitis C virus | Unknown | HDAC9 | (Chen et al. 2015) |
| Influenza A virus | PA-X | HDAC4 | (Galvin and Husain 2019) |
| Hepatitis B virus | HBx | HDAC1 | (Fu et al. 2018) |
Pathogens known to target different eukaryotic histone deacetylases.
| Pathogen | Effector | Target HDAC (complex) | Reference |
|---|---|---|---|
| Changes in HDAC activity | |||
| Anaplasma phagocytophilium | AnkA | HDAC1 | (Rennoll-Bankert et al. 2015) |
| Helicobacter pylori | Unknown | Unknown | (Ding et al. 2010) |
| Mycobacterium avium/tuberculosis | Unknown | HDAC1/2 (Sin3) | (Wang et al. 2005) |
| Listeria monocytogenes | LntA | HDAC1/2 (BAHD1) | (Lebreton et al. 2011) |
| Beta-herpesvirus | UL38 and UL29/28 | HDAC1/2 (Mi-2/NuRD) | (Terhune et al. 2010) |
| Gamma-herpesvirus | orf36 | HDAC1/2 | (Mounce et al. 2013) |
| Herpes simplex virus 1 | US3 | HDAC2 | (Walters et al. 2010) |
| Toxoplasma gondii | TgIST | HDAC1/2 (Mi-2/NuRD) | (Olias et al. 2016) |
| TgNSM | HDAC3 (NCoR/SMRT) | (Rosenberg and Sibley 2021) | |
| ROP18 | HDAC3 | (An et al. 2018) | |
| Changes in HDAC localization | |||
| Salmonella typhimurium | Unknown | SIRT1 | (Ganesan et al. 2017) |
| Listeria monocytogenes | InlB | SIRT2 | (Eskandarian et al. 2013) |
| Human immunodeficiency virus 1 | Vpr | HDAC1/3 | (Romani et al. 2016) |
| Vaccinia virus | C6 | HDAC4 | (Lu et al. 2019) |
| Changes in HDAC expression | |||
| Mycobacterium tuberculosis | Unknown | HDAC6/11 | (Wang et al. 2018) |
| Unknown | SIRT1 | (Cheng et al. 2017) | |
| Hepatitis C virus | Unknown | HDAC9 | (Chen et al. 2015) |
| Influenza A virus | PA-X | HDAC4 | (Galvin and Husain 2019) |
| Hepatitis B virus | HBx | HDAC1 | (Fu et al. 2018) |
| Pathogen | Effector | Target HDAC (complex) | Reference |
|---|---|---|---|
| Changes in HDAC activity | |||
| Anaplasma phagocytophilium | AnkA | HDAC1 | (Rennoll-Bankert et al. 2015) |
| Helicobacter pylori | Unknown | Unknown | (Ding et al. 2010) |
| Mycobacterium avium/tuberculosis | Unknown | HDAC1/2 (Sin3) | (Wang et al. 2005) |
| Listeria monocytogenes | LntA | HDAC1/2 (BAHD1) | (Lebreton et al. 2011) |
| Beta-herpesvirus | UL38 and UL29/28 | HDAC1/2 (Mi-2/NuRD) | (Terhune et al. 2010) |
| Gamma-herpesvirus | orf36 | HDAC1/2 | (Mounce et al. 2013) |
| Herpes simplex virus 1 | US3 | HDAC2 | (Walters et al. 2010) |
| Toxoplasma gondii | TgIST | HDAC1/2 (Mi-2/NuRD) | (Olias et al. 2016) |
| TgNSM | HDAC3 (NCoR/SMRT) | (Rosenberg and Sibley 2021) | |
| ROP18 | HDAC3 | (An et al. 2018) | |
| Changes in HDAC localization | |||
| Salmonella typhimurium | Unknown | SIRT1 | (Ganesan et al. 2017) |
| Listeria monocytogenes | InlB | SIRT2 | (Eskandarian et al. 2013) |
| Human immunodeficiency virus 1 | Vpr | HDAC1/3 | (Romani et al. 2016) |
| Vaccinia virus | C6 | HDAC4 | (Lu et al. 2019) |
| Changes in HDAC expression | |||
| Mycobacterium tuberculosis | Unknown | HDAC6/11 | (Wang et al. 2018) |
| Unknown | SIRT1 | (Cheng et al. 2017) | |
| Hepatitis C virus | Unknown | HDAC9 | (Chen et al. 2015) |
| Influenza A virus | PA-X | HDAC4 | (Galvin and Husain 2019) |
| Hepatitis B virus | HBx | HDAC1 | (Fu et al. 2018) |
Pathogens alter HDAC activity
Pathogens known to impact HDAC functions either target macromolecular complexes containing HDACs or alter HDAC activity directly. The first pathogen reported to target HDAC-containing macromolecular complexes is the intracellular bacterium Mycobacterium tuberculosis, the cause of severe pulmonary infections and Mycobacterium avium, a human pathogen in immune compromised patients (Pai et al. 2016; Wassilew et al. 2016). Infections with these bacteria lead to a repression of HLA-DR expression, a class II Major Histocompatibility Complex and a key player in cellular antigen presentation. HLA-DR repression is caused by an overexpression of mSin3A, a core component of the HDAC1/2-containing Sin3 complex (Adams et al. 2018), which leads to an enhanced deacetylation of the HLA-DRα promoter (Y. Wang et al. 2005). This inhibition of class-II MHC expression is thought to impair antigen presentation, thereby promoting the survival of intracellular bacteria, similar effects can be seen with the intracellular pathogen Toxoplasma gondii (Lüder et al. 2001). Another bacteria shown to interact with HDAC complexes is Listeria monocytogenes, a food-borne pathogen that may cause infections with symptoms ranging from fever and diarrhea to sepsis and meningitis (Schlech 2019). During infection, L. monocytogenes secretes a small basic protein called LntA (Listeria-nuclear-targeted protein A), which translocates into the host cell nucleus, where it interacts with BAHD1, a core component of the BAHD1 chromatin repressive complex (Lebreton et al. 2011). This interaction alleviates the binding of the BAHD1 complex to the promotor regions of Interferon-stimulated-genes (ISGs), which subsequently promotes the transcription of these genes. In addition, the deletion of LntA leads to drastic changes in the infection process as seen in a murine model (Lebreton et al. 2011, 2014).
The beta-herpesviruses, in particular the human cytomegalovirus, a virus linked to infections of fetuses, AIDS patients, and allograft transplant recipients (Griffiths, Baraniak and Reeves 2015), manipulates cellular HDACs in a similar way. The viral proteins UL38 and UL29/28 can interact with the HDAC1/2-containing nucleosome remodeling and deacetylase complex (NuRD). The viral proteins recruit the complex to the viral major immediate-early promoter, leading to an accumulation of viral RNA in the host cell. A deficiency of either protein, UL38 or 29/28, leads to a decrease in viral replication (Terhune et al. 2010).
Also, eukaryotic pathogens exploit host HDAC complexes. An example is the protozoan parasite Toxoplasma gondii, the causative agent of toxoplasmosis. In healthy adults, toxoplasmosis is often asymptomatic or causes mild flu-like symptoms, however, children and immune compromised individuals can develop a severe form that can be fatal. Recently, two secreted effectors have been described in this pathogen that target the expression of IFN-γ stimulated genes through different mechanisms. The first one, TgIST (Toxoplasma inhibitor of STAT1-dependent transcription), recruits the repressive Mi-2/NuRD complex to STAT1 (signal transducer and activator of transcription 1), thereby inhibiting its activation by IFN-γ and the expression of ISGs (Olias et al. 2016). The second effector, TgNSM (Toxoplasma NCoR/SMRT modulator), also targets the nucleus of the host cell, where it interacts with the NCoR/SMRT complex. The recruitment of NCoR/SMRT by TgNSM impedes the expression of interferon-regulated necroptotic genes, such as PRK (protein kinase K) and MLKL (mixed-lineage-kinase-domain-like pseudokinase). By blocking necroptosis, the parasite protects its intracellular replication niche. Moreover, TgIST is also involved in this process of inhibition, highlighting its importance for the survival of the parasite (Rosenberg and Sibley 2021).
One of the pathogens that were shown to alter HDAC activity directly is Helicobacter pylori, a leading cause of gastric diseases worldwide, from gastritis to ulcers and malignancies (Schulz et al. 2019). During H. pylori infection, lysine 23 of histone H3 (H3K23) is genome-wide deacetylated. This deacetylation is dependent on the presence of the cag pathogenicity island (cagPAI) encoded by many H. pylori strains; but interestingly, deacetylation is not dependent on CagA, the only known translocated effector encoded in the cagPAI (Ding et al. 2010). Thus, the exact mechanism and consequence of this deacetylation remains to be explored.
Another well-studied example is the type IV secreted effector AnkA of Anaplasma phagocytophilium. A. phagocytophilium is an obligate intracellular bacterium that is transmitted by ticks and can cause granulocytic anaplasmosis in humans (Atif 2015). AnkA contains several ankyrin repeats, which are commonly found in eukaryotic proteins, but are also present in some bacterial and archaeal proteins. Their main function is the mediation of protein-protein interactions (Mosavi et al. 2004). AnkA recruits HDAC1 in the host cell, promoting the deacetylation of specific promotors. This deacetylation leads to a repression of host defense genes, one of which is CYBB. This gene encodes the beta subunit of the NADPH oxidase 2, an enzyme crucial for the production of reactive oxygen species, which are paramount for the cellular defense against intracellular pathogens (Rennoll-Bankert et al. 2015).
Members of the Herpesviridae family encode protein kinases that can influence HDAC activity. Herpes simplex virus 1, the causative agent of oral herpes, and other members of the alpha-herpesviruses, encode the US3 kinase that can hyperphosphorylate HDAC2 on a conserved serine residue in the C-terminus of the protein. Mutants lacking the US3 kinase exhibit an impaired growth in different cell lines, but this growth defect can partially be rescued by the addition of HDAC inhibitors. This suggests that hyperphosphorylation impedes the activity of HDAC2, thereby promoting viral replication. However, the exact mechanism is not known (Walters et al. 2010). Another subfamily of the Herpesviridae, the gamma-herpesviruses, with its well-known representatives the Epstein-Barr virus, the causative agent of infectious mononucleosis (Nowalk and Green 2016), also encode for a conserved protein kinase, which is involved in the process of HDAC inhibition. This kinase, named orf36 in the mouse gamma herpesvirus 68 and its EBV homologue BGLF4, were shown to directly interact with HDAC1 and HDAC2. Moreover, the interaction orf36 with these HDACs impedes their binding to the promoter region of RTA, a crucial viral transcriptional activator. The deletion of orf36 disrupts viral replication and DNA synthesis. However, this phenotype can be rescued by knockdown of HDAC1 and HDAC2, meaning both of these HDACs seem to be involved in repressing viral DNA synthesis (Mounce et al. 2013).
In T. gondii, the secreted serine-threonine kinase ROP18 was identified as a major virulence factor (Taylor et al. 2006). Recently, it was shown that ROP18 targets the host protein RTN1-C, a key regulator of the stress response of the endoplasmic reticulum. Subsequently, this ER-stress response induces apoptosis, a common consequence of T. gondii infection. The phosphorylation of RTN1-C leads to a downregulation of HDAC3 activity, causing the accumulation of acetylated GRP78, a chaperone present in the ER (An et al. 2018). GRP78 is one of the main regulators of the unfolded-protein response in the ER, which can, under prolonged conditions, induce apoptosis (Madeo and Kroemer 2009).
Interfering with histone deacetylase activity or recruiting these enzymes to specific proteins or genetic regions is only one way, pathogens can manipulate host HDACs. Another possibility to influence the activity of HDACs is to change their intracellular localization (Fig. 1).
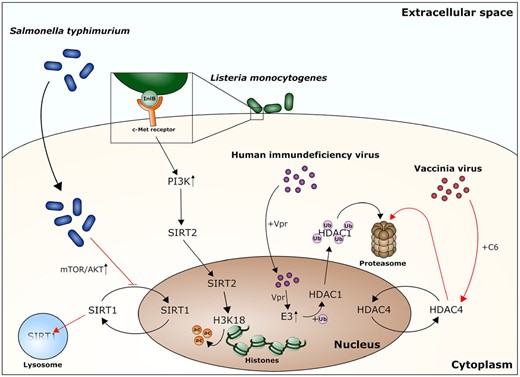
Pathogens interfering with HDAC localization often promote HDAC degradation. Pathogens interact with HDACs through a variety of different processes: Salmonella typhimurium infections lead to an upregulation of the mTOR/AKT pathway, which disrupts SIRT1 nuclear shuttling. The following cytoplasmic accumulation promotes the degradation of SIRT1 in a lysosome dependent manner. The surface protein InlB of L. monocytogenes interacts with the eukaryotic c-Met receptor, thereby promoting the uptake of the bacteria and activating phosphatidylinositol 3-kinase (PI3K), which then causes a nuclear translocation of SIRT2. In the nucleus, SIRT2 deacetylates lysine 18 of histone H3 (H3K18), leading to changes in gene expression. The HIV protein Vpr is a protein essential for the viral entry in the nucleus. Furthermore, it promotes E3 ubiquitin ligase activity. HDAC1 is ubiquinated through this process, travels to the cytoplasm and is degraded by the proteasome. The C6 protein of the Vaccinia virus also promotes proteasomal degradation of a eukaryotic histone deacetylase, HDAC4.
Pathogens change the localization of HDACs
As mentioned before, the activity of many histone deacetylases is tightly linked to their subcellular localization. Hence, this represents a major target for pathogens to manipulate cellular functions.
The histone deacetylase SIRT1 shuttles between the nucleus and the cytoplasm, a process regulated by the PI3K-AKT signaling pathway (Tanno, Miura and Horio 2007). Infection with Salmonella typhimurium, which causes nontyphoidal salmonellosis, a severe enteritis leading to diarrhea and fever, induces the activations of mTOR and AKT. This activation leads to the accumulation of SIRT1 in the cytoplasm, where it is degraded in a lysosome-dependent manner. This relocalization of SIRT1 allows the bacteria to evade the host autophagy response and it promotes their intracellular survival. The type III-secretion system is crucial for this process, however no specific effector connected to this mechanism has been identified (Ganesan et al. 2017). Another bacterium, L. monocytogenes, has its own method to manipulate the PI3K-AKT signaling pathway. One of its surface proteins, InlB, interacts with the host c-Met receptor, which subsequently leads to the uptake of the bacteria by non-phagocytic cells. In addition, binding of InlB to c-Met activates the PI3-kinase, causing the translocation of SIRT2 from the cytoplasm to the nucleus. Here, SIRT2 deacetylates lysine 18 of histone 3 (H3K18) at the transcriptional start sites of SIRT2-regulated genes, altering the host gene expression to the pathogen's advantage. Indeed, a lack of SIRT2, or its inhibition, impairs L. monocytogenes infection, demonstrating the importance of HDAC manipulation for this pathogen (Eskandarian et al. 2013).
In HIV-1, Vpr is an important virulence factor that was shown to interact with E3 ubiquitin ligases, promoting the degradation of specific proteins by the proteasome (Romani and Engelbrecht 2009). One class of proteins targeted for degradation are class I HDACs, in particular HDAC1 and HDAC3. In HIV-infected macrophages, the depletion of HDACs leads to a hyperacetylation of the viral LTR regions. The hyperacetylation promotes the expression of viral genes and helps the virus to infect more cells by overcoming latent infections (Romani et al. 2016). During vaccinia virus infection, HDAC4 is involved in regulating the interferon-α response of the cell to infection; furthermore, an overexpression of HDAC4 interferes with viral replication and spreading. To counteract the activity of HDAC4, the vaccinia virus encodes for protein C6, which was shown to interact with HDAC4 and to promote its degradation by the proteasome. However, the exact mechanism is yet to be understood (Lu et al. 2019).
The manipulation of histone deacetylases by obstructing their intracellular localization or promoting their degradation is a sophisticated mechanism that pathogens can employ to boost their own survival and replication. Yet, this process has its limits, since, like the changes in HDAC activity, it is restricted by the intrinsic protein levels in the host cell.
Pathogens induce changes in HDAC expression
Influencing the expression levels of histone deacetylases may affect the entire cell, however, pathogens targeting this process, seem to be able to exactly regulate and fine-tune the changes they cause.
As mentioned before, IL-10 is an important anti-inflammatory cytokine produced by immune cells. During M. tuberculosis infection, the production of IL-10 is highly increased in macrophages. This is caused by changes in the levels of HDAC6 and HDAC11. M. tuberculosis promotes, through an unknown process, an increase in HDAC6 gene expression and protein level as well as a decrease in HDAC11 protein levels. HDAC6 is subsequently recruited to the promoter regions of IL-10, inducing its overexpression, which subsequently supports the intracellular survival of the bacteria by dampening the immune response (X. Wang et al. 2018). M. tuberculosis infection also leads to a downregulation of SIRT1. Under normal conditions, SIRT1 promotes autophagy and phagosome-lysosome fusion, furthermore, its activation leads to a decrease in M. tuberculosis growth in a mouse model. Both processes are detrimental to the bacteria but by inhibiting SIRT1 expression during infection, the bacteria can evade the host response and facilitate their intracellular growth (Cheng et al. 2017).
Changes in HDAC expression have also been observed during viral infections. The hepatitis C virus (HCV), causing chronic hepatitis, upregulates HDAC9 expression, which is linked to metabolic changes, that can lead to hyperglycemia and type 2 diabetes mellitus. HDAC9 was shown to play a major role in regulating hepatic gluconeogenesis by deacetylating Forkhead box O1 (FoxO1), a transcription factor central to the regulation of gluconeogenic genes in hepatocytes. These results suggest that HDAC9 is a key regulator in gluconeogenesis connected to type 2 diabetes (Jizheng Chen et al. 2015).
Influenza A virus (IAV) infection decreases the protein levels of HDAC4, a histone deacetylase known to regulate antiviral responses (Q. Yang et al. 2019). HDAC4 downregulation is caused both, at the gene expression level, where the viral RNA-endonuclease PA-X strongly interferes with HDAC4 mRNA levels, and at the protein level, due to the proteolytic activity of caspase 3. However, the exact mechanism of caspase 3 activation by IAV remains to be elucidated. Furthermore, this decrease cannot be rescued by adding caspase inhibitors. This demonstrates that the virus employs two independent mechanisms to impede HDAC4 and its antiviral activity (Galvin and Husain 2019).
The hepatitis B virus (HBV) induces autophagy in infected hepatocytes, a step critical to promote its own DNA replication (Sir et al. 2010). One of the key activators of autophagy is the viral protein X (HBx), which was shown to inhibit binding of transcription factor SP1 to the promoter region of HDAC1, hence repressing HDAC1 expression. The lack of HDAC1 subsequently causes the accumulation of acetylated high mobility group box 1 (HMGB1), causing its translocation from the nucleus to the cytoplasm, where it acts as an enhancer of autophagy by directly binding Beclin1, a major regulator of autophagy. In addition, HBx is directly involved in the regulation of autophagy, by binding HMGB1 and promoting the complex formation with Beclin1 (Fu et al. 2018). This shows that a single viral protein not only changes expression levels of a specific HDAC, but it can also influence processes downstream of this histone deacetylase.
Pathogens may mimic HDAC enzymes
Pathogens can influence many steps in the regulation of HDAC activity, but some go even a step further by encoding HDAC-like proteins to manipulate the host cell.
HDAC-like proteins have been structurally described in different bacterial species, but were never assessed for their possible involvement in virulence (Finnin et al. 1999; Hildmann et al. 2004). However, this changed very recently as an HDAC-like protein, named Gc-HDAC, was described to play a role in the pathogenesis of Neisseria gonorrhoeae. Gc-HDAC seems to directly influence H3K9ac and thereby causes a down-regulation in the expression of specific genes like beta-defensin 1 and cathelicidin (Zughaier, Rouquette-Loughlin and Shafer 2020). Interestingly, homologues of this gene have been found in many commensal and pathogenic Neisseria species (Zughaier, Rouquette-Loughlin and Shafer 2020). However, Neisseria is probably not the only bacterial genus encoding HDAC homologues. For example, Legionella pneumophila is known to encode many eukaryotic-like proteins and proteins containing eukaryotic-like domains, acquired during their co-evolution with aquatic protozoa (Cazalet et al. 2004, 2010; de Felipe et al. 2005; Lurie-Weinberger et al. 2010). Indeed, Legionella is known to mimic a vast variety of eukaryotic protein that allow this pathogen to subvert many signaling pathways in the host (Mondino, Schmidt and Buchrieser 2020). Recently the genome sequences of the entire genus Legionella were analyzed, which revealed many new eukaryotic domains encoded in these bacteria, including known chromatin remodeling domains like SET or DOT1 (Gomez-Valero et al. 2019). Furthermore, it has been shown previously that L. pneumophila encodes a SET-domain containing protein that confers methyltransferase activity. In L. pneumophila strain Paris this protein was named RomA and was shown to methylate H3K14 during infection (Rolando et al. 2013). Thus, it was tempting to assume that Legionella might also mimic HDAC proteins.
To investigate if HDAC-like proteins are present in the genus Legionella, we searched for proteins containing a histone deacetylase domain in 80 Legionella genomes belonging to 58 different Legionella species (Gomez-Valero et al. 2019). Excitingly, when using the pfam database (Mistry et al. 2021) we identified in 51 of the 58 analyzed species, proteins predicted to encode an HDAC domain. The majority encode only one HDAC domain, whereas five species contain two and one species (L. gresilensis) contains as many as three HDAC domain encoding proteins. These Legionella HDAC proteins can be classified in two different orthologous groups according to OrthoMCL results (L. Li, Stoeckert and Roos 2003) (Fig. 2). Only in L. gresiliensis two inparalogs were found in the same orthologous group, which suggests a gene duplication event after the speciation. The proteins in group 1 are with an average length of 447 amino acids bigger than those of group 2, which are on average 309 amino acids long. Moreover, group 1 proteins also possess a longer HDAC domain than group 2 proteins. The HDAC domain in group 1 is on average 350 amino acids long, compared to 283 amino acids for group 2. Interestingly, although the second group contains smaller proteins with smaller HDAC domains, the domain covers almost the whole protein length (92%), in contrast, in the first group the HDAC domain represents on average 79% of the amino acid sequence of the protein.

Distribution of histone deacetylase proteins on the phylogeny of the genus Legionella. An HDAC domain search in 58 different Legionella species identified two different types of HDAC proteins named group 1 (orange), and group 2 (green). The phylogenetic tree shown is a species tree of the genus Legionella published in Gomez-Valero et al. 2019. Several strains (number noted next to species name) of the same species were collapsed due to high homology between their proteins. The length of the protein lines and domain symbols are proportional to the protein/domain size and the domain length is noted within the domain. In the species L. gresiliensis two proteins belonging to group 2 are present.
Interestingly, the two HDAC domains are matching differently with the HMM profile as the match of the group 2 HDAC domain against the HMM profile is complete, whereas the match of the group 1 HDAC domain is only partial. To understand their evolution within the genus Legionella, we mapped their distribution on the phylogenetic tree of the genus that we had constructed previously (Gomez-Valero et al. 2019). We found that the distribution of the two HDAC groups followed the phylogeny of the species. Their distribution on the Legionella phylogeny suggests that both HDAC types have followed an independent vertical evolution for a long time, with several loss events in specific strains/clusters. According to the most parsimonic scenario, proteins of group 1 were probably present in the ancestor of Legionella, as they are also present in the outgroup species L. geestiana and L. osnabrueckensis. The second group might have been acquired at the same time but was lost in the outgroup species or it was acquired in a more recent ancestor. However, additional analyses are necessary to determine the exact evolution of the two different HDAC domains. It will be particularly exciting to functionally analyze whether these HDAC domains confer histone deacetylase activity in the host cell and help Legionella to establish their intracellular infection cycle.
The examples discussed above further show that histone deacetylases are auspicious targets for pathogens, whether it is by using their activity for their own advantage, obstructing their intracellular localization or directly interfering with their expression on the genetic level. As promising as HDACs are for pathogens, the same can be said about their potential as therapeutic targets during infection.
HDAC inhibitors as promising therapeutics for infections
In recent years, it became clear that HDAC functions are not only exploited, or inhibited by pathogens, but may also be used as novel targets to fight infectious diseases (Table 2). Their influence on immune cells and the host immune response is multifaceted, underlining the importance they might have as therapeutic targets.
HDAC inhibitors shown to affect different pathogens.
| Inhibitor | Target HDAC | Pathogen | Reference |
|---|---|---|---|
| Suberanilohydroxamic acid | Class I, II and IV | Salmonella typhimurium | (Ariffin et al. 2015) |
| (SAHA/Vorinostat) | Escherichia coli | (Ariffin et al. 2015) | |
| Adenovirus | (Saha and Parks 2019) | ||
| Human papillomavirus | (Banerjee et al. 2018) | ||
| Cryptosporidium parvum | (Guo et al. 2018) | ||
| Leishmania infantum/donovani | (Corpas-López et al. 2019) | ||
| Fusarium solani | (X. Li et al. 2019) | ||
| Trichostatin A | Class I and II | Salmonella typhimurium | (Ariffin et al. 2015) |
| (TSA) | Escherichia coli | (Ariffin et al. 2015) | |
| Angiostrongylus cantonensis | (Zhang et al. 2019) | ||
| Tubastatin A | HDAC6 | Salmonella typhimurium | (Ariffin et al. 2015) |
| Legionella pneumophila | (X. Yang et al. 2021) | ||
| Sodium butyrate | Class I | Klebsiella pneumoniae | (Chakraborty et al. 2017) |
| Tenovin-1 | SIRT1 and SIRT 2 | Arboviruses | (Hackett et al. 2019) |
| Sirtinol | SIRT1 and SIRT2 | West Nile virus | (Hackett et al. 2019) |
| Inhibitor | Target HDAC | Pathogen | Reference |
|---|---|---|---|
| Suberanilohydroxamic acid | Class I, II and IV | Salmonella typhimurium | (Ariffin et al. 2015) |
| (SAHA/Vorinostat) | Escherichia coli | (Ariffin et al. 2015) | |
| Adenovirus | (Saha and Parks 2019) | ||
| Human papillomavirus | (Banerjee et al. 2018) | ||
| Cryptosporidium parvum | (Guo et al. 2018) | ||
| Leishmania infantum/donovani | (Corpas-López et al. 2019) | ||
| Fusarium solani | (X. Li et al. 2019) | ||
| Trichostatin A | Class I and II | Salmonella typhimurium | (Ariffin et al. 2015) |
| (TSA) | Escherichia coli | (Ariffin et al. 2015) | |
| Angiostrongylus cantonensis | (Zhang et al. 2019) | ||
| Tubastatin A | HDAC6 | Salmonella typhimurium | (Ariffin et al. 2015) |
| Legionella pneumophila | (X. Yang et al. 2021) | ||
| Sodium butyrate | Class I | Klebsiella pneumoniae | (Chakraborty et al. 2017) |
| Tenovin-1 | SIRT1 and SIRT 2 | Arboviruses | (Hackett et al. 2019) |
| Sirtinol | SIRT1 and SIRT2 | West Nile virus | (Hackett et al. 2019) |
HDAC inhibitors shown to affect different pathogens.
| Inhibitor | Target HDAC | Pathogen | Reference |
|---|---|---|---|
| Suberanilohydroxamic acid | Class I, II and IV | Salmonella typhimurium | (Ariffin et al. 2015) |
| (SAHA/Vorinostat) | Escherichia coli | (Ariffin et al. 2015) | |
| Adenovirus | (Saha and Parks 2019) | ||
| Human papillomavirus | (Banerjee et al. 2018) | ||
| Cryptosporidium parvum | (Guo et al. 2018) | ||
| Leishmania infantum/donovani | (Corpas-López et al. 2019) | ||
| Fusarium solani | (X. Li et al. 2019) | ||
| Trichostatin A | Class I and II | Salmonella typhimurium | (Ariffin et al. 2015) |
| (TSA) | Escherichia coli | (Ariffin et al. 2015) | |
| Angiostrongylus cantonensis | (Zhang et al. 2019) | ||
| Tubastatin A | HDAC6 | Salmonella typhimurium | (Ariffin et al. 2015) |
| Legionella pneumophila | (X. Yang et al. 2021) | ||
| Sodium butyrate | Class I | Klebsiella pneumoniae | (Chakraborty et al. 2017) |
| Tenovin-1 | SIRT1 and SIRT 2 | Arboviruses | (Hackett et al. 2019) |
| Sirtinol | SIRT1 and SIRT2 | West Nile virus | (Hackett et al. 2019) |
| Inhibitor | Target HDAC | Pathogen | Reference |
|---|---|---|---|
| Suberanilohydroxamic acid | Class I, II and IV | Salmonella typhimurium | (Ariffin et al. 2015) |
| (SAHA/Vorinostat) | Escherichia coli | (Ariffin et al. 2015) | |
| Adenovirus | (Saha and Parks 2019) | ||
| Human papillomavirus | (Banerjee et al. 2018) | ||
| Cryptosporidium parvum | (Guo et al. 2018) | ||
| Leishmania infantum/donovani | (Corpas-López et al. 2019) | ||
| Fusarium solani | (X. Li et al. 2019) | ||
| Trichostatin A | Class I and II | Salmonella typhimurium | (Ariffin et al. 2015) |
| (TSA) | Escherichia coli | (Ariffin et al. 2015) | |
| Angiostrongylus cantonensis | (Zhang et al. 2019) | ||
| Tubastatin A | HDAC6 | Salmonella typhimurium | (Ariffin et al. 2015) |
| Legionella pneumophila | (X. Yang et al. 2021) | ||
| Sodium butyrate | Class I | Klebsiella pneumoniae | (Chakraborty et al. 2017) |
| Tenovin-1 | SIRT1 and SIRT 2 | Arboviruses | (Hackett et al. 2019) |
| Sirtinol | SIRT1 and SIRT2 | West Nile virus | (Hackett et al. 2019) |
The pre-treatment of macrophages with broad spectrum histone deacetylase inhibitors (HDACi), such as suberanilohydroxamic acid (SAHA, also known as vorinostat) and trichostatin A (TSA) decreases bacterial phagocytosis in cells challenged with either S. typhimurium or Escherichia coli. In addition, co-treatment with these inhibitors on cells already infected by the bacteria leads to an increase of bacterial clearance. This observation could be explained by the enhanced production of reactive oxygen species from the mitochondria caused by the HDACi treatment (Ariffin et al. 2015). Especially tubastatin A, a specific HDAC6 inhibitor, seems to be very potent in reducing the intracellular survival of S. typhimurium and L. pneumophila (Ariffin et al. 2015; X. Yang et al. 2021). Another example of the possible usage of HDACi during infections are chronic lung infections, called non-resolving pneumonia. One of the most prevalent pathogens to cause this type of infection is Klebsiella pneumoniae. During this infection cell death induces a release of cardiolipin, the main lipid component of the inner mitochondrial membrane and a mitochondrial damage-associated molecular pattern. Amid the lung infection, the released cardiolipin causes the SUMOylation of nuclear receptor PPARγ, which consequently leads to the recruitment of a protein complex containing HDAC3 to the promoter region of IL-10, repressing its expression. This repression promotes a persistent inflammation in the lung. Inhibiting HDAC3 restores the IL-10 production and increases the survival in an in vivo mouse model (Chakraborty et al. 2017).
HDAC inhibition is not only a therapeutic option in fighting bacterial infections but can also be used in viral infections. Lately, several studies assessed if HDACi could be used to treat viral infections: e.g. SAHA drastically decreases human adenovirus replication by interfering with viral gene expression, protein production, and DNA replication (Saha and Parks 2019).
Also, Sirtuin-specific inhibitors show promising antiviral properties. One example is Tenovin-1 that displays antiviral activity against different members of the Arbovirus family (Hackett et al. 2019). Arboviruses include major human pathogens such as West Nile virus, Chikungunya virus, and Zika virus (Barzon 2018). The treatment of infected cell cultures with Tenovin-1 reduces the viral load of these three viruses. In addition, sirtinol, a SIRT1 and SIRT2 inhibitor, interferes with the formation of West Nile virus replication foci, a process where viral dsRNA accumulates in the host cell (Hackett et al. 2019). These results support the potential of HDACi as therapeutic options for the treatment of viral infections in the near future.
Pan HDAC inhibitors, such as vorinostat, not only show antiviral properties (Banerjee et al. 2018), but also can be used to treat protozoan infections. However, the success of HDAC inhibition to alleviate protozoal infections is often not based on the inhibition of the host HDAC, but rather on the inhibition of the parasite's HDACs. An example is the use of HDACi as treatment of Cryptosporidium infections, a genus closely related to Plasmodium and Toxoplasma. In a mouse model, treatment with low concentrations of vorinostat led to a decrease of Cryptosporidium parvum oocysts. Oocysts are formed during the life cycle of the parasite and are crucial for the spreading of the parasite from one host to another. In addition, low concentrations of vorinostat killed the majority of C. parvum parasites in a cell culture model (Guo et al. 2018).
The efficacy of a vorinostat derivative was also tested using a mouse model of visceral leishmaniasis, a severe infection with members of the genus Leishmania. Visceral leishmaniasis is characterized by a dissemination of the parasites to internal organs such as the liver, the spleen, and the bone marrow. The study demonstrates that the vorinostat derivative shows a high efficacy against the parasite in vivo at very low doses. This corroborates the potency of vorinostat, and its derivatives, as a possible treatment option for parasitic infections (Corpas-López et al. 2019). Very recently, HDACi have also been described to alleviate infections caused by multicellular pathogens such as nematodes and fungi. Trichostatin A contributes to the alleviation of eosinophilic meningitis caused by the nematode Angiostrongylus cantonensis in a mouse model. The main cause for this might be the reduction of the inflammatory response in the animals (Yanhua Zhang et al. 2019). Another mouse model, one for fungal keratitis caused by Fusarium solani, illustrated that HDAC1 is upregulated during infection, causing an overexpression of proinflammatory cytokines. This overexpression and the resulting keratitis can be counteracted using vorinostat (X. Li et al. 2019).
Taken together, the studies presented here show that the inhibition of histone deacetylases may be a potent mean to fight infectious diseases. However, further research is required to elucidate the mechanism of action of these inhibitors. Moreover, clinical studies in this field are scarce and many questions are yet to be answered. Additionally, the development of new HDACi derivatives might help to minimize negative side effects as well as potentiate their antimicrobial properties, so that these molecules might become novel options to treat infectious diseases.
CONCLUSIONS
The examples discussed in this review show the importance of histone deacetylases in the immune response, as targets of pathogens and as possible treatment options. However, there are still many unanswered questions surrounding HDACs and their function. For example, how bacterial, viral or protozoan pathogens exploit or mimic eukaryotic HDACs is a wide-open field of investigation and only little is known to date. Excitingly, as shown here, Legionella and probably also other bacterial pathogens encode HDAC mimics in their genome and might secrete them into the host cell to manipulate it to their advantage. Further analyses will elucidate in which ways these HDAC mimics function. This knowledge might lead to the discovery of new targets to fight bacterial infections. However, as already shown in cellular and animal models, inhibition of HDACs might also be a promising alterative to fight infections caused by antibiotic resistant bacteria. Future research will proof if this approach is applicable. We are convinced that the research on the function of eukaryotic and bacterial HDACs, as well as on the mechanisms employed by bacterial, viral or protozoan pathogens to subvert HDAC function will lead to a wealth of new knowledge. This will ultimately lead to a better understanding of many fundamental processes but may also lead to the development of new therapeutics.
FUNDING
Work in the CB laboratory is financed by the Institut Pasteur and funding has been received from the Agence National de recherche grants ANR-10-LABX-62-IBEID and ANR-15-CE17-0014-03 to CB and ANR-18-CE15-0005-01 to MR and the ‘Fondation de la Recherche Médicale’ grant EQU201903007847 to CB. DS was supported by the École Doctorale ‘ED515: Complexité du vivant’.
Conflict of interest
None declared.
REFERENCES

{kind=link}
{kind=link}