





Abstract
Grape proanthocyanidins (PAs) play a major role in the organoleptic properties of wine. They are accumulated mainly in grape skin and seeds during the early stages of berry development. Despite the recent progress in the identification of genes involved in PA biosynthesis, the mechanisms involved in subunit condensation, galloylation, or fine regulation of the spatio-temporal composition of grape berries in PAs are still not elucidated. Two Myb transcription factors, VvMybPA1 and VvMybPA2, controlling the PA pathway have recently been identified and ectopically over-expressed in an homologous system. In addition to already known PA genes, three genes coding for glucosyltransferases were significantly differentially expressed between hairy roots over-expressing VvMybPA1 or VvMybPA2 and control lines. The involvement of these genes in PA biosynthesis metabolism is unclear. The three glucosyltransferases display high sequence similarities with other plant glucosyltransferases able to catalyse the formation of glucose esters, which are important intermediate actors for the synthesis of different phenolic compounds. Studies of the in vitro properties of these three enzymes (Km, Vmax, substrate specificity, pH sensitivity) were performed through production of recombinant proteins in E. coli and demonstrated that they are able to catalyse the formation of 1-O-acyl-Glc esters of phenolic acids but are not active on flavonoids and stilbenes. The transcripts are expressed in the early stages of grape berry development, mainly in the berry skins and seeds. The results presented here suggest that these enzymes could be involved in vivo in PA galloylation or in the synthesis of hydroxycinnamic esters.
Introduction
Phenolic compounds constitute one of the most numerous and ubiquitously distributed groups of plant secondary metabolites. In grape, the most common phenolic acids include cinnamic and benzoic acids. Tartaric esters [i.e. caftaric (caffeoyl-tartaric), fertaric (feruloyl-tartaric), and coutaric acids (p-coumaroyl-tartaric)] of hydroxycinnamic acids are mainly found in fruits (Ribéreau-Gayon, 1964; Singleton et al., 1978). Hydroxycinnamic esters are distributed in the flesh and skin of the berry and are important because of their involvement in browning reactions (Singleton, 1972; Singleton et al., 1978), and because they are precursors of volatile phenols (Etiévant, 1981; Dubois, 1983; Chatonnet et al., 1993). The hydroxycinnamic esters are accumulated during the initial period of growth, exhibiting a peak of accumulation prior to veraison, the onset of ripening (Romeyer et al., 1983).
Flavonoids are widely distributed in plants. Among them, proanthocyanidins (PAs) are polymers of flavan-3-ols involved in several physiological and biochemical processes in plants (Beck and Schoonhoven, 1980; Coley et al., 1985). In grape, they also play a critical role for wine quality as they contribute to its astringency and colour stability. Monomeric, oligomeric, and polymeric flavan-3-ols are found in high concentrations in skins and seed tissues of grapevine (Kennedy et al., 2001). In seeds, PAs are based on (+)-catechin, (–)-epicatechin, and (–)-epicatechin 3-O-gallate, the gallic ester of (–)-epicatechin, with a polymerization degree below 10 and a galloylation rate between 10–20% (Prieur et al., 1994). In skins, proanthocyanidins are mainly based on (+)-catechin (primarily as terminal subunit), (–)-epicatechin, (–)-epigallocatechin, and (–)-epicatechin 3-O-gallate, with a polymerization degree around 30 and galloylation rates below 5% (Souquet et al., 1996). PA accumulation occurs immediately after fruit-set and maximum levels of accumulation are reached around veraison (Kennedy et al., 2001; Verriès et al., 2008).
PAs, like other flavonoids, are derived from the phenylpropanoid pathway. Genetic and biochemical characterization of the flavonoid pathways in several plant species have led to the cloning and identification of a series of key structure and regulating genes (Shirley et al., 1992; Winkel-Shirley, 2001). In grapevine, two MYBs factors, VvMybPA1 and VvMybPA2, were recognized as regulators of the PA pathway, respectively, in seeds and skins (Bogs et al., 2007; Terrier et al., 2009). To date, and in spite of this recent progress, little is known about the mechanisms involved in either polymerization or galloylation of PA units or in their transport into the vacuole, their final storage site (Zhao et al., 2010). In order to identify these missing actors, a transcriptomic analysis was performed on hairy-roots transformed with the two Myb factors known to regulate the PA pathway, VvMybPA1 and VvMybPA2 (Terrier et al., 2009). Several of the significantly induced genes were already known to belong to the flavonoid pathway. Other genes have miscellaneous or even unknown function, and among them, three glucosyltransferases (GTs).
In the literature, several types of GTs have been demonstrated to be involved in the phenylpropanoid metabolism. In addition to the grape gene encoding the enzyme UDP-glucose: flavonoid 3-O-glucosyltransferase (UFGT) that catalyses the O-glucosylation of anthocyanidins (Ford et al., 1998), two other grape genes (VvGT5, VvGT6) involved in flavonol glycoside metabolism were characterized and catalysed, respectively, the synthesis of flavonol 3-O-glucuronide and flavonol 3-O-glucoside/galactoside in grapevine (Ono et al., 2010). In Vitis labrusca, Hall and DeLuca (2007) identified a bifunctional glucosyltransferase, VlRSgt, able to form a glc-ester with phenolic acids in acidic conditions and O-glucosides with stilbenes and flavonols in more basic conditions, at much lower rates.
Recently, a GT, UGT72L1 was shown to be induced in Medicago truncatula hairy roots in parallel with PA biosynthesis (Pang et al., 2008). The protein encoded by this gene was shown to catalyse the formation of epicatechin 3′-O-glucoside (Pang et al., 2008), which may serve as an intermediate in PA polymerization.
GT activities toward metabolites of the general phenylpropanoid pathway have also been investigated in relation to sinapate-ester formation in the Brassicaceae family. Sinapoylmalate and sinapoylcholine are two of the major sinapate esters of Arabidopsis (Chapple et al., 1992). 1-O-sinapoylglucose (SG) is an intermediate in the sinapate ester biosynthesis and genes encoding those glc-esters forming enzymes were isolated and further characterized in Arabidopsis and rapeseed (Milkowski et al., 2000a, b, 2004; Lim et al., 2001). The synthesis of methyl and ethyl esters of cinnamate, important strawberry flavor compounds, also involves cinnamoyl-Glc-ester as intermediate, whose formation is catalysed by FaGT2 (Lunkenbein et al., 2006).
This report describes the isolation and functional validation of three grapevine genes encoding glucosyltransferases. Full-length cDNAs were amplified, cloned, and fully sequenced and a phylogenetic analysis of these sequences was performed. The expression of the genes was studied during berry development and in different parts of the berry. The proteins were heterologously expressed and functionally characterized in vitro. Being able to form a glc-ester with a wide range of phenolic acids and most particularly with gallic acid, they were called Vv galloyl Glucosyl Transferase (VvgGT). The glucose-ester formation and its role in PA and other phenolic metabolites biosynthesis will be discussed.
Materials and methods
Plant material and nucleic acid extraction
Grape berries (V. vinifera L. cv. Syrah) were collected at several developmental stages from plants grown in SupAgro-INRA vineyard (Montpellier- France) before and after the onset of fruit ripening (veraison) on days 11, 18, 35, 52, 65, 86, and 99 after anthesis. On day 18, veraison (day 52), and day 99, the skin, pulp, and seed were separated.
After sampling, berries were immediately frozen in liquid nitrogen, then ground to a fine powder with a Dangoumau blender (Dangoumill 300, Lonjumeau, France) and stored at –80 °C until use. Total RNA was extracted using the RNeasy Plant Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions, starting from 50 mg tissue.
Real-time PCR
Total RNA isolated from berries was quantified using the Ribogreen reagent (Molecular Probes, Leiden, Netherlands). 500 ng of total RNA was used for reverse transcription using the Superscript II RT kit in a total volume of 20 μl (Invitrogen, Fisher Bioblock, Illkirch, France). Gene expression was monitored in triplicate using 1 μl of cDNA with the gene-specific primers called VvgGTiexp F and R, where i=1–3 and F and R correspond to Forward and Reverse primers, respectively (Table 1). The PCR (Polymerase Chain Reaction) was performed using a model 7300 sequence Detection System (Applied Biosystems, Warrington, UK) and the Power SYBR-Green PCR Master-kit (Applied Biosystems, Applera France, Courtaboeuf, France). EF1α was used as reference for normalization of gene expression (Terrier et al., 2005; Reid et al., 2006). The difference between the cycle threshold (Ct) of the target gene and EF1α was used to obtain the normalized expression of the target gene, calculated as 2 exp–(Cttarget–CtEF1alpha).
Primers used for amplification: the Atg start codon is underlined, and added restriction sites are in italic letters
| Purpose | Primer name | Sequence 5′–3′ | Forward or reverse | Restriction site |
| Real-time PCR | VvgGT1expF | TTGAAGGGACACGTGTGAAG | F | |
| VvgGT1expR | GAAACAAACAGGTTGCGTGA | R | ||
| VvgGT2expF | ATTTTGATGTAGCGTGAAATAAG | F | ||
| VvgGT2expR | TGGGTGACAAAACTAGGGAAA | R | ||
| VvgGT3expF | TGAGTATGGTGCTGGTTTGC | F | ||
| VvgGT3expR | ACGCTACATCGTGACTCCTG | R | ||
| PGEX-4T2 cloning | VvgGT1pGEXstart | GGATCCATGGGATCTGAATCAAAGCTA | F | BamHI |
| for fusion protein | VvgGT1pGEXstop | GCGGCCGCTTAAATTTTTGTTTTGTAAATG | R | NotI |
| expression | VvgGT2pGEXstart | GGATCCATGGGGTCTGAATCAAAGCTAG | F | BamHI |
| VvgGT2pGEXrev | CTTATTTCACGCTACATCAAAAT | R | ||
| VvgGT2pGEXstop | GCGGCCGCTCAAATTTTCTTTGACTTGCA | R | NotI | |
| VvgGT3pGEXstart | GGATCCATGGGTTCTGAATCAAAGCTAG | F | BamHI | |
| VvgGT3pGEXstop | GCGGCCGCTCAAATTTTGTTTGACTGGCA | R | NotI |
| Purpose | Primer name | Sequence 5′–3′ | Forward or reverse | Restriction site |
| Real-time PCR | VvgGT1expF | TTGAAGGGACACGTGTGAAG | F | |
| VvgGT1expR | GAAACAAACAGGTTGCGTGA | R | ||
| VvgGT2expF | ATTTTGATGTAGCGTGAAATAAG | F | ||
| VvgGT2expR | TGGGTGACAAAACTAGGGAAA | R | ||
| VvgGT3expF | TGAGTATGGTGCTGGTTTGC | F | ||
| VvgGT3expR | ACGCTACATCGTGACTCCTG | R | ||
| PGEX-4T2 cloning | VvgGT1pGEXstart | GGATCCATGGGATCTGAATCAAAGCTA | F | BamHI |
| for fusion protein | VvgGT1pGEXstop | GCGGCCGCTTAAATTTTTGTTTTGTAAATG | R | NotI |
| expression | VvgGT2pGEXstart | GGATCCATGGGGTCTGAATCAAAGCTAG | F | BamHI |
| VvgGT2pGEXrev | CTTATTTCACGCTACATCAAAAT | R | ||
| VvgGT2pGEXstop | GCGGCCGCTCAAATTTTCTTTGACTTGCA | R | NotI | |
| VvgGT3pGEXstart | GGATCCATGGGTTCTGAATCAAAGCTAG | F | BamHI | |
| VvgGT3pGEXstop | GCGGCCGCTCAAATTTTGTTTGACTGGCA | R | NotI |
Primers used for amplification: the Atg start codon is underlined, and added restriction sites are in italic letters
| Purpose | Primer name | Sequence 5′–3′ | Forward or reverse | Restriction site |
| Real-time PCR | VvgGT1expF | TTGAAGGGACACGTGTGAAG | F | |
| VvgGT1expR | GAAACAAACAGGTTGCGTGA | R | ||
| VvgGT2expF | ATTTTGATGTAGCGTGAAATAAG | F | ||
| VvgGT2expR | TGGGTGACAAAACTAGGGAAA | R | ||
| VvgGT3expF | TGAGTATGGTGCTGGTTTGC | F | ||
| VvgGT3expR | ACGCTACATCGTGACTCCTG | R | ||
| PGEX-4T2 cloning | VvgGT1pGEXstart | GGATCCATGGGATCTGAATCAAAGCTA | F | BamHI |
| for fusion protein | VvgGT1pGEXstop | GCGGCCGCTTAAATTTTTGTTTTGTAAATG | R | NotI |
| expression | VvgGT2pGEXstart | GGATCCATGGGGTCTGAATCAAAGCTAG | F | BamHI |
| VvgGT2pGEXrev | CTTATTTCACGCTACATCAAAAT | R | ||
| VvgGT2pGEXstop | GCGGCCGCTCAAATTTTCTTTGACTTGCA | R | NotI | |
| VvgGT3pGEXstart | GGATCCATGGGTTCTGAATCAAAGCTAG | F | BamHI | |
| VvgGT3pGEXstop | GCGGCCGCTCAAATTTTGTTTGACTGGCA | R | NotI |
| Purpose | Primer name | Sequence 5′–3′ | Forward or reverse | Restriction site |
| Real-time PCR | VvgGT1expF | TTGAAGGGACACGTGTGAAG | F | |
| VvgGT1expR | GAAACAAACAGGTTGCGTGA | R | ||
| VvgGT2expF | ATTTTGATGTAGCGTGAAATAAG | F | ||
| VvgGT2expR | TGGGTGACAAAACTAGGGAAA | R | ||
| VvgGT3expF | TGAGTATGGTGCTGGTTTGC | F | ||
| VvgGT3expR | ACGCTACATCGTGACTCCTG | R | ||
| PGEX-4T2 cloning | VvgGT1pGEXstart | GGATCCATGGGATCTGAATCAAAGCTA | F | BamHI |
| for fusion protein | VvgGT1pGEXstop | GCGGCCGCTTAAATTTTTGTTTTGTAAATG | R | NotI |
| expression | VvgGT2pGEXstart | GGATCCATGGGGTCTGAATCAAAGCTAG | F | BamHI |
| VvgGT2pGEXrev | CTTATTTCACGCTACATCAAAAT | R | ||
| VvgGT2pGEXstop | GCGGCCGCTCAAATTTTCTTTGACTTGCA | R | NotI | |
| VvgGT3pGEXstart | GGATCCATGGGTTCTGAATCAAAGCTAG | F | BamHI | |
| VvgGT3pGEXstop | GCGGCCGCTCAAATTTTGTTTGACTGGCA | R | NotI |
Cloning and vectors
cDNA fragments corresponding to VvgGT1, VvgGT2, and VvgGT3 were amplified from V. vinifera L. cv. Maccabeu, using high fidelity Taq polymerase (Advantage®–HF 2 PCR kit, Clontech, California, USA). Genebank accession numbers are JN164679, JN164680, and JN164681, respectively. For heterologous expression of the proteins, forward (VvgGT1-to-3-pGEXstart) and reverse (VvgGT1-to-3-pGEXstop) primers, including start and stop codons, respectively, and adequate restriction sites (Table 1) were used. To ensure specificity of the amplification for VvgGT2 and 3, a first round of amplification was performed with specific reverse primers called VvgGT2pGEXrev and VvgGT3expR located in their divergent 3′NC region (Table 1). After cloning into pGEMT-Easy (Promega, Madison, Wisconsin, USA) and digestion with the adequate restriction enzymes, the resulting fragments were ligated into plasmid pGEX-4T2 digested with the same restriction enzymes (GE Healthcare, Chalfont St Giles, UK).
All types of constructions were inserted into DH5α Escherichia coli competent cells for propagation. Transformants with the inserted sequences were identified by colony PCR using the same primers as for amplification and were verified by plasmid sequencing.
Sequence analysis
Full-length amino-acid sequences of glucosyltransferases from several species were retrieved from public databases. Multiple sequence alignment was performed with ClustalW2 alignment algorithm with default parameters (Thompson et al., 1994). Phylogenetic analysis was performed from the ClustalW alignment using the Neighbor–Joining method in the MEGA4 package (Tamura et al., 2007).
Heterologous expression of the three VvgGTs
Escherichia coli BL21(DE3) (Stratagene, La Jolla, California, USA) cells containing the VvgGT cDNA fused with the GST sequence in PGEX-4T2 vector were incubated overnight in LB (Luria-Bertani) medium with ampicillin 100 μg ml−1 at 37 °C. The next day, 3 ml of E. coli cells were inoculated into 120 ml of YTA 2X containing ampicillin (100 μg ml−1) and incubated with shaking at 37 °C until the OD600 reached 0.8. 0.1 mM final IPTG (isopropyl-1-thio-β-D-galactopyranoside) was then added and the culture was incubated overnight (17–24 h) at room temperature (24 °C) to initiate the induction of GST-VvgGT1 to three fusion proteins. After 24 h of induction, cells were pelleted by centrifugation at 4 °C, 8000 g, and resuspended in 2 ml of ice-cold PBS (phosphate buffer saline), 0.1% (v/v) β-mercaptoethanol, 1% (v/v) Triton X-100, and lyzosyme (final concentration of 1 mg ml−1). Cells were disrupted by sonication and the lysate was centrifuged at 12 000 g for 5 min. The supernatant was then incubated with Glutathione–Sepharose™ 4B (GE Healthcare, Chalfont St Giles, UK) for 30 min at room temperature with gentle mixing. The beads were washed several times with 1× PBS buffer and heterologous proteins were liberated from their GST-fusion part using thrombin. Bradford method (Bradford, 1976) was used to determine protein concentration with a bovine serum albumin calibration curve. The purified recombinant proteins were also analysed by SDS-polyacrylamide gel electrophoresis on a 14% gel according to Laemmli’s method (Laemmli, 1970) (results not shown). The purified recombinant protein stock was aliquoted and stored at –20 °C with glycerol 10% (v/v) till their use for activity assays.
Enzyme assays
Reactions were carried out in an assay mix (200 μl) containing a final concentration of 20 μg ml−1 of recombinant protein, 2.5 mM UDP-Glc (Sigma-Aldrich, St Louis, MO, USA) as sugar donor, and 1 mM (except for Km determination) of phenylpropanoid substrate prepared using adequate buffers. To determinate optimum pH, reactions were performed at 30 °C for 10 min at different pH values using acetate buffer (100 mM) for assays at pH 4.5, MES-HCl (100 mM) for assays at pH 5.5/6.5 and TRIS-HCl (100 mM) for those at pH 7.5 and 8.5. All reactions contained 0.1% (v/v) β-mercaptoethanol. Reactions were stopped by mixing 60 μl of the reaction medium with 54 μl of 100% methanol and 6 μl of benzoic acid (5 mM, 10% v/v methanol) (Sigma-Aldrich, St Louis, MO, USA) used as internal standard and stored at –20 °C prior to the electrophoresis analysis. Reactions without protein extract were systematically performed as controls. The specific enzyme activity was expressed in pmol of phenylpropanoid glucosylated per mg of protein and per second.
For substrate specificity, initial screening for activity of the three proteins was performed against 11 potential substrates including hydroxybenzoic acids (C6C1): PHBA (4-hydroxybenzoic acid, 4-HBA), protocatechuic acid (3,4-dihydroxybenzoic acid, 3,4-diHBA), gallic acid (3,4,5-trihydroxybenzoic acid, 3,4,5-triHBA), syringic acid (2,3-dimethoxy,4-hydroxybenzoic acid, 3,5 diM-4-HBA), hydroxycinnamic acids (C6C3): p-coumaric acid (4-hydroxycinnamic acid, 4-HCA), caffeic acid (3,4-dihydroxycinnamic acid, 3,4-diHCA), sinapic acid (3,5-di methoxy, 4-hydroxycinnamic acid, 3,5-diM-4-HCA), a stilbene (trans-resveratrol), and flavonoids (quercetin, cyanidin, catechin). Reactions were carried out at pH 6.5, using identical assay conditions of temperature and reaction time. Quercetin, cyanidin, catechin, and trans-resveratrol were purchased from Extrasynthèse (Genay, France). All other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA).
The kinetic parameters of the recombinant VvgGT enzymes were obtained from hyperbolic Michaelis–Menten saturation curves for acceptor substrates under optimal conditions. They were measured using 2.5 mM UDP-Glc and over a range of 2 μM–2 mM final concentrations of gallic acid and caffeic acid at pH 6.5 under identical assay conditions of temperature (30 °C) and reaction time (10 min). Kinetic parameters were obtained by non-linear fitting of the experimental data. All assays were repeated in triplicate.
Identification of the VvgGTs products
Products of the enzymatic reactions were identified by HPLC mass spectrometry analysis using a Waters Millenium HPLC-DAD system (Milford, MA) coupled with a Thermo Finnigan Advantage LCQ ion trap spectrometer fitted with an electrospray (ESI) interface (San Jose, USA).
Separations were performed by reversed phase HPLC on a (250×2 mm i.d.) Atlantis dC18 column (Waters, Milford, MA; 5 μm) with a guard column, operated at 30 °C. The mobile phase consisted of water (eluent A) and water/acetonitrile (20/80, by vol.) (eluent B). Flow rate was 0.25 ml min−1. The elution programme was as follows: starting with 0% B, linear gradients from 0–2% B (2–10 min), from 2–8% B (10–20 min), from 10–20% B (30–40 min), from 20–30% B (40–45 min), and from 30–40% B (45–50 min), followed by washing and reconditioning of the column.
Mass spectrometry experiments were conducted in the negative ion mode. The scan range was 100–2000 a.m.u. The desolvation temperature was 300 °C, high spray voltage was set at 3000 V, nitrogen was used as the dry gas at a flow of 5 and 35 units for the auxiliary and the sheath gas, respectively. An MS/MS analysis was performed with collision energy of 35%.
Alkaline hydrolysis was performed in 1 N (final concentration) NaOH at room temperature for 1 h and neutralized by 3 M (final concentration) sodium acetate, pH 5.2.
Enzymatic hydrolysis was performed after drying the reaction medium and adding 12 units of β-glucosidase from almond (Sigma-Aldrich, St Louis, MO, USA) in acetate buffer 50 mM pH 5, incubated for 3 h at 37 °C. HPLC analysis of reaction products (before and after β-glucosidase hydrolysis) were performed as described in Verries et al. (2008).
Quantification by capillary electrophoresis analysis
Enzymatic activity was assessed by measuring UDP by capillary electrophoresis (CE), performed using a Beckman PACE 5500 CE unit equipped with a diode array detector. Beckman capillary tubing was for 75 cm×75 μm I.D×375 μm O.D. Samples were hydrodynamically injected for 5 s in the CE equipment using a laboratory-made programmable arm controlled by a micro-computer via an electronic interface. The running buffer used was a solution of 25 mM of H3BO3 at 8.5 (adjusted with NaOH). The applied voltage was 30 kV, the average current 40.1 μA, the temperature 30 °C, and the selected wavelength at 214 nm. Capillary surface was regenerated once a day by consecutive washing with water (3 min), methanol, 1 M sodium hydroxide (2 min), 0.1 M sodium hydroxide (2 min), 0.1 N HCl (2 min), and water (2 min). Calibration graphs were obtained by injecting standard solutions in the range of 0.1–1 μg ml−1. In all quantifications, benzoic acid was used as internal standard (0.25 mM final concentration), added after the enzyme reaction had been stopped by the addition of methanol 100%. The eluting peaks were processed via the 32 Karat™ Software, version 5.0 (Beckman-Coulter, Fullerton, CA, USA) and quantification was performed by evaluating the normalized area (NA) of UDP formed versus the internal standard area.
Results
Identification of three putative glucosyltransferases
The full-length VvgGT1, VvgGT2, and VvgGT3 cDNAs were amplified and the transcripts were 1437, 1437, and 1434 nucleotides long, coding for polypeptides of 479, 479, and 478 amino acid residues, respectively, with a predicted molecular mass of 53.95 kDa, 53.79 kDa, and 53.65 kDa. According to the 12X version of the Vitis vinifera grape genome, the three genes are clustered on chromosome 3. VvgGT1 corresponds to GSVIVT01037921001, located on position 6338114 to 6339547; VvgGT2 corresponds to GSVIVT01003157001, located on position 6124678 to 6126114; VvgGT3 corresponds to GSVIVT01003143001, located on position 6282074 to 6283510.
The three glucosyltransferases share more than 90% identity at the nucleotide level, a 92% amino acid identity and a similarity ranging between 97.5% and 98.7% to each other. Phylogenetic study was performed by comparing the three VvgGT complete protein sequences to other plant sequences already identified as glucosyltransferases involved in phenylpropanoid metabolism (Fig. 1). High similarity in amino acid sequences were observed when comparing our three VvgGTs to those clustered into the subgroup of glucosyltransferases able to catalyse the formation of glucose-esters. High divergences were observed when comparing our three VvgGTs to those forming O-glucosides from various substrates, including flavonoids.
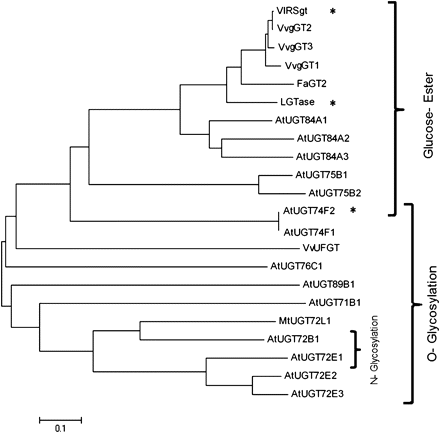
Phylogenetic relationship of three VvgGTs cloned from Vitis vinifera with 19 other functionally characterized glucosyltransferases from several other plant species (MtUGT72L1 (ACC38470) in Pang et al. (2008), VlRSgt (ABH03018) in Hall and De Luca (2007), LGTase (Q9MB73) in Kita et al. (2000), FaGT2 (AAU09443) in Lukenbein et al. (2006), VvUFGT (AAB81682) in Ford et al. (1998), AtUGT84A1 (Z97339), AtUGT75B2 (AC005106), AtUGT75B1 (AC005106), AtUGT74F1 (AC002333), AtUGT74F2 (AC002333), AtUGT78D2 (AL391141), AtUGT71C1 (AC005496), AtUGT71C4 (AC067971), AtUGT71B1 (AB025634), AtUGT72B1 (AC067971), AtUGT73B3 (AL161584), AtUGT73B4 (AC006248), AtUGT89B1 (AC016662), and AtUGT89A2 (AL162751) in Lim et al. (2002), AtUGT72E2 (AB018119), AtUGT72E3 (AF077407), AtUGT84A2 (AB019232) and AtUGT84A3 (Z97339) in Lim et al. (2001), AtUGT72E1 (AL049862) in Lim et al. (2005), AtUGT76C1 (AB017060) in Hou et al. (2004). The dendrogram was created using the Neighbor–Joining method in the MEGA4 package. Lengths of lines indicate the relative distance between nodes. Asterisks indicate O-glucosylation activity of the enzyme despite being clustered with glucose-ester forming enzymes.
Expression of VvgGT1, VvgGT2, and VvgGT3
In order to study spatial and temporal expression pattern of the three VvgGT genes, a quantitative real-time PCR approach was performed throughout berry development in the pericarp and in the different parts of the berries (skin, pulp, and seeds) at three development stages (middle of the green stage, véraison, and the ripe stage). At the developmental level, the three VvgGTs were strongly expressed at the beginning of the green stage and their expression decreased thereafter until reaching very low values after véraison (Fig. 2; 1A, 2A, 3A).
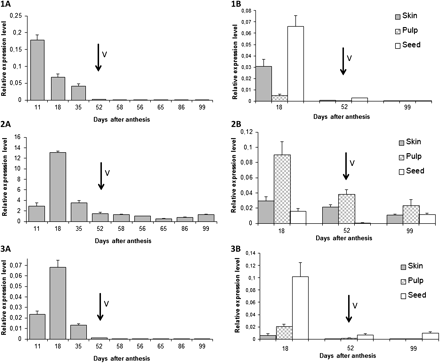
Quantitative real-time PCR expression, normalized with the expression of EF1α, of VvgGT1 (1), VvgGT2 (2), VvgGT3 (3) during berry pericarp development (1A, 2A, 3A) and at three development stages in different grape berry tissues (1B, 2B, 3B). Véraison (V) is marked with arrows. All data are means of three replicates, with error bar indicating SD.
Figure 2 (1B, 2B, 3B) shows that the expression of the three genes is divergent among the different parts of the berry. VvgGT1 is expressed in green tissues in both skin and seeds, VvgGT3 expression is almost seed specific, while VvgGT2 is predominantly expressed in the pulp.
Enzymatic activity of the recombinant enzymes
The three recombinant proteins were generated in Escherichia coli cells as GST fusion proteins. Purification was followed by using Glutathione Sepharose as an affinity matrix and then the GST portion of the recombinant fusion protein was cleaved by digestion with thrombin. The three purified VvgGTs were screened for glucosyltransferase activity.
To characterize the substrate specificity, the three VvgGTs were analysed for activity against seven phenolic derivatives of hydroxybenzoic (C6C1) and hydroxycinnamic (C6C3) acids differing by the kind of aromatic substitution (hydroxy, dihydroxy, trihydroxy, hydroxydimethoxy) (Fig. 3), some flavonoids (quercetin, catechin, cyanidin), and a stilbene (resveratrol). While most of Glc conjugates are found in the vacuole, glycosylation usually takes place in the cytosol (Vogt and Jones, 2000, Felle, 2005; Schulte et al., 2006). The assays were thus performed in presence of UDP-Glc as the sugar donor at pH 6.5 to be closer to cytoplasmic conditions.
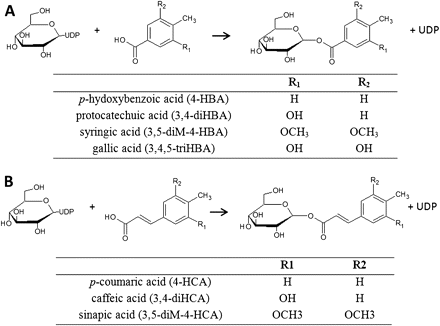
Chemical structures of some substrates and products of VvgGTs. VvgGTs converts (A) hydroxybenzoic acids, (B) hydroxycinnamic acids to the corresponding Glc esters, using UDP-activated Glc.
CE analysis of the reaction media showed that, among the 11 substrates tested, all phenolic acids were accepted as potential substrates by the three recombinant enzymes, UDP and an additional product being detected in the electrophoresis. On the other hand, no significant activity was detected toward stilbene and flavonoid substrates (Fig. 4). Under those conditions, VvgGT1 and VvgGT3 exhibited a slightly higher activity for hydroxybenzoic acids than for hydroxycinnamic acids, whereas this difference was less marked for VvgGT2. For both C6C1 and C6C3 substrates, the best activity was obtained with dihydroxysubstituted phenol rings (Fig. 4A, B, C).
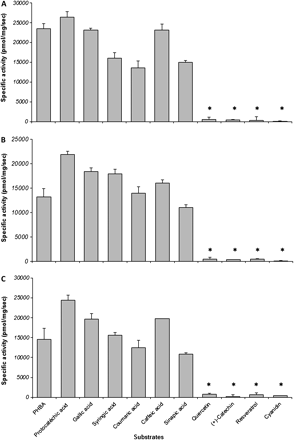
Substrate specificity of the three recombinant VvgGTs tested in vitro at pH 6.5. (A) VvgGT1; (B) VvgGT2; and (C) VvgGT3. Each bar represents the mean of three assays ± standard deviation. Asterisks indicate values not significantly different from zero (P=0.05).
To define the optimal pH further, recombinant VvgGT activities were assayed toward gallic acid between pH 4.5 and 8.5, by evaluating the UDP released by CE. The recombinant enzymes were active over a broad pH range. The maximal glucosyltransferase activity was observed between 5.5 (for VvgGT1 and VvgGT3) and 6.5 (for VvgGT2) and this activity decreased at higher pH (Fig. 5).
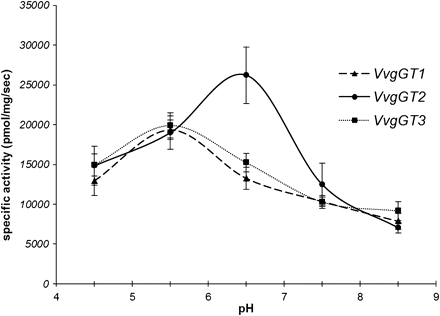
Effect of pH on in vitro activity of the three VvgGTs with gallic acid as substrate. VvgGT1, dashed line and triangles; VvgGT2, continuous line and circles; VvgGT3, dotted line and squares. Each point represents the mean of three assays ± standard deviation. Reaction product is mostly produced between pH 5.5 and 6.5.
The products formed from the 11 substrates in the presence of the three VvgGTs were analysed by HPLC-DAD-ESI-IT-MS and compared with authentic standards (Table 2). Figures 6 and 7 present the results obtained for p-hydroxybenzoic and caffeic acids as substrates for VvgGT3. Identical results were obtained with the two other enzymes (results not shown). It was confirmed that the three VvgGTs catalysed the formation of glucose conjugates as described in Fig. 3, eluting earlier than the corresponding aglycone, as expected from their relative polarities, and showing an excess of 162 a.m.u compared with the m/z signal of the acid. Fragmentation patterns were characteristic of the glucose-ester: loss of the glucose (–162 and –180), loss of the glucose and carboxylic group (–162 and –44) and cleavage of the glucoside (–660 and –120), and similar to these observed by Lunkenbein et al. (2006). No glucosylated molecule was detected when flavonoids or stilbene were used as substrates with HPLC-DAD-ESI-IT-MS in our conditions in contrast with the results shown in Hall and DeLuca (2007).
Retention times and molecular ion and fragment ions of glc-esters formed by VvgGT3 and the corresponding phenolic acids.
| Compound | Retention time (min) | MS [M–H]– | MS2 [M–H]– |
| Galloyl-D-glc | 7.4 | 331 | 271-211-169-125 |
| Protocatechoyl-D-glc | 11.7 | 315 | 255-195-153-109 |
| p-Hydroxybenzoyl-D-glc | 17.2 | 299 | 239-209-179-137 |
| Caffeoyl-D-glc | 23.3 | 341 | 203-179-161-135 |
| Syringoyl-D-Glc | 24.1 | 359 | 299-289-239-211-197-153 |
| p-Coumaroyl-D-Glc | 26.5 | 325 | 145-163-187 |
| Sinapoyl-D-glc | 30.0 | 385 | 247-223-205 |
| Gallic acid | 7.6 | 169 | 125 |
| Protocatechuic acid | 13.2 | 153 | 109 |
| p-Hydroxybenzoic acid | 20.1 | 137 | 93 |
| Caffeic acid | 26.8 | 179 | 135 |
| Syringic acid | 27.0 | 197 | 182-153-169-138 |
| p-Coumaric acid | 31.9 | 163 | 119 |
| Sinapic acid | 36.5 | 223 | 208-179-164 |
| Compound | Retention time (min) | MS [M–H]– | MS2 [M–H]– |
| Galloyl-D-glc | 7.4 | 331 | 271-211-169-125 |
| Protocatechoyl-D-glc | 11.7 | 315 | 255-195-153-109 |
| p-Hydroxybenzoyl-D-glc | 17.2 | 299 | 239-209-179-137 |
| Caffeoyl-D-glc | 23.3 | 341 | 203-179-161-135 |
| Syringoyl-D-Glc | 24.1 | 359 | 299-289-239-211-197-153 |
| p-Coumaroyl-D-Glc | 26.5 | 325 | 145-163-187 |
| Sinapoyl-D-glc | 30.0 | 385 | 247-223-205 |
| Gallic acid | 7.6 | 169 | 125 |
| Protocatechuic acid | 13.2 | 153 | 109 |
| p-Hydroxybenzoic acid | 20.1 | 137 | 93 |
| Caffeic acid | 26.8 | 179 | 135 |
| Syringic acid | 27.0 | 197 | 182-153-169-138 |
| p-Coumaric acid | 31.9 | 163 | 119 |
| Sinapic acid | 36.5 | 223 | 208-179-164 |
Retention times and molecular ion and fragment ions of glc-esters formed by VvgGT3 and the corresponding phenolic acids.
| Compound | Retention time (min) | MS [M–H]– | MS2 [M–H]– |
| Galloyl-D-glc | 7.4 | 331 | 271-211-169-125 |
| Protocatechoyl-D-glc | 11.7 | 315 | 255-195-153-109 |
| p-Hydroxybenzoyl-D-glc | 17.2 | 299 | 239-209-179-137 |
| Caffeoyl-D-glc | 23.3 | 341 | 203-179-161-135 |
| Syringoyl-D-Glc | 24.1 | 359 | 299-289-239-211-197-153 |
| p-Coumaroyl-D-Glc | 26.5 | 325 | 145-163-187 |
| Sinapoyl-D-glc | 30.0 | 385 | 247-223-205 |
| Gallic acid | 7.6 | 169 | 125 |
| Protocatechuic acid | 13.2 | 153 | 109 |
| p-Hydroxybenzoic acid | 20.1 | 137 | 93 |
| Caffeic acid | 26.8 | 179 | 135 |
| Syringic acid | 27.0 | 197 | 182-153-169-138 |
| p-Coumaric acid | 31.9 | 163 | 119 |
| Sinapic acid | 36.5 | 223 | 208-179-164 |
| Compound | Retention time (min) | MS [M–H]– | MS2 [M–H]– |
| Galloyl-D-glc | 7.4 | 331 | 271-211-169-125 |
| Protocatechoyl-D-glc | 11.7 | 315 | 255-195-153-109 |
| p-Hydroxybenzoyl-D-glc | 17.2 | 299 | 239-209-179-137 |
| Caffeoyl-D-glc | 23.3 | 341 | 203-179-161-135 |
| Syringoyl-D-Glc | 24.1 | 359 | 299-289-239-211-197-153 |
| p-Coumaroyl-D-Glc | 26.5 | 325 | 145-163-187 |
| Sinapoyl-D-glc | 30.0 | 385 | 247-223-205 |
| Gallic acid | 7.6 | 169 | 125 |
| Protocatechuic acid | 13.2 | 153 | 109 |
| p-Hydroxybenzoic acid | 20.1 | 137 | 93 |
| Caffeic acid | 26.8 | 179 | 135 |
| Syringic acid | 27.0 | 197 | 182-153-169-138 |
| p-Coumaric acid | 31.9 | 163 | 119 |
| Sinapic acid | 36.5 | 223 | 208-179-164 |
![Characterization of the VvgGT protein product (p-hydroxybenzoyl-glucose) with p-hydroxybenzoic acid as substrate. (A) Chromatogram at 280 nm of the reaction medium after 10 min of incubation at pH 6.5 of VvgGT3 with UDP-Glc and p-hydroxybenzoic acid. Mass spectrum (B) and MS2 spectrum (C) of p-hydroxybenzoyl-glucose (Rt=17.2 min). Molecular ion [M-H]– at m/z=299, and major fragments corresponding to the aglycone (–162; m/z=137) and to degradation of the glucose group (–60: m/z=239; –120: m/z=179).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jxb/63/3/10.1093_jxb_err340/2/m_jexboterr340f06_ht.gif?Expires=1750513846&Signature=hKXmlk5~xvOLyN-g3KoBxggOEKRTqF-GY6PEFPGxYwKsty4XfOFF7Xlh7FDeaAsLk-NHpsgPXvcGlfKvNeRGZNxnuFPmyx-fkqFCkLgp~7wClfDZgkqNRsLVCpAsTuCJcyFPzVc8hrWCRlCajwrs2oCnbGv4hSpZRPyI1gtK2v5SYBBy4-blX2YEaTN3xRbo-SELnc2PBFL6TXGAFmLKx5Fhtxbml48BWAFkcmweJ-TLGl1j15rKsiydTMtErqgJ3AppciGVFYOyGE1MhsdUijzryYU4qxTQdjH9dsPZpoQiTKwpBcV4j7BEqxZTQydTkIu4F1sLmQCYRy5vQcLA3Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characterization of the VvgGT protein product (p-hydroxybenzoyl-glucose) with p-hydroxybenzoic acid as substrate. (A) Chromatogram at 280 nm of the reaction medium after 10 min of incubation at pH 6.5 of VvgGT3 with UDP-Glc and p-hydroxybenzoic acid. Mass spectrum (B) and MS2 spectrum (C) of p-hydroxybenzoyl-glucose (Rt=17.2 min). Molecular ion [M-H]– at m/z=299, and major fragments corresponding to the aglycone (–162; m/z=137) and to degradation of the glucose group (–60: m/z=239; –120: m/z=179).
![Characterization of the VvgGT protein product (caffeoyl-glucose) with caffeic acid as substrate. (A) Chromatogram at 280 nm of the reaction medium after 10 min of incubation at pH 6.5 of VvgGT3 with UDP-Glc and caffeic acid. Mass spectrum (B) and MS2 spectrum (C) of caffeoyl-glucose (Rt=23.3 min). Molecular ion [M-H]– at m/z=341, and major fragments corresponding to the aglycone (–162; m/z=179) aglycone –H2O (–180; m/z=161) aglycone –carboxylic group (–206; m/z=135).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jxb/63/3/10.1093_jxb_err340/2/m_jexboterr340f07_ht.gif?Expires=1750513846&Signature=XMHWYeFNS9uWpZE3ghf2uDdk33JGmQsQIDnUptRLyV2VllKarSdom7znvMkKHN~kXTqHEPslin8GgnBPCdtDrIDmViDhF9lhg0p5kbYMYN~IVkLlxd5E32CtgTC7N4ncCl06oDShwSTN2GZpFS-4pNDczlxSBIGU9OpkrnX5tbwvmiBIm-tVTxSEqCsKz9EiFp4WgGo92XGsaOhSokyZAq-3wqTkDMD8T6VzPIEutmIFdaqaeOxyKtfqImvPm9JDlT8Hm3Vgm5ULDL0p49UA~6SbftqaijziJSwn8IOq4DlG8h~YPILIWjA-I1CcAgwoAYEPmX81XjY8F0vvnrTumg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characterization of the VvgGT protein product (caffeoyl-glucose) with caffeic acid as substrate. (A) Chromatogram at 280 nm of the reaction medium after 10 min of incubation at pH 6.5 of VvgGT3 with UDP-Glc and caffeic acid. Mass spectrum (B) and MS2 spectrum (C) of caffeoyl-glucose (Rt=23.3 min). Molecular ion [M-H]– at m/z=341, and major fragments corresponding to the aglycone (–162; m/z=179) aglycone –H2O (–180; m/z=161) aglycone –carboxylic group (–206; m/z=135).
Alkaline hydrolysis of reaction products converted them back into the aglycone indicating that the glucose-ester of hydroxybenzoic and hydroxycinnamic acids have been produced and thus demonstrating the ability of the three VvgGTs to glucosylate hydroxycinnamic and hydroxybenzoic acids on their carboxyl group to form glucosyl esters of the phenolic acids. Products were hydrolysed after incubation with almond β-glucosidase (see Supplementary Fig. S1 at JXB online), suggesting a β-configuration of glucosidic linkages for the esters formed.
The control, using the GST alone produced by E. coli carrying the empty plasmid as enzymatic extract, was unable to glucosylate any of the phenylpropanoids (results not shown).
Kinetic parameters of the three recombinant proteins
The kinetic parameters of the three VvgGTs were examined using two potential in vivo substrates as suggested by in vitro substrate specificity studies and grape phenolic composition. Kinetic and affinity assays were thus performed toward gallic acid and caffeic acid, representative of the hydroxybenzoic and hydroxycinnamic acid series, respectively, as acceptor substrates and UDP-Glc as donor substrate. Kinetic parameters were obtained using hyperbolic Michaelis–Menten saturation curves.
As shown in Table 3, the resulting plots for the two substrates yielded values between 105 and 165 μM as Km for caffeic acid, while with gallic acid, Km values varied between 510 μM and 884 μM. These results suggest that VvgGTs exhibit higher affinity for caffeic acid (C6C3) compared with gallic acid (C6C1). Affinity for gallic acid was higher for VvgGT1 and VvgGT3 than for VvgGT2, while the opposite was observed for caffeic acid. For the three VvgGTs, maximal reaction rates were obtained with gallic acid.
Michaelis–Menten Kinetics (Km, Vmax values and catalytic efficiency Kcat and Kcat/Km) of the three recombinant VvgGTs toward gallic acid (A), and caffeic acid (B) at pH 6.5
| Km (μM) | Vmax (nkat mg−1) | Kcat (s−1) | Kcat/Km (s−1 M−1) | |
| (A) Gallic acid | ||||
| VvgGT1 | 510±32 | 40.1±5.3 | 2.16±0.28 | 4231 |
| VvgGT2 | 884±188 | 52.3±4.5 | 2.82±0.24 | 3194 |
| VvgGT3 | 566±190 | 40.6±1.7 | 2.19±0.09 | 3873 |
| (B) Caffeic acid | ||||
| VvgGT1 | 165±67 | 21.6±2.2 | 1.16±0.12 | 7069 |
| VvgGT2 | 105±30 | 27.5±1.8 | 1.48±0.09 | 14142 |
| VvgGT3 | 142±57 | 19.9±1.9 | 1.07±0.10 | 7567 |
| Km (μM) | Vmax (nkat mg−1) | Kcat (s−1) | Kcat/Km (s−1 M−1) | |
| (A) Gallic acid | ||||
| VvgGT1 | 510±32 | 40.1±5.3 | 2.16±0.28 | 4231 |
| VvgGT2 | 884±188 | 52.3±4.5 | 2.82±0.24 | 3194 |
| VvgGT3 | 566±190 | 40.6±1.7 | 2.19±0.09 | 3873 |
| (B) Caffeic acid | ||||
| VvgGT1 | 165±67 | 21.6±2.2 | 1.16±0.12 | 7069 |
| VvgGT2 | 105±30 | 27.5±1.8 | 1.48±0.09 | 14142 |
| VvgGT3 | 142±57 | 19.9±1.9 | 1.07±0.10 | 7567 |
Michaelis–Menten Kinetics (Km, Vmax values and catalytic efficiency Kcat and Kcat/Km) of the three recombinant VvgGTs toward gallic acid (A), and caffeic acid (B) at pH 6.5
| Km (μM) | Vmax (nkat mg−1) | Kcat (s−1) | Kcat/Km (s−1 M−1) | |
| (A) Gallic acid | ||||
| VvgGT1 | 510±32 | 40.1±5.3 | 2.16±0.28 | 4231 |
| VvgGT2 | 884±188 | 52.3±4.5 | 2.82±0.24 | 3194 |
| VvgGT3 | 566±190 | 40.6±1.7 | 2.19±0.09 | 3873 |
| (B) Caffeic acid | ||||
| VvgGT1 | 165±67 | 21.6±2.2 | 1.16±0.12 | 7069 |
| VvgGT2 | 105±30 | 27.5±1.8 | 1.48±0.09 | 14142 |
| VvgGT3 | 142±57 | 19.9±1.9 | 1.07±0.10 | 7567 |
| Km (μM) | Vmax (nkat mg−1) | Kcat (s−1) | Kcat/Km (s−1 M−1) | |
| (A) Gallic acid | ||||
| VvgGT1 | 510±32 | 40.1±5.3 | 2.16±0.28 | 4231 |
| VvgGT2 | 884±188 | 52.3±4.5 | 2.82±0.24 | 3194 |
| VvgGT3 | 566±190 | 40.6±1.7 | 2.19±0.09 | 3873 |
| (B) Caffeic acid | ||||
| VvgGT1 | 165±67 | 21.6±2.2 | 1.16±0.12 | 7069 |
| VvgGT2 | 105±30 | 27.5±1.8 | 1.48±0.09 | 14142 |
| VvgGT3 | 142±57 | 19.9±1.9 | 1.07±0.10 | 7567 |
The catalytic efficiency (Kcat/Km) values for the two substrates suggested that gallic acid is a better substrate for VvgGT1 and VvgGT3 than for VvgGT2. Caffeic acid is the best substrate for the three enzymes, but especially for VvgGT2.
The kinetic properties of the three VvgGTs with both caffeic and gallic acid as acceptor substrates values were comparable to the properties of previously characterized glucose ester-forming enzymes from Fragaria ananassa and Vitis labrusca (Lunkenbein et al., 2006; Hall and De Luca, 2007).
Discussion
VvgGTs are glucose-ester forming enzymes
In plant secondary metabolism, glycosylation is a frequent chemical modification involved in intermediate or terminal metabolic steps of natural product biosynthesis (including transport, detoxification processes) and contributes to the diversity of these compounds (Vogt and Jones, 2000). Single or multiple glycosylation of the acceptors can occur at -COOH, -OH, -NH2, -SH, and -CH groups. These reactions, respectively leading to the production of glc-esters and O-, N-, S-, and C-glycosides, have been extensively reviewed (Pflugmacher and Sandermann, 1998; Ikan, 1999; Vogt and Jones, 2000; Jones and Vogt, 2001; Lim et al., 2002; Bowles et al., 2006).
The sequences of the three VvgGTs are most closely related to ester-forming GTs from Arabidopsis (Milkowski et al., 2000a, Sato et al., 2000; Jackson et al., 2001; Lim et al., 2001) and from rapeseed (Milkowski et al., 2000b) as well as FaGT2 from strawberry (Lunkenbein et al., 2006) and the VLRSgt identified by Hall and De Luca (2007) showing a bi-functional activity toward resveratrol to form O-glucosides at elevated pH and hydroxycinnamic acids to form glc-esters in more acidic conditions. Interestingly, the sequences are located within the same branch in the phylogenetic tree, and separately from the GTs involved in the O-glucosylation of anthocyanins (Harborne and Williams, 2000) or the formation of epicatechin 3′-O-glucoside (Pang et al., 2008). Noticeable divergence was also measured with several multifunctional glucosyltransferases catalysing the formation of O-, N- or C-glucosides such as the Arabidopsis UGT72B1 (Bazier-Hicks et al., 2007) and UGT72 E1 involved in N- and O- glucosylation (Lim et al., 2002), UGT72E2 and UGT72E3 characterized as O-sinapoyl glucosyltransferases (Lim et al., 2001) and UGT71B1 involved in formation of O-glucosylation in the 2-OH position of benzoate substrates and UGT74F1 highly specific for O-glucoside formation (Lim et al., 2002).
However, sequence homology is far from being a definitive argument to describe the precise enzymatic activity of the VvgGTs, as illustrated by A. thaliana UGT74F1 and UGT74F2 exhibiting 82% similarity but catalysing, respectively, the formation of glucose–ether and –ether plus –ester with salicylic acid as substrate (Lim et al., 2002). Some glucosyltransferases can be involved in both O-glucoside and glucose-ester formation, consistent with bi-functional role in substrate modification and depending on the reaction condition or not (Hall and De Luca, 2007; Dean and Delanay, 2008). The three VvgGTs are assigned to the same group in the phylogenetic tree as Satsuma mandarina known to produce limonoid O-glycosides (Kita et al., 2000). This knowledge emphasizes the necessity of complementary assays for the characterization of the three GTs enzymes.
Substrate specificity studies with the three VvgGTs showed that they can recognize multiple substrates and that all tested hydroxybenzoic and hydroxycinnamic acids are used as substrates. The three VvgGTs exhibited a strict regio-specificity for the acceptor site as described in Vogt and Jones (2000), being able to glycosylate substrates by attaching the glucose to their carboxyl group.
VvgGT1 and VvgGT3 showed a higher catalytic efficiency for benzoic acids than for hydroxycinnamic acids and, therefore, their substrate specificity is similar to those of glucosyltransferases involved in the formation of glucose esters of hydroxybenzoic acids in Fagaceae, Pinaceae, Brassicaceae, Solanaceae, and Lamiaceae species (Gross, 1983; Klick and Herrmann, 1988). On the other hand, VvgGT2 displayed a slight preference for hydroxycinnamic acids, proposing similarity in function with other glucosyltransferases described in Lee and Raskin (1999), Milkowski et al. (2000a), Lim et al. (2001), Lunkenbein et al. (2006), and Kita et al. (2000). The three VvgGTs showed no glycosyl-transfer activity toward compounds containing no carboxylic group even at elevated pH. These results are in contrast to those presented by Hall and De Luca (2007) concerning both flavonoid and trans-resveratrol glycosylation activity, despite high homology between protein sequences (more than 99% of identity between VvgGT2 and VLRSgt). However, in the earlier study (Hall and DeLuca, 2007), the catalytic properties of the heterologously produced VLRSgt were studied at more elevated non-physiological pH, without detaching the enzyme from its GST tag, and the enzymatic products were not analysed by mass spectrometry.
The optimal pH of the three VvgGTs (pH 5.5–6.5) is consistent with the results obtained for other glucose ester-forming enzymes, showing maximum activity around pH 6 (Lim et al., 2002; Milkowski et al., 2000a, b; Hall and DeLuca, 2007), a bit lower than the optimal pH of enzymes forming O-glucosides with phenolic acids (Lim et al., 2003).
The VvgGTs could be involved in the production of hydroxycinnamic esters and PA galloylation using glc-esters as intermediates
The in vivo role of glc-esters of phenolic acids and therefore of the enzyme catalysing their formation remains to be established. Glucosides of gallic acid were detected in grape pomace but identified as O-glucosides by NMR (Lu and Foo, 1999). Glc-esters of coumaric acid were detected in the ripe berries of Vitis labrusca in trace amounts (Hall and DeLuca, 2007), like, more generally, those of phenolic acids in the pericarp of Vitis vinifera berries (Reschke and Herrmann, 1981; Monagas et al., 2005). Moreover, transgenic hairy-roots where these VvgGTs are induced are not affected for glucosylated final end-products (Terrier et al., 2009). It can therefore be assumed that the enzymes characterized in this work catalyse the formation of glucose esters involved as intermediates in the phenylpropanoid pathway.
Glucose has often been described as a recognizable ‘tag’ for membrane transporters. In Arabidopsis, p-aminobenzoate (pABA) is synthesized in chloroplasts and then transported and accumulated in the vacuole in its glc-ester form (Eudes et al., 2008). The predominant forms of salicylic acid in tobacco leaves are its O-glucosides, stored in the vacuole and it has been shown that the vacuolar uptake of the glucosylated molecule involves a H+-antiport mechanism (Dean et al., 2005).
Transport experiments demonstrated that MATE1 or TT12, MATE-type transporters from Medicago and Arabidopsis, respectively, mediate epicatechin 3′-O-glucoside transport (Zhao and Dixon, 2009), while neither catechin nor epicatechin are transported by these proteins (Marinova et al., 2007; Zhao and Dixon, 2009). Epicatechin 3′-O-glucoside was detected as a transient component of Medicago seed during PA accumulation (Pang et al., 2008) and significant amounts of this molecule were encountered in immature seeds of tt12 mutants, suggesting that the glucosylated form of epicatechin would be an intermediate metabolite, substrate of the MATE transporters (Kitamura et al., 2010).
Despite not yet being localized, it is possible that VvgGTs are cytoplasmic enzymes, as demonstrated for other glucosyltransferases (Werner and Matile, 1985; Jones and Vogt, 2001) even though the glucose conjugate products are stored in the vacuole (Oba et al., 1981; Rataboul et al., 1985; Werner and Matile, 1985; Yazaki et al., 1995; Taguchi et al., 2000). Glc-ester formation in grape cells would appear as a first modification step, allowing the transport of these molecules in intracellular organelles for further metabolic events.
On the other hand, evidence is growing in the literature that glc-esters of phenolic acids can act as acyl donors in trans-esterification reactions. In Brassicaceae, 1-O-sinapoyl-β-glucose serves as an acyl donor for the formation of sinapoylcholine, sinapoylmalate, and disinapoylglucose (Dahlbender and Strack, 1986; Lehfeldt et al., 2000; Shirley and Chapple, 2003). Li and Steffens (2000) have also characterized a 1-O-β-acylglucose-dependent acyltransferase functioning in glucose polyester biosynthesis in Lycopersicon pennelli. More recently, the synthesis of avenacin A-1 in oats, an acylated compound involved in plant defence, has been demonstrated to arise from trans-acylation between des-acyl avenacin A and N-methyl anthraniloyl-O-glucose, a glucose ester (Mugford et al., 2009). Glc-esters would serve as activated intermediates in the biosynthesis of other secondary metabolites, as described for the synthesis of chlorogenic acid and its derivatives in sweet potato (Villegas and Kojima, 1986) or gallotannins in oak (Gross, 1983). However, in both of the last cases, gene identification is still lacking.
In the case of grape, the three VvgGTs are induced by the over-expression of VvMybPA1 and VvMybPA2 recognized as specific regulators of the PA pathway, respectively, in seeds and skins (Bogs et al., 2007; Terrier et al., 2009). Moreover, the three genes are expressed predominantly in the early development stages before veraison, in parallel with PA and hydroxycinnamate ester accumulation (Kennedy et al., 2001; Verriès et al., 2008). Furthermore, whereas VvgGT1 and VvgGT3 expression are important in seeds and to a lesser extent in skins in accordance with an implication in the galloylated PA biosynthesis, VvgGT2 is mainly expressed in pulp, which would fit with an involvement in the hydroxycinnamate pathway. This putative involvement in those pathways is also in accordance with substrate preferences of VvgGT1 and VvgGT3 towards C6C1 acids and VvgGT2 towards C6C3 acids. Therefore it can be hypothesized that glc-esters of gallic acid and hydroxycinnamic acids could be metabolic intermediates necessary for the galloylation of PAs and the synthesis of tartaric esters of hydroxycinnamic acids, respectively. It appears unlikely that those enzymes are involved in the in vivo glucosylation of stilbenes as hypothesized for the orthologue of VvgGT2, VLRSgt (Hall and DeLuca, 2007), since these genes are expressed in the early stages of berry development unlike piceids, whose accumulation in healthy berries is restricted to the ripening stages (Gatto et al., 2008), and that significant activity of resveratrol glucosylation was detected in vitro for VLRSgt only in pH conditions far from physiological ones (Gout et al., 1992; Hall and DeLuca, 2007).
The remaining question is how these precursors give rise to the final biosynthetic products. Each time the protein involved in trans-esterification with glc-ester as the acyl donor was identified, that is avenacin synthesis in oats, glc-polyester in wild tomato, sinapate esters in Brassicaceae, the genes encoding glucose-acyltransferases were shown to belong to the serine carboxypeptidase-like (SCPL) protein family (Mugford et al., 2009; Li and Steffens, 2000; Lehfeldt et al., 2000; Shirley and Chapple, 2003; Milkowski et al., 2004). Future studies for deciphering the PA galloylation steps and the formation of hydroxycinnamic esters will include the study of the SCPL genes already identified in the transcriptomic screening of grapevine hairy roots over-expressing VvMybPA1 and VvMybPA2 (Terrier et al., 2009).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments