





Abstract
Mass spectrometry-based imaging techniques applied to small molecules complement the growing research field of metabolomics and can be used to interpret many important biological processes occurring in plants. In untargeted imaging applications, chemical identification is a critical step since it cannot take advantage of separative techniques applied to neutral molecules (e.g. liquid chromatography). The use of high resolution spectrometers is of great help, but fragmentation experiments are often necessary. In many cases, the information on ion fragmentation is embedded in the data sets, because analytes break up during ionization, but the extraction of this information is not easy considering the complexity of the imaging data files. Here an approach is proposed for applying conventional untargeted MALDI (matrix-assisted laser desorption ionization) profiling and advanced data analysis to perform imaging of metabolites in apple tissues. The pipeline, based on intensity correlation analysis, is used to extract fragmentation information from untargeted, high resolution, wide range mass spectra and to reconstruct compound-specific images which can be used for interpretation purposes. The proposed approach was used to investigate the distribution of glycosylated flavonols and dihydrochalcones in Golden Delicious apples. The results indicate that the method is effective, showing a high potential for ascertaining detailed metabolite localization.
Introduction
Assessment of the spatial and temporal distribution of metabolites in tissues is important for rationalizing many biological processes occurring in plants. Qualitative or semi-quantitative imaging techniques are thus ideal for complementing the expanding field of metabolomics, when studying the tissue-specific distribution of metabolic biomarkers.
Of the various imaging techniques, mass spectrometry (MS)-based technologies would seem to be particularly promising, due to their sensitivity, broad response, and speed. Matrix-assisted laser desorption ionization MS imaging (MALDI-MSI) (Caprioli et al., 1997) in particular has become one of the most important molecular histology methods for understanding the spatial complexity of biological samples, and has been demonstrated to be applicable to different kinds of animal and plant tissues (Vrkoslav et al., 2010).
As far as the imaging of small molecules is concerned, the performance of MALDI is hindered by the presence of ions resulting from matrix ionization at the lower end of the mass spectrum (Krutchinsky and Chait, 2002). There are different approaches to dealing with the matrix background produced by MALDI or related techniques, mainly relying on specific laser-absorbing media (such as graphite or colloidal silver) (Zhang et al., 2007; Cha et al., 2008, 2009; Jun et al., 2010) or on matrix-free approaches (Holscher et al., 2009). However, due to the robustness and widespread use of conventional MALDI protocols, there is definitive interest in developing tools to use them in metabolic imaging applications.
In these MS-based technologies the unique chemical identification of the ions is a major challenge, in the absence of separative techniques applied to neutral molecules (e.g. liquid chromatography). The use of high resolution spectrometers is of great help to reduce the set of possible molecular formulae (Vrkoslav et al., 2010), but more detailed structural information has to be obtained from fragmentation experiments (Zhang et al., 2007; Cha et al., 2008).
In fragmentation experiments, however, primary ions have to be selected according to their mass-to-charge ratio, so acquisition time, isolation width, and progressive matrix depletion represent a limit to the information yield of MS/MS which can be performed on a specific tissue section. Of these factors, analysis time is particularly relevant, considering that a MALDI imaging run composed of a full scan spectrum and a few fragmentation experiments can easily last 20–30 h.
In conclusion, although full scan MALDI would be suitable for untargeted profiling, in practice it is often necessary to make an a priori choice of the molecules of interest, thus reducing the profiling potential of the technique.
Due to the dynamics of the MALDI process, however, in-source fragmentation is ubiquitous and fragmentation information is already embedded in a full scan spectrum. The development of tools optimized for its extraction would then represent a key step for the full exploitation of MALDI profiling. To take advantage of such information it is necessary to develop and validate a framework to compare the ‘localization’ of primary and fragment ions. A visual approach, indeed, is not optimal because its results are likely to depend very much on the experimenter, and manual inspection cannot be applied over an extensive database of biomarkers as should be required in untargeted experiments. All these considerations push towards the development of bioinformatic tools to treat MALDI-MSI data sets, but the topic is particularly challenging, considering the size and the complexity of the data files. To date, bioinformatic algorithms have been developed to perform reduction and image segmentation for biomarker discovery (McCombie et al., 2005) and to measure the correlation of different ion images (McDonnell et al., 2008).
Here an approach is proposed for applying conventional untargeted MALDI profiling and advanced data analysis to perform imaging of metabolites in apple tissues, developing a unique framework taking advantage of the in-source fragmentation information to increase the chemical selectivity of the technique. With the proposed pipeline, co-localization between parent and fragment ions is assessed, validated, and exploited to reconstruct compound-specific images which can be used for interpretation purposes.
In the present experiment, MALDI-MSI data were acquired using a high resolution mass spectrometer. An algorithm based on intensity correlation analysis (ICA) was developed to study the co-localization between parent and fragment ions, thus visualizing the distribution of specific metabolites in tissues.
To perform the experiment, a simple sample preparation protocol was set up to optimize the sectioning of the apple tissues, while minimizing the negative effects of oxidation of the molecules concerned.
The proposed approach was applied to study the distribution of some relevant glycosylated flavonols and dihydrochalcones in apple sections. This latter class of molecules is challenging from the spectrometric point of view. Indeed, polyphenol glycosides easily fragment in nearly all ionization sources, losing primarily their sugar subunits. Furthermore, they show relevant UV absorption in the wavelength region typical of nitrogen lasers and, for this reason, they have been proposed as MALDI matrices (Petkovic et al., 2010), and have been analysed in imaging applications in matrix-free conditions (Holscher et al., 2009). In this class of molecules, then, ionization and fragmentation happen at the same time in the laser desorption process, making them suitable for the application of the proposed bioinformatic approach.
The distribution of this particular class of metabolites in plant tissues can also be visualized by optical/UV microscopy, possibly in combination with histological staining (Hutzler et al., 1998; Abeynayke et al., 2011). Several classes of phenolics such as hydroxycinnamic acids, coumarins, stilbenes, and styryl pyrones, for example, are suitable for direct analysis, being strongly autofluorescent when irradiated with UV or blue light (Hutzler et al., 1998). Other phenolics, such as the FLAVANOLS, can be visualized via light and electron microscopy (Amrani-Joutei et al., 1994; Lees et al., 1995) and can be stained with chromogenic reagents such as 4-dimethylamino-cinnamaldehyde (Feucht et al., 1992) or vanillin-HCl (Liu et al., 2009). Staining reagents lack the specificity required for characterization of single compounds: for example, the above-mentioned staining reagents for FLAVANOLS react with both the catechins and the oligomeric proanthocyanidins. In terms of spatial resolution, however, these methods show a definitive advantage over MS-based approaches. In fact, with optimized sample preparation protocols, it is possible to achieve subcellular resolution (Abeynayke et al., 2011), still beyond the capabilities of the majority of MS-based imaging platforms.
In terms of generality and molecular selectivity, however, MS shows some relevant advantages. Metabolites are identified on the basis of their mass spectra, often allowing the reconstruction of molecule-specific pictures. In addition, these techniques can be applied to the analysis of many different classes of metabolites, permitting a more comprehensive profiling of the tissue sections. Another reason in favour of MS imaging technologies is that these are gradually evolving in the direction of single cell metabolomics (Svatos, 2011). In any case, all these different imaging techniques should be seen as complementary and can be combined to tackle the chemical complexity of the different metabolites.
From a biological point of view, glycosylated flavonols and dihydrochalcones are known to have important and diverse key functions in plant growth, development, and protection, while antioxidant activity is relevant to food and nutrition (Andersen, 2006). The tissue-specific synthesis of glycosylated polyphenols in apples has already been proposed on the basis of high performance liquid chromatography diode array detection (HPLC-DAD) studies and has been suggested by a graphite-assisted laser desorption ionization study (Zhang et al., 2007). The availability of this background knowledge is of fundamental importance for the overall validation of the proposed methodology.
Materials and methods
Reagents
α-Cyano-4-hydroxycinnamic acid (CHCA) and 2,5-dihydrobenzoic acid (DHB) were purchased from Sigma-Aldrich (Steinheim, Germany) and L(+)-ascorbic acid from VWR international (Geldenaaksebaan, Leuven, Belgium), while acetonitrile was of LC-MS Chromasolv grade (Sigma-Aldrich, Steinheim, Germany).
Preparation of apple sections
Apples (Golden Delicious) were purchased locally. Metabolite localization was tested in three types of sections; their position within the fruit is presented in Fig. 1, with a schematic view of the anatomy of the fruit (Tukey and Young, 1942; Trentham, 2008). The central part of a longitudinal section of the fruit was used to investigate the pericarp (20 mm×20 mm), while two other different types of cross-sections were selected to study the transition region between the skin and the hypanthium (∼6.6×6.6 mm). In relation to the skin, sectioning was performed orthogonally and obliquely (Fig. 1). In this second type of sectioning, cell layers just below the skin were cut with a favourable projection, enhancing the histological resolution of the MSI images.
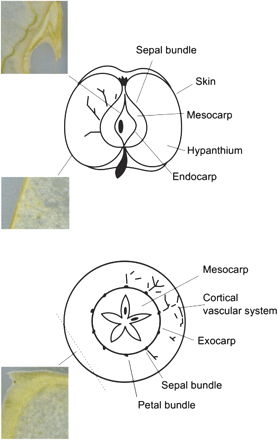
Graphic representation of the different sections considered in the study. The schematic view of the apple anatomy is reconstructed from Trentham (2008) and Tukey and Young (1942). The pericarp is the fruit wall developed from the ovary; it can be subdivided into endocarp, mesocarp, and exocarp. (This figure is available in colour at JXB online.)
Apple sections for MALDI-MSI were obtained by manual cutting of slices (∼0.5 mm thick) using a razor blade. The sections were mounted on a glass slide and immediately sprayed with ascorbic acid (10 mmol l−1) to prevent oxidation during handling and dehydration. After vacuum drying (12 h), the sections were regularized using a razor blade. Regularization was necessary due to shrinkage differences within the tissue during drying.
Vacuum drying was necessary considering the high water content of the sections; during this stage volatile molecules are likely to be removed from the section, but the same would happen inside the low vacuum MALDI interface. However, the majority of metabolites are not expected to be affected by the same phenomenon due to their low vapour pressure. This observation is also confirmed by the consistency of MALD-MSI results of experiments performed over a wide range of drying conditions and MALDI acquisition times.
Optical images of apple sections were obtained using a photo scanner (HP Scanjet G4050) at 600 dpi. High resolution scans were acquired using the optical scanner embedded in the MS instrument via the xy stage, holding the MALDI-MSI sample and the camera to examine the sample optically.
MALDI imaging
MALDI spectra were acquired by using CHCA as the UV-absorbing matrix. Standard MALDI conditions were preferred over matrix-free solutions for two reasons: (i) to avoid an almost complete fragmentation of the UV-absorbing metabolites; and (ii) to increase the profiling efficiency of the acquisition, by increasing the efficency towards non-UV-absorbing metabolites. According to the ImagePrep protocols, both CHCA and DHB were tested as possible MALDI matrices. The former was selected since its regular crystallization produces higher quality images. Matrix solution (7 g l−1 CHCA in 50% acetonitrile) was sprayed uniformly over the sections using an ImagePrep station (Bruker Daltonics, Germany) adopting the following program: 22 spray cycles, 75% sprayer power, 0% modulation, 1 s spray, 2 s incubation, and 70 s dry time. The matrix deposition program was optimized to minimize the wetting of the samples, because this would result in metabolite delocalization and possibly in the reactivation of oxidative degradation. The ImagePrep station performs the entire spraying process under nitrogen flux, which prevents oxidation of the sections during matrix deposition.
MALDI-MSI analysis was performed using a MALDI LTQ Orbitrap XL mass spectrometer [Thermo Fisher Scientific (Bremen), GmbH, Germany] with a resolution of 60 000 [at m/z 400, full width half maximum (FWHM)]. Positive and negative ion modes were evaluated. The latter was preferred because, in negative mode, the ions resulting from the ionization of the matrix were fewer in quantity and less intense.
Fourier transform MS (FTMS) full scans from m/z 120 to m/z 1000 were acquired using the Instrument Control Software (LTQ Tune Plus 2.5.5 SP1) and Xcalibur 2.1. The same software was used to raster the laser beam automatically across the sectioned surface in x and y dimensions. The Automatic Gain Control mode of the spectrometer was used to optimize the number of laser shots for every pixel. Typical values varied from 10 to 80 laser shots depending on the ion yield. In the case of the skin sections, each spectrum was obtained by averaging two full scan spectra, while only one spectrum was acquired for the pericarp section presented herein. Due to this setting, the acquisition time is different for every pixel, while the scan time for the spectrometer is determined by the resolution at ∼0.7 s.
The laser step size between adjacent x/y positions was set at 150, 100, and 75 μm. The pericarp section presented herein was analysed with a step of 150 μm, leading to an image of 125×136 pixels (17 000 steps). In the case of the skin region, the two sections were acquired with a step of 100 μm and 75 μm, resulting in images with 90×69 (6210 steps) and 67×68 (4556) pixels. Total acquisition times for the images were 9 h for the pericarp section, and 6 h and 7 h for the skin sections, respectively.
The Orbitrap was calibrated prior to mass analysis by external calibration using standard peptide mixtures (ProteoMass MALDI calibration kit for LTQ XL and LTQ hybrids, Sigma-Aldrich) for the normal mass range. To increase the accuracy of the acquisition, the spectra were internally calibrated at m/z 333.088, the matrix dimer having undergone a loss of CO2 ([2CHCA-CO2-H]−] ).
Data analysis
Data files were converted into open-source CDF format using Xcalibur Software. The analysis and visualization pipeline was developed in Python version 2.5 (http://www.python.org/). To correct any uneven responses in the sample, the total ion current of each pixel was normalized to one. Extracted ion chromatograms (XICs) were extracted from the full scan data with different levels of tolerance depending on the mass range. Mass-selected images were reconstructed from these traces. Plots were produced by using the interactive Ipython shell (http://ipython.scipy.org) with Scipy (http://www.scipy.org/) and Matplot (http://matplotlib.sourceforge.net/) libraries.
ICA—originally developed to assist the analysis and interpretation of immunohistochemical images (Li et al., 2004)—was selected as a strategy to compare the co-distribution of ions in the image. The starting point of ICA is comparison of the intensities of two ions within the same pixel. For each pixel i in the image, the intensity correlation factor (ICi) between two ions at m/z a and b is calculated according to the following relationship:
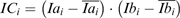
Here an ion of m/z ratio a has an intensity Iai at position pixel i and an ion of m/z ratio b has an intensity Ibi at pixel i, while and are the averages of the intensities of ions a and b over the whole rastered area.
The rationale behind this approach is that ICi is positive for a given pixel i, if both intensities are on the same side of their respective mean values on the image. If the ions at m/z a and m/z b are correlated—as in the case of a fragmentation occurring during ionization—their intensities should vary ‘in tune’ in almost every pixel, resulting in a set of positive intensity correlation (IC) factors.
The IC framework was adapted to the specific characteristics of MS images acquired using an FTMS instrument. First of all, the intensity range of ion signals spanned several orders of magnitude (up to 4 or 5) so logarithmic scaling of the normalized intensities was performed. This type of scaling also had the positive effect of making the distribution of the intensities less skewed. Co-localization is only meaningful when both species are present, so ICA was only performed on pixels where the intensities of m/z a and m/z b were different from zero.
A strategy to validate the results of ICA was also developed to evaluate the probability of obtaining only by chance a given set of IC factors. If one considers how IC factors are calculated, their distribution for two uncorrelated species should be symmetric around zero. Any asymmetry in the distribution towards positive values is therefore an indication of IC between the two ion traces, which indicates the presence of significant co-localization. To validate the asymmetry in ICs it is then possible to consider the distance between the medians of the distributions of positive and negative IC values [(med+)–(med–)]. The significance of this parameter was tested by checking its variability in a series of random permutations of pixel intensities for either m/z a or m/z b. In this way, it was possible to probe the intrinsic variability of the distribution of IC factors, calculating the likelihood of obtaining the actual asymmetry by chance. This validation strategy also had the advantage of being non-parametric, not requiring any a priori hypotheses regarding the distribution of IC factors.
If ICA is performed on two ion traces which can be the result of an in-source fragmentation process—such as glycoside and its corresponding aglycone—the presence of significant IC confirms the chemical identification of the parent ion. On the other hand, the presence of correlation between ions relative to two different metabolites suggests that the two species could be biologically related, since, on average, they show the same trend in relation to their means.
The results of the ICA were used to reconstruct maps showing the spatial distribution of highly correlated pixels. These images were then used for interpretation purposes.
Results and Discussion
In this section, the results of applying the proposed framework to the MALDI-MSI of Golden Delicious apple are presented. In the first part, the effectiveness of the sample preparation protocol and high resolution MS analysis is assessed by studying the distribution of some metabolites known to be present in apple. Subsequently, ICA will be applied to study the distribution of flavonoid glycosides in apples, focusing on the pericarp and on the skin.
Imaging of test metabolites
The reconstructed images for malic acid (C4H6O5), detected as C4H5O5 [(M-H)− ions at m/z=133.014], are shown in Fig. 2B.
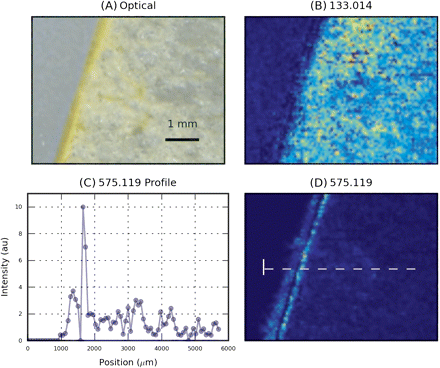
Spatial distribution of some metabolites known to be present in apples. MS images have been acquired with a raster step of 100 μm. (A) Optical image. (B) Image reconstructed from XIC at m/z 133.014, identified as malic acid. (C) Intensity profile of m/z 575.119 along the dotted line. (D) Image reconstructed from XIC at m/z 575.119, identified as a procyanidin-related ion. (This figure is available in colour at JXB online.)
Malic acid was detected inside apple hypanthium with an almost constant concentration. Malic acid, as its name suggests, accounts for 90% of total organic acids in apple (Ackermann et al., 1992). Figure 2D shows the same graphic representation for an ion detected at m/z=575.119. Among the possible molecular formulae for this ion, C30H23O12 can be related to procyanidin B [C30H26O12 detected as (M-H2-H)−]. The observed ion is compatible either with the direct ionization of procyanidin B or with the fragmentation of higher order procyanidins. Procyanidins—polymeric flavonoids made up of catechin/epicatechin units—are known to be abundant in apples, where they can account for a significant fraction of overall phenolic content (Vrhovsek et al., 2004). The ion image presented in Fig. 2D is reconstructed from a trace extracted with a 10 ppm window, corresponding to 0.005 mDa.
Figure 2C displays the intensity profile of the signal along a line crossing the skin. The images indicate that the metabolite was more concentrated in the cell layers just below the cuticle and that its concentration decreased inside the fruit. This trend agrees with the relatively high content of procyanidin B2 in Golden Delicious hypanthium reported in a previous HPLC-DAD study (Tsao et al., 2003) where a 1–2 mm thick ‘peel’ fraction was analysed. The intensity of the 575.119 ion decreased by a factor of ∼5 moving from the external hypanthium cells towards the inner hypanthium (Fig. 2C). This particular localization could explain the much lower content of procyanidin B2 in ‘pulp’ reported by Chinnici et al. (2004), since in this experiment a 5 mm thick ‘peel’ fraction was sampled, which also included the procyanidin-rich cells present below the skin. This example demonstrates the capability of MSI to provide semi-quantitative information, complementary to that obtained from the analysis of extracts or homogenates.
Imaging of selected flavonols and dihydrochalcones
The polyphenol glycosides detected in different apple sections are listed in Table 1. The table also displays the molecular formulae and their main ions observed in the spectra. In all tested sections, diglycosides were not detected or were close to the detection limit, so they were not taken into account.
Polyphenol glycosides considered, with their respective aglycones 6S stands for six-carbon sugars, excluding rhamnose. Likewise, 5S stands for all the five-carbon sugars.
| Name | Molecular formula | Ion observed | Theoretical m/z |
| Quercetin | C15H10O7 | [M-2H]− | 300.0276 |
| Kaempferol | C15H10O6 | [M-H]− | 285.0405 |
| Phloretin | C15H14O5 | [M-H]− | 273.0768 |
| Quercetin-hexoside [querc-6S] | C21H20O12 | [M-H]− | 463.0882 |
| Quercetin-rhamnoside [querc-rham] | C21H20O11 | [M-H]− | 447.0933 |
| Kaempferol-hexoside [kaemp-6S] | C21H20O11 | [M-H]− | 447.0933 |
| Phloretin-hexoside [phlor-6S] | C21H24O10 | [M-H]− | 435.1297 |
| Quercetin-pentoside [querc-5S] | C20H18O11 | [M-H]− | 433.0776 |
| Kaempferol-pentoside [kaemp-5S] | C20H18O10 | [M-H]− | 417.0827 |
| Name | Molecular formula | Ion observed | Theoretical m/z |
| Quercetin | C15H10O7 | [M-2H]− | 300.0276 |
| Kaempferol | C15H10O6 | [M-H]− | 285.0405 |
| Phloretin | C15H14O5 | [M-H]− | 273.0768 |
| Quercetin-hexoside [querc-6S] | C21H20O12 | [M-H]− | 463.0882 |
| Quercetin-rhamnoside [querc-rham] | C21H20O11 | [M-H]− | 447.0933 |
| Kaempferol-hexoside [kaemp-6S] | C21H20O11 | [M-H]− | 447.0933 |
| Phloretin-hexoside [phlor-6S] | C21H24O10 | [M-H]− | 435.1297 |
| Quercetin-pentoside [querc-5S] | C20H18O11 | [M-H]− | 433.0776 |
| Kaempferol-pentoside [kaemp-5S] | C20H18O10 | [M-H]− | 417.0827 |
Polyphenol glycosides considered, with their respective aglycones 6S stands for six-carbon sugars, excluding rhamnose. Likewise, 5S stands for all the five-carbon sugars.
| Name | Molecular formula | Ion observed | Theoretical m/z |
| Quercetin | C15H10O7 | [M-2H]− | 300.0276 |
| Kaempferol | C15H10O6 | [M-H]− | 285.0405 |
| Phloretin | C15H14O5 | [M-H]− | 273.0768 |
| Quercetin-hexoside [querc-6S] | C21H20O12 | [M-H]− | 463.0882 |
| Quercetin-rhamnoside [querc-rham] | C21H20O11 | [M-H]− | 447.0933 |
| Kaempferol-hexoside [kaemp-6S] | C21H20O11 | [M-H]− | 447.0933 |
| Phloretin-hexoside [phlor-6S] | C21H24O10 | [M-H]− | 435.1297 |
| Quercetin-pentoside [querc-5S] | C20H18O11 | [M-H]− | 433.0776 |
| Kaempferol-pentoside [kaemp-5S] | C20H18O10 | [M-H]− | 417.0827 |
| Name | Molecular formula | Ion observed | Theoretical m/z |
| Quercetin | C15H10O7 | [M-2H]− | 300.0276 |
| Kaempferol | C15H10O6 | [M-H]− | 285.0405 |
| Phloretin | C15H14O5 | [M-H]− | 273.0768 |
| Quercetin-hexoside [querc-6S] | C21H20O12 | [M-H]− | 463.0882 |
| Quercetin-rhamnoside [querc-rham] | C21H20O11 | [M-H]− | 447.0933 |
| Kaempferol-hexoside [kaemp-6S] | C21H20O11 | [M-H]− | 447.0933 |
| Phloretin-hexoside [phlor-6S] | C21H24O10 | [M-H]− | 435.1297 |
| Quercetin-pentoside [querc-5S] | C20H18O11 | [M-H]− | 433.0776 |
| Kaempferol-pentoside [kaemp-5S] | C20H18O10 | [M-H]− | 417.0827 |
Most of the molecules concerned were detected as [M-H]− ions, while quercetin was mainly detected as the [M-2H]−· radical anion. The production of such an anion in the ionization was checked in the MALDI analysis of a standard. The formation of radical anions in the fragmentation of flavonoid glycosides has already been reported (Hvattum and Ekeberg, 2003). Even though the two experiments cannot be directly compared due to the different ionization interfaces, the electrospray ionization (ESI) experiments suggest that the collision-induced fragmentation of the flavonoid glycoside parent ions can produce radical anions. These phenomena can also occur during the extraction of the ions within the MALDI source.
Table 1 shows the expected m/z value for the metabolites of interest. These values have been used to extract the XICs necessary for ICA (10 ppm tolerance). The experimentally measured m/z values for the metabolites identified in each tissue section are presented in Supplementary Table S1 available at JXB online. The values obtained in the different sections are slightly different, as can be expected for acquisitions running over several days; however, measured values are always very close to the theoretical values.
As discussed in the Introduction, the presence of the mass in Table 1 in the spectra is not sufficient to assess the distribution of the corresponding metabolite in the tissue. To increase the chemical selectivity of the analysis it is necessary to run a co-localization analysis between the ions in Table 1 and a characteristic fragment.
It has already been pointed out that this class of molecules undergoes fragmentation during ionization so aglycones show generally high intensity profiles, and were therefore selected as characteristic fragments for the running of the ICA. The aglycones of dihydrochalcones (i.e. phloretin) are absent in apple tissues, and only traces of those of flavonols are present (Vrhovsek et al., 2004), thus supporting this specific choice. In Table 1 the abbreviation 6S refers to the six-carbon sugars, excluding rhamnose, which is not isobaric as it is a deoxyhexose, while 5S indicates all the pentoses attached to the specific polyphenols. Isomeric forms are indeed difficult to distinguish by MS. Full scan information alone cannot help, but MS/MS or higher order MS studies possibly could provide structural and linkage information helping to differentiate isoforms. The only phloretin hexoside in apples is the phloretin-2-glucoside (phloridzin), while the main known quercetin-hexosides in Golden Delicious apples are (in decreasing order) quercetin 3-galactoside and quercetin 3-glucoside. The quercetin-pentosides are quercetin 3-arabinoside and quercetin 3-xyloside (Vrhovsek et al., 2004). Kaempferol is not usually reported in apples. It was recently reported as 3-glucoside (Feliciano et al., 2010) or as aglycone.
Apple pericarp
The distribution of masses corresponding to flavonoid hexosides in the apple pericarp is presented in Fig. 3.
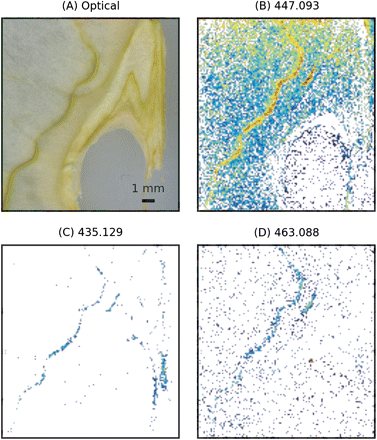
Spatial distribution of flavonoid glycosides (Table 1) in apple pericarp. The intensities were calculated as the logarithm of the normalized ion trace. MS images have been acquired with a raster step of 150 μm. (A) Optical image. (B) Image reconstructed from XIC at m/z 447.093. This mass can be assigned either to quercetin-rhamnoside or to kaempferol-6S. (C) Image reconstructed from XIC at 435.129 (phloretin-6S). (D) Image reconstructed from XIC at 463.088 (quercetin-6S). (This figure is available in colour at JXB online.)
The experimental results indicated that the ion at m/z 447.093 was distributed over the tissue with a maximal intensity on the sepal bundle. The ion at m/z 435.129 was detected at both locations, the sepal bundle and the rims of the endocarp. The yield of the ion at m/z 463.088 was low, showing a slight increase towards the sepal bundle. Table 1 indicates that the ion at m/z 447.093 can be related to the presence of both quercetin-rhamnoside and kaempferol-6S, while the ions at m/z 435.129 and m/z 463.088 can be assigned to phloretin-6S and quercetin-6S, respectively. To discriminate between the two contributions in the ion trace of m/z 447.093 and to confirm the chemical origin of the ions, ICA of these ions and the relative aglycones was performed. The results of this analysis for m/z ratios 447.093, 300.028, and 285.040 are summarized in Fig. 4. The results of ICA are presented in the two plots on the left (Fig. 4A, B): each point in the plot represents a pixel of the image in which two masses were related, m/z 447.093 and m/z 300.028 in the case of Fig. 4A. IC factors are displayed on the horizontal coordinate, while the mean centred value of the ion intensity is used as vertical coordinate (Li et al., 2004). In this representation, pixels showing positive intensity correlation show up in the right-hand plane, with the high intensity pixels occupying the top right quadrant. A significant skew in the distribution of the IC factors towards the right therefore indicates that the two m/z traces are positively intensity correlated, and this allows the conclusion that the two ions are significantly co-localized. This is clearly visible on the first scatter plot (Fig. 4A), in which the high correlation tail is clearly directed towards the top right. Validation was performed with the permutation test discussed in the previous section and the results are summarized in Fig. 4C. The two histograms show the variability of the (med+)–( med–) parameter over 1000 permutations, while the vertical lines indicate the values obtained from the measured values. For both ICAs, validation indicated that the null hypothesis should be discarded at a 0.001 level of confidence, indicating significant co-localization of the two ion traces.
![Graphic representation of the ICA for m/z 447.093/300.028 and m/z 447.093/285.040. (A and B) Position of each pixel in the IC/intensity plane. The intensity of mass 447.093 was mean centred. (C) Validation of the ICA between m/z 447.093/300.028 (grey) and m/z 447.093/285.040 (black). The histograms show the variability of the [(med+)-( med–)] parameter with 1000 permutations of the intensities. Vertical lines indicate the measured values.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jxb/63/3/10.1093_jxb_err327/3/m_jexboterr327f04_ht.gif?Expires=1750271618&Signature=AiQ3x~lwOhtA-A4Ai92PgpRHhiiR9fEDz1aqbc0SuCfOQ0lGTjApKDqtsksJWBCmwJ84q6lnIih-ZEa9oRw2yeSybEnbozzYcNzfwfiCRWIn-cYGfAA5eCPF6d22gTs3f6y~7dpLbQl6rSkXnupP9-ZPRVa4B1lm81Ad1TaoXFXuAmS43TcMgb6M6GmoeMseZA9cWNCmD3QCrkoCjtkfwOxsWoLYbgIqOpgTB1XUTnWbHvzaVZYe4uerUJVE9DDN8tDysh1g1kyCHt2lIRRui8npyjzdCuVvGRdfH-Ja5VQmlHYSW14RP5X-WGFNN5Th67IYbogjcwzpzYfA63U62w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Graphic representation of the ICA for m/z 447.093/300.028 and m/z 447.093/285.040. (A and B) Position of each pixel in the IC/intensity plane. The intensity of mass 447.093 was mean centred. (C) Validation of the ICA between m/z 447.093/300.028 (grey) and m/z 447.093/285.040 (black). The histograms show the variability of the [(med+)-( med–)] parameter with 1000 permutations of the intensities. Vertical lines indicate the measured values.
For interpretative purposes, it is helpful to reconstruct the spatial distribution of the pixels showing positive IC. This image is also included in Fig. 5C and D, where positive IC pixels are displayed with colours ranging from grey to red depending on the IC value. The image shows that, for pixels where m/z 447.093 was present, a part—mainly distributed in the central part of the endocarp—could not be co-localized either to m/z 300.028 or to m/z 285.040, thus suggesting that, in this region, the signal at m/z 447.093 was likely to come from a neutral molecule not listed in Table 1. Of the other pixels, maximal co-localization—in correspondence with the pixels with the highest IC factors—was found almost exclusively along the sepal bundle. This was mainly true for m/z 300.028, while correlation with the ion at m/z 285.040 was less strong. Due to the choice of ions—glycoside and aglycones here—the presence of high intensity correlation confirmed the chemical assignment of the ion at m/z 447.093, while the spatial distribution of the high IC pixels showed the highly localized presence of both metabolites in the tissue.
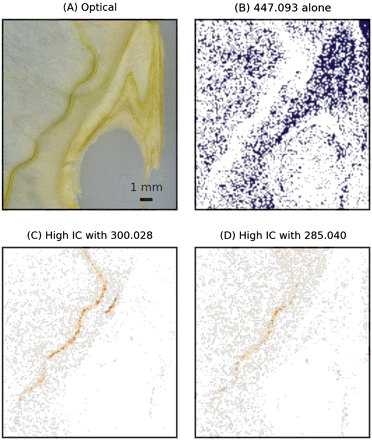
Results of the ICA on the ion at m/z 447.093. (A) Optical image. MS images have been acquired with a raster step of 150 μm. (B) Mask showing the distribution of the ion at m/z 447.093 not correlated with m/z 300.028 or m/z 285.040. (C and D) Images reconstructed from the pixels showing positive IC factors between m/z 447.093/300.028 (C) and m/z 447.093/300.028 (D). (This figure is available in colour at JXB online.)
The same pipeline was applied to analyse the couple of masses at m/z 435.129 and 273.076, which correspond to phloretin-6S and phloretin, respectively. The results are presented in Supplementary Fig. S1 available at JXB online. Here, the distribution of intensity correlation factors peaks noticeably towards the right side of the plot, indicating that the intensities of these two masses were correlated almost everywhere, and thus indicating an almost perfect co-localization. This result would be expected if the smaller m/z ion is produced in the fragmentation of the larger one, so it is substantially confirming that the trace of the ion at m/z 435.129 shows the distribution of phloretin-6S.
The results of the correlation analysis for the masses corresponding to pentosides (m/z 417.082 with m/z 285.040 and m/z 433.077 with m/z 300.028) are also included in Supplementary Fig. S1. Distribution of IC factors for the couple m/z 417.082/285.040 shows a significant asymmetry in the permutation test, but lacks a clear tail with higher IC. Furthermore, the higher IC pixels do not show a definite spatial pattern. Thus the assignment of m/z 417.082 to kaempferol-5S is not conclusive on this basis. On the other hand, quercetin-5S (m/z 433.077/300.028 couple) shows a markedly asymmetric IC distribution, with higher IC factors (meaning higher correlation) being observed towards the sepal bundle.
On the basis of the chemical assignment obtained from ICA, the spatial distribution of flavonoid glycosides in the tissue section was assessed. Quercetin-6S (m/z 463.088 co-localized with m/z 300.028) was not abundant in the apple pericarp, only showing up with a low intensity on the sepal bundle. Quercetin-rhamnoside (m/z 447.093 co-localized with m/z 300.028) was instead present, mainly within this bundle. Kaempferol-6S (m/z 447.093 co-localized with m/z 285.040) was probably also present in the same position, albeit at a low concentration. In the pericarp, phloretin-6S (m/z 435.129 co-localized with m/z 273.076) was mainly present in the sepal bundle, but was also detectable on the papery rim of the endocarp near the seed locules. Finally, quercetin-5S (m/z 433.077 co-localized with m/z 300.028) was present mainly on the sepal bundle.
Skin region
The skin is the outher part of the apple and it is composed of cuticle, epidermis, and hypodermal layer (Tukey and Young, 1942; Trentham, 2008). The skin is known to be rich in secondary metabolites. In particular, flavonoid glycoside accumulation in this area guarantees the protection of the fruit from UV radiation (Andersen, 2006). In keeping with the analysis pipeline applied for the core data, MS images for the ions at m/z 447.093 and m/z 463.088 are shown in Fig. 6, with the distribution of pixels with high IC between m/z 463.088 and m/z 300.028. The results indicate that m/z 447.093 was present in low concentration in the tissue sections of the skin region. In contrast, the signal at m/z 463.088 was intense, showing a decrease towards the hypanthium. Intensity correlation with m/z 300.028 was higher in the region just below the cuticle, with an increase in correspondence with one of the bundles of the cortical vascular system, visible as a high intensity spot within the apple flesh.

MS imaging of the oblique section of the skin region. MS images have been acquired with a raster step of 75 μm. (A) Optical image. (B and C) Images reconstructed from XIC at m/z 447.093 (assigned to quercetin-rhamnoside) and m/z 463.088 (assigned to quercetin-6S). Logarithmic scaling. (D) Images reconstructed from the pixels showing positive intensity correlation between m/z 463.088 and m/z 300.028. (This figure is available in colour at JXB online.)
In this area of the apple, the ion at mass m/z 435.129 was not detected, while the intensity of the ion at m/z 273.076 was low, showing a decreasing profile towards the hypanthium (see Supplementary Fig. S2 at JXB online). As far as the m/z 433.077/300.028 couple is concerned, ICA identified the co-localization region right below the skin. As before, the situation for the pair at m/z 417.082/285.040 was not clear cut, and ICA resulted in a relatively symmetric distribution. Pixels showing higher correlation were located in the skin region but, interestingly, they were not found where the intensities of the ions at m/z 417.082 and m/z 285.040 were higher. In this specific case, the results of ICA were not conclusive for assignment of the ion at m/z 417.082 to kaempferol-5S, in agreement with the fact that its presence in apple has never been reported to date.
The same analysis pipeline was applied to the orthogonal section: results for masses at m/z 447.093 and m/z 463.088 are presented in Supplementary Fig. S3 at JXB online. In this case, MS images show a picture consistent with the one just discussed. The same consideration applies to the ions at m/z 273.076, 417.082, and 433.077 observed by MALDI-MSI of the orthogonal section.
The experiments performed to characterize the apple skin region indicate that quercetin-6S (m/z 463.088 co-localized with m/z 300.028) was strongly concentrated in the region just below the cuticle. With a rastering step of 75 μm, the width of the high concentration region can be estimated to be ∼150 μm. This evidence is supported by previous HPLC analysis which indicated quercetin-galactoside as the main glucoside present in the skin (Chinnici et al., 2004). In contrast to the pericarp, quercetin-rhamnoside was less abundant. In none of the skin sections was a clear signature of phloretin-glycosides detected, though a weak phloretin signal was detectable. Considering that the signals of aglycone fragment ions are more intense than the corresponding glycosides, this observation did not rule out the presence of a low concentration of phloretin-glycosides below the cuticle. This hypothesis is supported by the observation that the phloretin signal decreased in the hypanthium, in accordance with what was observed for phloridzin in HPLC-DAD measurements (Chinnici et al., 2004).
As far as pentosides were concerned, the results of ICA made it possible to conclude that quercetin-5S was present in higher concentrations just below the cuticle, dropping rapidly in the hypanthium. Interestingly, flavonoid glycosides also showed a higher concentration in correspondence with the cortical vascular system.
Conclusions
In a biological context, the present study supports the idea of tissue-specific biosynthesis of flavonoid glycosides in apple, in agreement with what has already been proved in Arabidopsis at the genetic expression level (Pourcel et al., 2005). Major differences were found in the distributions of quercetin-6S—present mainly below the cuticle—and quercetin-rhamnoside and phloretin-6S, detected in the pericarp.
In the skin region, glycosides were more concentrated in the cell layers lying just below the cuticle—the thickness of the polyphenol-rich layer was ∼150 μm—while in the pericarp they were located mainly in correspondence with the vascular system.
As a general rule, the distribution of glycosides was very much localized. This makes the study of their spatial tissue-specific biosynthesis a challenge using more conventional analytical methods. In particular, great care must be taken in studies involving the analysis of peel and pulp after mechanical separation, in the light of the critical concentration of some important metabolites in tissues close to the cuticle. This specific observation also has important practical implications, in view of the industrial production of foods with an increased content of beneficial compounds.
The experimental results discussed so far indicate that the proposed pipeline based on the application of ICA to study co-localization in high resolution MALDI spectra is effective for studying the distribution of polyphenol glycosides in apples. Substantial agreement with previous HPLC studies validated this approach, which showed a higher potential for ascertaining detailed metabolite localization. ICA proved to be of great help in assessing co-localization and extracting and interpreting fragmentation profiles from untargeted MALDI profiles, by increasing the selectivity of high resolution spectra.
The authors thank Dr Ron Wehrens for helpful discussions and for critical reading of the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments