





Abstract
We sequenced part of the mitochondrial control region and the cytochrome b gene in 72 specimens from 32 gull species (Laridae, Larini) and 2 outgroup representatives (terns: Laridae, Sternini). Our control region segment spanned the conserved central domain II and the usually hypervariable 3′ domain III. Apart from some heteroplasmy at the 3′ end of the control region, domain III was not more variable than domain II or the cytochrome b gene. Furthermore, variation in the tempo of evolution of domain III was apparent between phyletic species groups. The lack of variation of the gull control region could not be explained by an increase in the proportion of conserved sequences in these birds, and the gull control region showed an organization similar to those of other avian control regions studied to date. A novel invariant direct repeat was identified in domain II of gulls, and in domain III, two to three inverted, sometimes imperfect, repeats are able to form a significantly stable stem-and-loop structure. These putative secondary structures have not been reported before, and a comparison between species groups showed that they are more stable in the group with the more conserved control region. The unusually slow rate of evolution of control region part III of the gulls could thus be partly explained by the existence of secondary structures in domain III of these species.
Introduction
In most species, the mitochondrial DNA control region (D-loop region) is the most variable part of the mitochondrial DNA (mtDNA) molecule, presumably because of the lack of coding constraints (see review in Simon 1991 ; Baker and Marshall 1997 ). Sequence variation consists not only of substitutions but also of indels of various lengths and of variation in copy number of tandem repeats (Sbisà et al. 1997 ). In spite of this, a common organization has been retained in a wide range of vertebrates. A comparatively conserved central segment, domain II, is flanked by two regions where most sequence variability is usually found: domain I and domain III (names follow Baker and Marshall 1997 ). Base composition also differs between the C-rich domain I, the G-rich domain II, and the AT-rich domain III. In addition, several blocks have been identified where sequence is highly conserved in various vertebrate orders (Baker and Marshall 1997 ; Faber and Stepien 1997 ; Sbisà et al. 1997 ).
This common organization and the existence of conserved blocks indicate that selective constraints must act on the evolution of the control region, some of these constraints being certainly related to the known functions of the control region. Domain I contains the terminus of the H-strand synthesis (Doda, Wright, and Clayton 1981 ). Domain III contains the origin of replication of the H strand (OH), several short conserved sequence blocks (CSBs), and some thermodynamically stable cloverleaf-like structures thought to be associated with the control of DNA synthesis (Brown et al. 1986 ; Nass 1995 ; Sbisà et al. 1997 ). In addition, it contains in birds one bidirectional transcription promoter (L'Abbé et al. 1991 ; Nass 1995 ; Randi and Lucchini 1998 ). Domain II contains three conserved blocks (F, D, and C boxes) and is generally the least variable part of the control region, although no function has been identified to date (but see Gemmell et al. [1996] and Sbisà et al. [1997] for the existence of open reading frames).
A certain amount of variation in evolutionary rate also exists between control regions of different organisms. A slower-than-expected rate has been found, for example, in salmonid fishes (Shedlock et al. 1992 ; Bernatchez and Danzmann 1993 ), rats (Brown et al. 1986 ), and butterflies of the Jalmenus genus (Taylor et al. 1993 ). Unusually low evolutionary rates probably result from increased functional constraints, but these constraints have generally not been identified. In birds, many previous studies have found avian control regions to be variable, as expected from other vertebrate studies (see review in Baker and Marshall 1997 ). However, in partridges of the Alectoris genus (Randi and Lucchini 1998 ), the bluethroat (Luscinia svecica) (Questiau et al. 1998), and the Toxostoma thrashers (Zink et al. 1999 ), the control region demonstrated no more variation than coding genes.
In the course of a molecular evaluation of phylogenetic relationships within the tribe Larini (gulls), we observed a lower-than-expected interspecific divergence among control regions of gulls. Using cytochrome b sequences from the same individuals as a yardstick allowed us to identify peculiarities specific to the control region as opposed to factors affecting the whole mitochondrial genome, such as historical and demographic processes or the substitution rate of the mitochondrial DNA (see, e.g., Martin and Palumbi 1993 ; Rand 1994 ; Mindell et al. 1996 ; Page et al. 1998 ). Variation in the rate of control region evolution was apparent between species groups. We sought explanations for this by (1) identifying conserved sequence blocks in the gull control region and comparing them with those identified in other vertebrate taxa and (2) investigating the thermodynamic stability of putative secondary structures in the control region. We especially tried to relate structural properties of specific control region sequences to their rates of evolution.
Materials and Methods
Specimens
One individual from each species completely sequenced in Crochet, Lebreton, and Bonhomme (2000 ; see for sample origin) was used in the present work. These sequences have been deposited in GenBank (accession numbers AF268527–AF268560 for the control region and AF268493–AF268526 for the cytochrome b gene). Dunlin Calidris alpina (accession number L20137) and guillemot Cepphus carbo (accession number AF027220) control region sequences were obtained from GenBank. Genomic DNA was extracted from ethanol-preserved tissues (muscle or feather bases) or dried feather bases by complete digestion with 20 μl of proteinase K at 57°C in 400 ml of 5% Chelex 100 (Biorad), followed by a 10-min boiling. Extraction of genomic DNA from blood (in buffer, in ethanol, or dried on paper) and from skin or feather bases collected on long dead bodies found in the field was performed using the Qiaamp tissue extraction kit (Qiagen) following the supplier's procedure. Museum specimens (skin from the underside of the foot) were processed either using the Qiaamp method or, for most specimens, using a silica bead method adapted from Taberlet and Fumagalli (1996) , adding proteinase K to the extraction buffer in the sample digestion step.
Amplification and Sequencing
Polymerase chain reaction (PCR) amplifications were carried out in 50-μl volumes containing 1 × amplification buffer/1 U of Taq DNA polymerase, 1.5 mM MgCl2, 0.2 mM of each dNTP, and 0.4 μM of each primer. The amplification primers for control region domains II and III were L438 (5′-TCACGTGAAATCAGCAACCC-3′) (Wenink, Baker, and Tilanus 1993 ) and H1248 (5′-CATCTTCAGTGCCATGCTTT-3′). For museum specimens, three overlapping segments of the control region were amplified separately, using primers L438 and HDLI3 (5′-GTATTCCTGAGGGCCAAACT-3′), L699 (5′-ATAAACCCCTCCAGTGCACC-3′) and HHTR (5′-ATCGCTGTTGTTGACATGTA-3′), and L892 (5′-GTGTAGTGCTCAATGGACATG-3′) and H1248. Primer H1248 was designed from homologies between gull sequences and other sequences available in GenBank. We designed primers HDLI3, L699, and L892 from our own gull sequences and HHTR from published gull sequences (Berg, Moum, and Johansen 1995 ). To check the origin of our control region sequences (see Discussion), we also used primers H1343 (5′-CACTGGGATGCGGATACTTGCATG-3′) (Berg, Moum, and Johansen 1995 ) and H1561 (5′-CGGTTAATTTGGGTCTCTTG-3′) (designed by alignments of sequences available in GenBank). A 280-bp segment of the cytochrome b gene was amplified and sequenced using primers L15008 (5′-AACTTCGGATCTCTACTAGG-3′) and H15326 (5′-GAATAAGTTGGTGATGACTG-3′). L refers to light strands and H refers to heavy strands, and the numbers refer to the position of the 3′ nucleotide of the primer in the white Leghorn chicken (Gallus gallus) mtDNA sequence (Desjardins and Morais 1990 ). Primers HHTR and HDLI3 could not be aligned with the chicken sequence.
PCR products were purified using Gene Clean II kits (Bio 101, Inc.) according to the manufacturer's suggested protocol. Direct sequencing was performed using a Thermosequenase sequencing kit with dye primers (Amersham Pharmacia Biotech). The products were run on an ALF sequencer (Amersham Pharmacia Biotech) following recommended procedures. After checking the accuracy of our sequencing procedure by repeating the extraction and amplification steps for the same individuals, we sequenced only one strand for each specimen, often producing largely overlapping segments. The sequencing primers were L438, L699, L892, L15008, and H15326.
Sequence Analysis
Sequences were aligned manually. Sequence nucleotide composition, frequency and distribution of substitutions and indels, and percentage of sequence divergence (Kimura two-parameter model) were obtained from MEGA (Kumar, Tamura, and Nei 1993 ). The variation of substitution rates across nucleotide sites was quantified by assuming a gamma distribution and estimating the shape parameter α of the distribution (cf. Yang and Kumar [1996] for a summary on this issue). The average number of substitutions per site, the α parameter of the gamma distribution, and the transition/transversion ratio were estimated using the maximum-likelihood method of PAML (Yang 1995 ). The attribution of gull species to their species groups and the construction of the maximum-likelihood tree (fig. 1 ) used as the input tree in PAML are explained in Crochet, Lebreton, and Bonhomme (2000) . Briefly, the tree depicted in figure 1 was obtained by using a maximum-likelihood model (DNAML in the PHYLIP package, version 3.57c; Felsenstein 1993 ), combining the cytochrome b and control region segments into a single composite sequence.
We used three approaches to identify variation in sequence evolution rates among gulls. First, we compared the likelihood of the best trees obtained with (option DNAMLK) and without (option DNAML) the assumption of a molecular clock in PHYLIP for the control region and cytochrome b segments separately. Significance of the difference in likelihood was evaluated by a likelihood ratio test (LRT) as proposed by Felsenstein (1993) . Since the topologies of the trees obtained with and without the molecular-clock assumption differed in both cases, the LRT performed did not exactly test deviation from the molecular clock (see Felsenstein 1993 ), but gave an indication of the behavior of the sequences. Trees of different topologies were compared using the Kishino and Hasegawa (1989) test as implemented in PHYLIP. We compared maximum-likelihood trees obtained with and without enforcement of the molecular clock for the cytochrome b and control region segments separately. Finally, we specifically tested for rate heterogeneity among gulls by performing relative-rate tests (see Robinson et al. 1998 ) using the program RRTree (Robinson-Rechavi and Huchon 2000; available at http://pbil.univ-lyon1.fr/software/rrtree.html).
We searched for potential secondary structures at 38°C using RNAdraw (Matzura and Wennborg 1996 ). To test whether these structures were likely to result from a particular sequence organization, we permuted all base positions in the sequences and counted how many of these permuted sequences gave secondary structure at least as stable (≤ΔG) as those observed with the original data. The significance level of the original structures was estimated by the proportion of randomized (permuted) sequences giving secondary structures as stable as, or more stable than, these original structures. A given structure is significant if it is more stable than 95% of the structures generated by permuted sequences.
Results
Control Region Primary Structure
Control region and cytochrome b sequences were obtained from 32 gull and two tern species (see fig. 1 ). Control region sequences began between positions 445 and 480 of the chicken mitochondrial genome (Desjardins and Morais 1990 ). Based on alignment with the dunlin control region sequence, about 65 bp were missing from the beginning of domain II. Our sequences spanned most of the central conserved domain II and the complete domain III. We could not align the end of our sequences with the chicken sequence. Due to heteroplasmy at the 3′ end of the control region (see below), it was usually not possible to reach the tRNAPhe, except when one haplotype clearly outnumbered others or when no heteroplasmy was apparent. Figure 2 gives the alignment obtained by selecting one sequence per species group (see fig. 1 ).
Figure 3 illustrates the distribution of variable sites across the control region segment. The border between domain II and domain III was placed before CSB1 according to Marshall and Baker (1997) and Sbisà et al. (1997) . The amount of variation differs little between these two domains, but base composition is strikingly different (table 1 ). Considerable heterogeneity in the amount of sequence variation is apparent across the control region, with some invariant and some hypervariable portions. Two invariant regions in domain II correspond to the C (from position 121 in fig. 2 ) or the D (from position 61 in fig. 2 ) boxes. The C box shows a similarity of 59% and 48% to the corresponding snow goose (in Quinn and Wilson 1993 ) and chicken (in Desjardins and Morais 1990 ) sequences, respectively. The D box is more conserved, sharing 92% (dunlin, most gulls, terns) or 100% (Hydrocoleus minutus) similarity with the snow goose D box and 84% (dunlin, most gulls, terns) or 92% (H. minutus) similarity with the chicken D box. CSB1 is also present in the gulls, the terns, and the dunlin, but it is more variable among these species. Similarity to the snow goose and chicken CSB1 sequences varies between 89% (Sterna maxima, chicken) and 68% (gull species groups 1 and 2, snow goose). We could not identify CSB2, but the sequence 5′-AAGAAACTCCCCTAAAAAACA-3′ (slightly variable among gulls) between positions 658 and 678 (fig. 2 ) is strongly reminiscent of CSB3 (see Sbisà et al. 1997 ). An invariant segment in domain III (from position 541 in fig. 2 ) is made of a direct 11-bp repeat, with two copies in all gulls but only one in the two tern species, and does not correspond to any previously described conserved sequence.
This marked heterogeneity between sites in the amount of variation is apparent in the values of the α parameter of the gamma distribution, which is well below 1 for both domain II (α = 0.1951) and domain III excluding tandem repeats (α = 0.3292).
We failed to identify sequences around CSB1 that were analogous to the sequence that encompasses H-strand initiation in the chicken (see Nass 1995 ) and were conserved among other Galliformes or Anseriformes (Nass 1995 ; Randi and Lucchini 1998 ). Nor could we find sequences analogous to the LSP/HSP bidirectional promoter.
Heteroplasmic Tandem Repeats
In all gulls and the two terns examined, the end of control region domain III contains variable numbers of tandem repeats (usually CAACAAA in the light strand) identical to those described by Berg, Moum, and Johansen (1995) in other Ciconiiformes. Most individuals were heteroplasmic for tandem repeat number, as determined from the numerous peaks (reproducible for each individual and absent when sequencing fragments which did not include the end of domain III) visible in the fluorigrams. Some variation existed in the core motif of the tandem repeats, and intraspecific variation was found even in Chroicocephalus ridibundus. Due to the particular nature of sequence variation in the tandem repeat arrays, they were excluded from subsequent analyses. The following is thus based on 660 nucleotide positions from the control region (positions 23–687 in fig. 2 ) available for the 31 gull and 2 tern species, ending at the beginning of the tandem repeat array.
Intraspecific Diversity
Intraspecific diversity was assessed by sequencing the cytochrome b segment and the control region segment (up to the tandem repeats) of 12 Larus cachinnans, 12 C. ridibundus, and 8 L. argentatus. Only one haplotype was found in the 12 L. cachinnans. Three haplotypes were found in L. argentatus, one shared by six individuals, one differing by one substitution in the control region, and the last one differing by another substitution in the control region and by four substitutions in the cytochrome b gene. Among the 12 C. ridibundus, six haplotypes were found, all differing by 1–7 substitutions and 0–1 indels located in the control region segment. The percentage of polymorphic sites (p) was thus 0 for both DNA regions in C. cachinnans. For L. argentatus, p = 0.32% for the control region and p = 1.45% for the cytochrome b gene. In contrast, the control region of C. ridibundus is more variable (p = 1.23%) than the cytochrome b gene (p = 0%).
Rate Heterogeneity Among Species Groups
Within gulls, domain II and domain III have, on average, the same substitution rate as cytochrome b (fig. 4 ). Of course, additional variation occurs in the control region segment due to indels that are not incorporated in distance calculations. When comparing gulls and terns, the control region is found to be 1.45 times more divergent on average than the cytochrome b gene (around 15% divergence for cytochrome b versus 21% for the control region). This might well be due to saturation of the less numerous variable sites of the cytochrome b gene, since divergence between the two tern species is higher for cytochrome b (3.36%) than for the control region (1.92%).
To investigate differences in the mode and rate of control region evolution between lineages, comparisons were restricted to pairs of species belonging to the same species group (fig. 1 ). Clear differences appear among species groups regarding relative rates of control region and cytochrome b gene differentiation. Group 8 (C. ridibundus and related species) and the fuscus subgroup represent extreme cases. Group 8 shows a clear tendency for the control region to be more differentiated than the cytochrome b gene (12 pairwise comparisons out of 15), whereas within the fuscus subgroup, the control region is usually more conserved than cytochrome b (15 comparisons out of 21). Four species of the fuscus subgroup (L. fuscus, L. argentatus, L. marinus, and L. cachinnans) have exactly the same control region haplotype but differ by one to five substitutions in the cytochrome b segment. On the other hand, the group 8 members Chroicocephalus cirrocephalus, Chroicocephalus scopulinus, and C. ridibundus, which share the same cytochrome b haplotype, differ by 9–35 substitutions and 0–4 indels in the control region segment. This pattern is concordant with the differences that were observed at the level of intraspecific diversity (see above).
To determine whether the differences in ratio of cytochrome b and control region divergence were mainly due to variation in the evolution rate of the control region, we evaluated the clocklike behavior of both genes. The likelihood of trees obtained without the molecular-clock assumption was higher than it was with the molecular-clock assumption. For the cytochrome b segment, the difference in likelihood was not significant (twice the difference in ln(likelihood) approximately follows a χ2 distribution with 30 df; ln(likelihood) without molecular clock: −1,266; ln(likelihood) with molecular clock: −1,285; P > 0.1). For the control region segment, the same difference was significant ln(likelihood) without molecular clock: −2,991; ln(likelihood) with molecular clock: −3,014; P < 0.03). The Kishino-Hasegawa test produced the same results: for the control region, the molecular clock–enforced tree was significantly worse than the nonclock tree; for cytochrome b, the clock tree was not significantly worse than the nonclock tree. The evolution of the gull cytochrome b segment did not significantly deviate from that expected under the assumption of a molecular clock. In contrast, the control region segment of the gulls did not behave in a clocklike fashion across all taxa.
The program RRtree (see Materials and Methods) was used to test for significant differences in control region evolution rate between the fuscus subgroup and group 8, using the terns as an outgroup. For cytochrome b, the fuscus subgroup (plus L. canus and L. delawarensis) and group 8 did not show any significant difference in rate of evolution (weighted mean distance (K) for the fuscus subgroup: 0.15093; K for group 8: 0.14802; difference in K values (dK) = 0.002914, dK/SD = 0.02091, P =0.83). For the control region, the differences in rate of evolution between the fuscus subgroup (plus L. canus and L. delawarensis) and group 8 were very close to significance (K for fuscus subgroup: 0.230901; K for group 8: 0.207106; dK = 0.023795, dK/SD = 1.84518, P = 0.065). The variations in relative rates of the control region and cytochrome b segments between the fuscus subgroup and group 8 are thus mainly due to variation in the rate of evolution of the control region. The control region segment of the species in the fuscus subgroup and their close relatives evolves slower than the control region segment of the species of group 8.
Secondary Structures
In most species, CSB1 and the adjacent segment of domain II were able to form a long stemlike structure (fig. 5A ). This structure was more stable in species group 1 (ΔG < −20 kcal/mol for all species) than in species group 8 (ΔG > −16 kcal/mol for all species), with other species groups having intermediate values. It should be noted that this structure was highly variable among species and was not significantly more stable than in sequences generated by random permutations. Nevertheless, it also tended to be more significant in species group 1 (only 20% of permuted sequences gave a more stable structure) than in group 8 (>60% of the permuted sequences gave a more stable structure). Similar structures were identified in the dunlin and tern control regions but were also weakly supported.
In domain III, a strongly significant structure exists in all gull species (generally, no permuted sequence gives a more stable structure, always <5% of permuted sequences give more stable results) (fig. 5B ). This structure originates from two (groups 2, 3, and 8) or three (group 1 except L. heermanni) inverted repeats (perfect or imperfect repeats) situated between positions 570 and 630 in figure 2 . It comprises two or three stem-and-loop formations, depending on the number of repeats. These structures do not exist in the terns or the dunlin, but in the guillemot four imperfect tandem repeats also form a very significant and stable secondary structure, including four stem-and-loop structures.
Discussion
Mitochondrial Origin of Control Region Sequences
One possible explanation for the low rate of evolution of the control region in gulls is that we amplified nuclear homologs of mitochondrial DNA (called numts hereinafter; see Quinn 1997 ). Nuclear copies of the control region in guillemots evolve at a slower rate than their mitochondrial homologs (Kidd and Friesen 1998 ). Several arguments support our sequences being of mitochondrial origin. First, the sequences of the 3′ end of the control region and the tRNAPhe that we obtained for L. cachinnans are identical to sequences published by Berg, Moum, and Johansen (1995) for L. fuscus, which were obtained from purified mitochondrial DNA. When numts have been compared with their mitochondrial homologs, differences have always been apparent (Quinn 1997 ; Kidd and Friesen 1998 ). Second, we amplified our target control region segments using four different primers in the heavy strand: HHTR (designed from mitochondrial control region sequences of gulls published by Berg, Moum, and Johansen [1995] ), H1248, H1343, or H1561 (all three designed from available mitochondrial DNA bird sequences and, respectively, situated in the tRNAPhe and 12S rRNA genes). In each case, the amplified products gave perfectly clear sequences. It is extremely unlikely that four different primers designed from mitochondrial avian sequences would only amplify numts. Third, the observed pattern of heteroplasmy in the 3′ end of the control region is similar to the pattern described for other avian species of the order Ciconiiformes (Berg, Moum, and Johansen 1995 ) and many vertebrates (see Kidd and Friesen [1998] for references). Observing heteroplasmy in numts would require simultaneous amplification of numerous (up to 30) different copies of mitochondrial inserts. In addition, mode of sequence evolution (e.g., strong heterogeneity among sites in rate of variation), variation in G content among regions, and presence of conserved blocks are all consistent with a mitochondrial origin of these sequences.
Evolution of Central Conserved Domain II
The pattern and rate of sequence evolution in gull mitochondrial control region domain II are both typical of what is known in other avian taxa. Highly unequal variation rates among sites in domain II have been documented in finches (Marshall and Baker 1997 ; α = 0.1563), guillemots (Kidd and Friesen 1998 ), and the dunlin (Wenink, Baker, and Tinalus 1994 ). Control region domain II differs little in this aspect from mitochondrial genes, for which α values are typically much less than 1 (Klicka and Zink 1998 ; Arbogast and Slowinski 1998 ). The gull multispecies alignment is also characterized by a mixture of conserved and highly variable blocks. Invariant regions correspond to the D and C boxes and to a strictly conserved segment adjacent to the D box (see fig. 2 ). Frequency of indels relative to substitutions and differences in G content between domain II and the rest of the control region are similarly typical of the avian taxa studied to date (Baker and Marshall 1997 ).
In interspecific comparisons between Fringilla finches (Marshall and Baker 1997 ), Polioptila gnatcatchers (Zink and Blackwell 1998 ), and Pipilo towhees (Zink, Weller, and Blackwell 1998 ), domain II was found to evolve at a rate similar to or slower than that of protein-coding mitochondrial genes. When comparing haplotypes detected within the dunlin, only slightly more variable sites were situated in domain II than in a similar-sized cytochrome b segment (Wenink, Baker, and Tilanus 1993 ). The similar rates of evolution observed in domain II and cytochrome b in gulls do not appear to be unusual for birds.
Slow Evolution Rate of Domain III
In contrast to what was observed in domain II, domain III in gulls displayed several unusual features. First, domain III evolves at the same rate as domain II (and cytochrome b). In most other vertebrates studied, domain III was found to be more variable than domain II. In comparisons between mice and rats (Brown et al. 1986 ), domain III was the most highly variable part of the control region. This result was also found for salmonid fish (Shedlock et al. 1992 ). In comparisons between 4 gadid fish and between 10 cichlid fish (Lee et al. 1995 ), more variable sites were located in domain III than in domain II. The same pattern was detected in birds (see Baker and Marshall 1997 for a review). Marshall and Baker (1997) found that for Fringilla finches, the frequency of substitutions and gaps is higher in domain III and lowest in domain II. More variable sites also occur in domain III than in the other domains in comparisons between the turnstone and the dunlin (Wenink, Baker, and Tilanus 1994 ). In gnatcatchers (Polioptila) (Zink and Blackwell 1998 ) and thrashers (Toxostoma) (Zink et al. 1999 ), most variation occurred in domain III. In towhees (Pipilo), domain III was nearly twice as variable as domain I or domain II (Zink, Weller, and Blackwell 1998 ). In guillemots, however, domain III was hardly more variable than domain II. Second, the strongly skewed shape of the gamma distribution in domain III of gulls indicated a mixture of variable and conserved sites (see also fig. 2 ). In contrast, the Fringilla finches or the dunlin domain III have equally distributed rates among sites (Wenink, Baker, and Tilanus 1994 ; Marshall and Baker 1997 ).
Gulls have an unusually conserved domain III in relation to the occurrence of low-variability fragments in this domain. This suggests the probable existence of constraints acting on these sites. These conserved fragments are not homologous to the CSBs present in many vertebrates, at least one of which can also be unambiguously identified in the gull control region, and they were not included in the putative secondary structures that we identified.
Secondary Structures
The secondary structures formed by CSB1 and the adjacent parts of domain II strongly differed from those documented in the same region of other vertebrates (compare fig. 5A with fig. 4 in Brown et al. [1986] or fig. 5A in Nass [1995] ). They were not significantly more stable than in sequences generated by random permutations, although for most species groups, only around 20% of permuted sequences gave equally or more stable structures. They were especially poorly supported in species group 8, where most permuted sequences produced more stable structures.
The strongly significant secondary structures identified in the middle of domain III in gulls have not previously been detected in the chicken (Nass 1995 ) or in other vertebrates, although they are present in the guillemot (our results). They are very variable in form, and the sequences responsible for their formation are not the same in all species groups, but they are always more stable than structures generated from permuted sequences. In species groups where they originate from two motifs, they form two stem-and-loop structures, each one very similar to some of the structures described in Brown et al. (1986) .
In the chicken and in several mammals, OH is situated within the secondary structures formed by CSB1 and the adjacent segment of domain II. The sequence encompassing OH in the chicken occurs slightly modified in the quail and the goose (Nass 1995 ) and in Alectoris partridges (Randi and Lucchini 1998 ), but we could not identify this sequence in gulls. The CSB1-linked structure of the gulls differed substantially from a similar structure published for chicken (see, e.g., Nass 1995 ), which is very similar to structures found in several mammals and Xenopus. This suggests that OH could be in gulls at a different position than in mammals and chickens. However, it should also be noted that our analysis of the chicken control region failed to obtain the CSB1-encompassing secondary structures documented by either Quinn and Wilson (1993) or Nass (1995) . These structures could only be obtained when calculations were constrained to a less stable structure. This suggests that putative secondary structures identified by available programs are poor predictors of actual structures.
Because the control region is a noncoding portion of DNA, constraints on its evolution are most likely related to specific functions, some of which are presumably regulated or mediated by secondary structures. On the one hand, there is no direct relationship between secondary structure conservation and primary sequence conservation within the control region. In gulls, secondary structures were formed by moderately to highly variable segments of the control region, while no potential structure was found involving the more conserved blocks. For finches, Marshall and Baker (1997) have also suggested that conservation of secondary structure may be independent of sequence similarity. It should also be noted that invariant segments of the control region in gulls do not necessarily correspond to motifs that are conserved across vertebrate evolution, suggesting that short-term sequence conservation originates from different constraints than does long-term sequence conservation.
On the other hand, our data suggest a relationship between the presence and stability of secondary structures in and around domain III and its evolutionary rate. First, no clear secondary structures are apparent in the dunlin or Fringilla finches domain III, which evolves faster than the corresponding central domain. In contrast, highly significant secondary structures were identified in the gull or guillemot domain III, which evolves at a rate similar to that of the central domain. Second, among gulls, species from group 1, which have a highly conserved domain III (the average rate of evolution is only half the rate of the central domain), have a three-stem-and-loop structure in domain III (two-stem-and-loop structures for all other gulls) and more stable CSB1-associated structures than any other species groups. Faster-evolving control regions from group 8 species have the least stable CSB1-associated structures.
In order to investigate these putative relationships between structural conservation and low evolutionary rate suggested by our results, more extensive data sets are required. If these data sets confirm that there is indeed a trend for a reduction in evolutionary rates in regions where secondary structures exist, functional mechanisms must be sought, because there is apparently no direct causal link between secondary-structure maintenance and sequence conservation. In addition, the existence of conserved segments that do not seem to be involved in the formation of secondary structures indicates that constraints related to unidentified functions act on these segments. Some functions of the control region might not be realized through secondary-structure interactions. Nevertheless, one must also consider the possibility that models used to reconstitute putative secondary structures are limited in their ability to retrieve actual in vivo structures.
Rodney Honeycutt, Reviewing Editor
Present address: Department of Animal Ecology, Evolutionary Biology Center, Uppsala, Sweden.
Keywords: Laridae birds control region mitochondrial DNA secondary structure sequence variation
Address for correspondence and reprints: Pierre-André Crochet, Department of Animal Ecology, Evolutionary Biology Center, Norbyvägen 18D, S-752 36 Uppsala, Sweden. E-mail: [email protected].
Table 1 Average Base Compositions of Control Region Domains II and III and the Cytochrome b Segment in the Gull Species for Which Sequences of the End of the Control Region Were Available

Table 1 Average Base Compositions of Control Region Domains II and III and the Cytochrome b Segment in the Gull Species for Which Sequences of the End of the Control Region Were Available
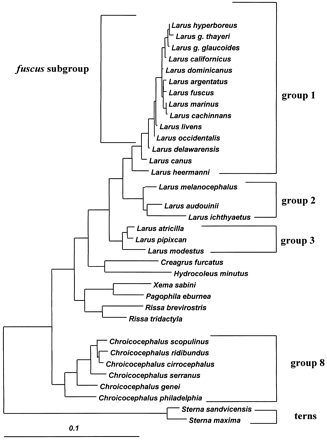
Fig. 1.—Maximum-likelihood tree of the 32 gull and 2 tern species analyzed in this work. This tree is based on a complete data set including sequences of 660 nucleotide sites from the control region and 275 sites from cytochrome b as detailed in Crochet, Lebreton, and Bonhomme (2000) . The numbers to the right indicate species groups used in the analyses. Branch lengths are proportional to maximum-likelihood distances
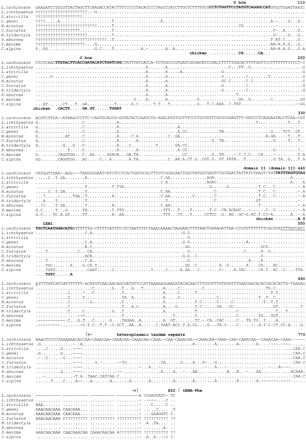
Fig. 2.—Multiple alignment of the L-stand of control region domain II (minus ≈65 bp) and domain III of eight gulls, one tern, and the dunlin. The C and D boxes and CSB1 are in bold, with the chicken sequence (Desjardins and Morais 1990 ) aligned for these segments. The domain boundary is indicated by a vertical dash above the fist base of the domain. Arrows between vertical dashes at the end of the alignment mark limits of the heteroplasmic tandem repeats. A repeated segment is underlined or in italics (first repeat). Base 1 corresponds to position 466 of the chicken sequence (Desjardins and Morais 1990)
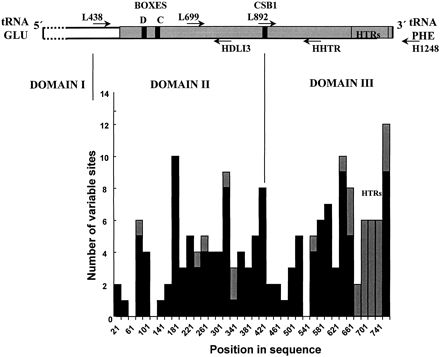
Fig. 3.—Above, Schematic representation of the avian control region. The gray area is the part sequenced in this study. Arrows indicate primer positions. Conserved sequences identified in gulls are also depicted. Domain boundaries are marked by vertical lines. Below, Plot of gull control region variability in nonoverlapping 20-base windows. Substitutions are indicated with solid back bars, and indels are indicated with gray ones. Positions in the sequence are numbered according to the alignment in figure 2 . HTRs = heteroplasmic tandem repeats. Based on the same gull species as used in figure 2
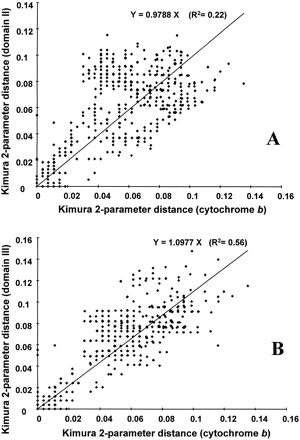
Fig. 4.—Pairwise distances calculated from control region sequences (domain II: A; domain III: B) plotted against pairwise distances calculated from cytochrome b sequences for all comparisons between the 32 gull species. Regression lines are forced to intercept at 0
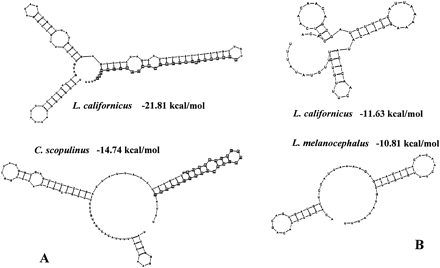
Fig. 5.—Examples of secondary structures obtained from CSB1 and the end of domain II (A) or domain III (B). CSB1 sites are marked by squares. The first nucleotide is at position 349 (A) or at position 571 (B) in fig. 2
We are extremely grateful to the numerous persons who sent us samples from all around the world and to the Zoological Institute of the University of Copenhagen, Louisiana State University Museum of Natural Science, and Muséum National d'Histoire Naturelle de Paris for the loan of biological material. The names of all contributors can be found in Crochet, Lebreton, and Bonhomme (2000) . A. J. Baker, F. Bonhomme, N. Galtier, and J. Nylander variously but significantly contributed to this work. E. Douzery, A. Härlid, R. L. Honeycutt, J.-D. Lebreton, B. C. Sheldon, and two anonymous referees greatly helped to improve the manuscript.
literature cited
Arbogast, B. S., and J. S. Slowinski.
Baker, A. J., and H. D. Marshall.
Berg, T., T. Moum, and S. Johansen.
Bernatchez, L., and R. G. Danzmann.
Brown, G. G., G. Gadaleta, G. Pepe, C. Saccone, and E. Sbisà.
Crochet, P.-A., J.-D. Lebreton, and F. Bonhomme.
Desjardins, P., and R. Morais.
Doda, J. N., C. T. Wright, and D. A. Clayton.
Faber, J. E., and C. A. Stepien.
Felsenstein, J.
Gemmell, N. J., P. S. Western, J. M. Watson, and J. A. Marshall Graves.
Kidd, M. G., and V. L. Friesen.
Kishino, H., and M. Hasegawa.
Klicka, J., and R. M. Zink.
Kumar, S., K. Tamura, and M. Nei.
L'Abbé, D., J. F. Duhaime, B. F. Lang, and R. Morais.
Lee, W.-J., J. Conroy, W. H. Howell, and T. D. Kocher.
Marshall, H. D., and A. J. Baker.
Martin, A. P., and S. R. Palumbi.
Matzura, O., and A. Wennborg.
Mindell, D. P., A. Knight, C. Baer, and C. J. Huddleston.
Nass, M. M. K.
Page, R. D. M., P. L. M. Lee, S. A. Becher, R. Griffiths, and D. H. Clayton.
Questiau, S., M.-C. Eybert, A. R. Gaginskaya, L. Gielly, and P. Taberlet.
Quinn, T. W.
Quinn, T. W., and A. C. Wilson.
Rand, D. M.
Randi, E., and V. Lucchini.
Robinson, M., M. Gouy, C. Gautier, and D. Mouchiroud.
Robinson-Rechavi, M., and D. Huchon.
Sbisà, E., F. Tanzariello, A. Reyes, G. Pesole, and C. Saccone.
Shedlock, A. M., J. D. Parker, D. A. Crispin, T. W. Pietsch, and G. C. Burmer.
Simon, C.
Taberlet, P., and L. Fumagalli.
Taylor, M. F. J., S. W. McKechnie, N. Pierce, and M. Kreitman.
Wenink, P. W., A. J. Baker, and M. G. J. Tilanus.
———.
Yang, Z.
Yang, Z., and S. Kumar.
Zink, R. M., and R. C. Blackwell.
Zink, R. M., D. L. Dittmann, J. Klicka, and R. C. Blackwell-Rago.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}