





Abstract
Despite being the number one fruit crop in the world, very little is known about the phylogeny and molecular biology of banana (Musa spp.). Six banana rbcS gene families encoding the small subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase from six different Musa spp. are presented. For a comprehensive phylogenetic study using Musa rbcS genes, a total of 57 distinct rbcS sequences was isolated from six accessions that contained different combinations of the A and B ancestral/parental genomes. As a result, five of the six members of the rbcS gene family could be affiliated with the A and/or B Musa genomes and at least three of the six gene families most likely existed before Musa A and B genomes separated. By combining sequence data with quantitative real-time PCR it was determined that the different Musa rbcS gene family members are also often multiply represented in each genome, with the highest copy numbers in the B genome. Expression of some of the rbcS genes varied in intensity and in different tissues indicating differences in regulation. To analyse and compare regulatory sequences of Musa rbcS genes, promoter and terminator regions were cloned for three Musa rbcS genes. Transient transformation assays using promoter–reporter–terminator constructs in maize, wheat, and sugarcane demonstrated that the rbcS-Ma1, rbcS-Ma3, and rbcS-Ma5 promoters could be useful for transgene expression in heterologous expression systems. Furthermore, the rbcS-Ma1 terminator resulted in a 2-fold increase of transgene expression when directly compared with the widely used Nos terminator.
Introduction
Banana (Musa spp.) is the world's number one fruit crop in terms of production and provides (after rice, wheat, and maize) also the most important staple food. Conventional breeding is difficult as commercial cultivars (e.g. ‘Grand Nain’ and ‘Williams’) are highly sterile. New cultivars are continually needed to maintain genetic diversity as well as to improve disease resistance and yield of this monocrop. Although the Musa genome project was initiated in 2001 and continues to date (Gewolb, 2001), little sequence information is publicly available with currently around 12 000 submissions in GenBank (February 2007). Despite the economic importance of banana, previously to this study, only one Musa acuminata cDNA sequence was available in GenBank (AF008214) encoding for ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCo), the most abundant protein on earth.
The chloroplast-encoded large subunit of RuBisCo RbcL has been widely used for phylogenetic analyses in plants as it is highly conserved and offers useful comparisons between distant lineages (Savolainen and Chase, 2003; Gontcharov et al., 2004; Kim et al., 2004). However, the small subunit RbcS is nuclear-encoded and less conserved, and therefore appears more suitable for phylogenetic analyses between closely related species, such as the different Musa spp. RuBisCo catalyses the primary step in carbon fixation and, structurally, is a hetero-16-mer, composed of eight small subunits and eight large subunits (Coen et al., 1977). The small subunit (SSU) is encoded by a gene family (rbcS), ranging from as many as 22 or more copies in wheat (Sasanuma, 2001) to as few as two in the green alga Chlamydomonas reinhardtii. These genes continue to serve as a model for nuclear gene families that frequently exist in plant genomes. The rbcS gene family holds great potential to resolve plant phylogeny within the classifications of family and order. The conserved amino acid sequences of rbcS proteins are useful for comparisons of all species within an order. Silent mutations in the coding sequence or in the non-coding DNA sequence (including introns and untranslated regions) offer an additional approach for resolving family phylogeny. The neutral rate of mutations in the non-coding regions provides good resolution for distinguishing between closely related species, whilst the exons provide robust alignments in more distantly related species. Using this combined phylogenetic approach potential common ancestral genes can be identified.
All RuBisCo promoters, like most other genes, contain the core promoter region, which contains the TATA box and the initiator. Several positive and negative cis-acting elements were identified within the 5’ flanking sequences of the rbcS genes (Kuhlemeier et al., 1987) and the expression of rbcS genes is regulated by light in a developmental- and tissue-specific manner (Timko et al., 1985; Terzaghi et al., 1995). Sequence elements found repeatedly in light-regulated gene promoters include the G-box, the GT-1 site, I-boxes, related motifs, and A/T-rich elements. Many other elements were described for rbcS regulatory sequences, but are less well characterized (Miles et al., 1993; Conley et al., 1994; Terzaghi et al., 1995). RbcS expression is restricted to the photosynthetic tissue of plants and is usually highest in leaves, lower in stems and petals, and not or barely detectable in roots (Coruzzi et al., 1984). Members of the same rbcS gene family were shown to be expressed differentially both with respect to the level of expression and developmental and/or organ specificity (Sugita and Gruissem, 1987). Sequence elements involved in this typical rbcS expression pattern are located in the promoter region while no distinct regulatory elements are known within the rbcS coding sequence. Introns, however, seem to affect transcription in some cases. In petunia, for example, removal of intron sequences from SSU301, the highest expressed petunia rbcS gene, decreased the steady-state mRNA levels 5-fold in transgenic tobacco (Dean et al., 1989). Addition of heterologous intron sequences to gene fusions was shown to increase their expression after electroporation in cultured maize protoplasts (Callis et al., 1987). It was suggested that the presence of introns affects post-transcriptional processes in maize cells. Differential stability of the nuclear transcripts with and without introns may cause the differences in mRNA levels (Leys et al., 1984).
The RbcS transit peptide, as most other transit peptides, is characterized by a high proportion of basic amino acids; this is thought to be important in the interaction of the transit peptide with the chloroplast envelope (Dean et al., 1989). Comparison of the transit peptide of RbcS precursors from various plant species reveals variation in both amino acid sequence and length of the transit peptide. Most rbcS genes in petunia, pea, tomato, tobacco, potato, wheat, maize, and Lemna have transit peptide sequences of 46–48 amino acids, while soybean genes have transit peptides of 55 amino acids (Dean et al., 1989). Despite the variation in amino acid sequences, there is a relatively strong conservation in the position of prolines and charged amino acid residues (Broglie et al., 1983; Coruzzi et al., 1984). This conservation may be of functional significance as RbcS precursors from one species can be imported and processed correctly by chloroplasts of another (Ostrem et al., 1989). By contrast to this evidence, it has been shown in Arabidopsis that peptide length was the most significant factor in determining the transport efficiency, and amino acid substitutions had very little effect on the overall efficiency (Lee et al., 2002). Translocation of foreign genes into chloroplasts can easily be achieved by fusion of the coding gene to sequences encoding the RbcS transit peptide. The transit peptide sequence alone is sufficient for translocation, since it contains all the sequence information required for this process (Vandenbroeck et al., 1985; Lee et al., 2002).
Except for the polyadenylation signal, a few other regulatory elements were found in the rbcS 3’ non-coding region of certain species. In petunia, for example, sequences downstream to the coding region contribute to quantitative differences in expression of SSU301 and SSU911 (the highest and weakest expressed petunia rbcS genes, respectively). Nuclear run-on transcription experiments indicated that these 3’ sequences affect transcription rates of the rbcS genes (Dean et al., 1989). In the maize rbcS-m3 gene, 3’ sequences are part of the photoreceptor–signal transduction system. These sequences co-operate with a 463 bp 5’ region to accomplish light-regulated rbcS expression in mesophyll cells (Viret et al., 1994).
A strategy for improving and regulating the expression of foreign genes in transgenic plants is the use of sequences that not only provide high levels of expression, but also show precise temporal and spatial regulation in specific plant organs. The RuBisCo small subunit gene promoter is an excellent promoter that confers both tissue-specific and light-regulated gene expression. Promoters, untranslated regions, and terminators of rbcS have been employed to generate transgenic plants in a number of species (e.g. Nomura et al., 2000; Outchkourov et al., 2003; Dai et al., 2005; Walker et al., 2005; Garg et al., 2006; Patel et al., 2006).
In this paper are presented six new members of the rbcS gene family in Musa acuminata Colla and Musa balbisiana Colla, including one pseudogene, and a total of 57 new rbcS sequences. This paper reports the first analysis of Musa rbcS genes and regulatory sequences and, as far as is known, it also presents the first sequence-based phylogenetic study of Musa spp.
Materials and methods
DNA and RNA extractions
DNA extractions from banana tissues were performed using a modified method of Stewart and Via (1993). Fifty milligrams of sample, ground in liquid nitrogen, were homogenized with 600 μl hot (70 °C) of CTAB buffer [cetyl trimethylammonium bromide 2% (w/v), NaCl 1.42 M, EDTA 20 mM, TRIS-HCl (pH 8.0) 100 mM, PVP-40 2% (v/v), L-ascorbic acid 5 mM, diethyldithiocarbamate 4 mM] and 3 μl of β-mercaptoethanol. Homogenates were extracted with 500 μl of chloroform–isoamyl alcohol (24:1v/v) and then shaken for 5 min, centrifuged at 1000 g in a 1.5 ml tube and, after addition of 0.7 vol of isopropanol and inverting to mix, left to precipitate at room temperature for 5 min. After centrifugation at 14 000 g for 20 min at room temperature, the supernatant was poured off, the pellet air-dried, and resuspended in 100 μl of TE or MilliQ H2O and 0.5 μl of RNase A (10 mg ml−1). Alternatively, total DNA and RNA extraction from banana leaves and roots was carried out by a CTAB method described by Graham et al. (1994). Modifications of this method included the addition of 1% (v/v) PVP-40 and the omission of β-mercaptoethanol. The pH of the CTAB buffer was modified with HCl to pH 5.0 after preparation in order to limit the oxidation caused by polyphenol oxidases. DNase I (20 units 100 μl−1 of DNA solution) was used instead of RNase for total RNA extractions.
Degenerate PCR
Two partial rbcS gene sequences (DQ088101 and DQ088125) were initially amplified from banana (Musa acuminata ‘Williams’) using degenerate primers (A5 5′-WCAARAARTTYGARAC-3′ and B8 5′-ARCTTCCACATIGTCCARTA-3′) that were designed in conserved amino acid regions with low coding redundancy following an alignment of RbcS sequences from rice, wheat, maize, and Lemna (data not shown) and assuming that G-T binding will occur. Degenerate PCR was carried out using 2–20 ng of CTAB-isolated genomic DNA from young banana leaves as template. Primers A5 and B8 were used at an annealing temperature of 50 °C and at final concentrations of 40 μM and 5 μM, respectively, taking into account their degeneracy (32-fold and 8-fold, respectively). PCR products were separated by agarose gel electrophoresis and cloned into pCR®2.1 (Invitrogen) for sequence analysis. This allowed the design of more specific primers A5.2 (5′-GAGACCCTCTCCTACCTTCCT-3′) and B8.1 (5′-TTCCACATGGTCCAGTASCGC-3′) spanning over the second intron in conserved regions of the two new Musa rbcS sequences. These primers were used at an annealing temperature of 60 °C and at final concentrations of 2 μM leading to an additional partial rbcS sequence from ‘Williams’ (DQ088100).
Inverse PCR
To isolate the regions upstream and downstream to the three fragments, the primers A8.1 (5′-GCGSTACTGGACCATGTGGAA-3′) that is complementary to primer B8.1, and B5.3 (5′-AGGAAGGTAGGAGAGGGTCTC-3′) as reverse primer downstream to primer A5.2 were used for inverse PCR on Musa (total) DNA. Templates for this PCR reaction were produced by digestion of 20 μg DNA (isolated from young leaves of ‘Williams’) with the compatible restriction enzymes PstI and NsiI (100 units each) at 37 °C for 4 h, followed by gel electrophoresis, extraction of DNA fragments of 1–9 kb in length (Qiagen), and overnight self-ligation in a large volume of 300 μl. Inverse PCR on these circular DNA fragments using primers A8.1 and B5.3 (1 μl template; 60 °C annealing temperature; 2.5 min extension at 72 °C in a 100 μl reaction) using the proofreading enzyme from Expand High Fidelity PCR System (Roche) according to the manufacturer's instructions resulted in two products of approximately 1.4 kb and 2.4 kb. These were cloned into pCR®2.1 (Invitrogen) and both strands were fully sequenced using universal M13 primers and a range of internal primers specifically designed for sequencing. To independently confirm the sequences obtained, primers I25.2A (5′-GGACTCTGTCAATGTCAACGC-3′) and I25.1B (5′-ATGTGTCTCTCGGCCCGG-3′) as well as 351A (5′-GCGTCGGTTGCATAAGCGAT-3′) and 352B (5′- TGGAAGCATTTAAGCTTTTAGATG-3′) were designed at the very 5’ and 3’ end (at or close to the PstI/NsiI ligation site) of the new Musa sequences. These were used for PCR (Expand High Fidelity PCR System) on genomic DNA (‘Williams’) to obtain coherent sequences for both genes and their flanking regions. Cloning and full-length sequencing of these fragments independently confirmed the sequences previously obtained by inverse PCR (DQ088100 and DQ088101).
Genomic library screening
A genomic DNA library produced from Musa acuminata ‘Grand Nain’ in Lambda ZAP® II (Stratagene) was kindly supplied by Dr Gregory May (Texas A&M University, Houston, TX, USA, currently Samuel Roberts Noble Foundation, Ardmore, OK, USA). Host preparation, infection [addition of 0.2% (w/v) maltose] and plating of the λ-phage vector, were conducted as described by Sambrook et al. (1989). The KW251 strain of Escherichia coli was used as the λ-phage host. Infected bacteria (forming approximately 40 000 pfu plate−1) were plated on eight 1.3% (w/v) large LB agar plates, in a 0.6% (w/v) LB agarose overlayer. Plaque lifts were prepared using a nylon membrane (Hybond-N; GE Healthcare). The λ-phage DNA was fixed to the membrane by placing the membrane phage-side down on wet 3MM filter paper (Whatman) soaked in denaturing solution (0.5 M NaOH, 1.5 M NaCl) for 5 min and neutralizing solution [1 M TRIS-HCl (pH 7.5), 1.5 M NaCl] for 5 min, before placing in 2× SSC, 0.1% (w/v) SDS for 15 min. The wet membranes were then exposed to 150 mJ of UV radiation. Hybridization in DIG Easy Hyb buffer (Roche) was carried out according to the manufacturer's instructions using a mixture of three Musa rbcS probes (based on DQ088100, DQ088101, and DQ088125). These probes were generated by PCR using primers A5.2 and B8.1 (2 μM each), 10 μl of DIG-labelled dNTPs (Roche), 2 ng total DNA from young banana leaves (‘Williams’) in a reaction volume of 100 μl and at an annealing temperature of 60 °C, according to the manufacturer's instructions (Roche). The sensitivity and specificity of the probes was previously tested using dot blot analyses (DIG-labelling and detection system; Roche) with a dilution series of the three cloned Musa rbcS sequences (DQ088100, DQ088101, and DQ088125). The detection limit was found to be at approximately 5 pg for all three sequences (data not shown). Detection of hybridization signals (anti-DIG alkaline phosphatase Fab fragment; CSPD substrate; Roche) was carried out according to the manufacturer's instructions following the DIG-labelling and detection system for colony hybridizations (Roche).
To obtain confidence in hybridization signals either a second plaque lift-off was performed on the same plate or a repeat hybridization was carried out on stripped filters. DIG-labelled probes were stripped from the membranes according to Dubitsky and Defiglia (1995) by incubation with 0.2 M NaOH containing 0.1% (w/v) SDS for 30 min at 37 °C under gentle agitation, followed by washing the membrane in 2× SSC containing 0.1% (w/v) SDS for 10 min at room temperature. After air-drying the membranes were stored at room temperature between 3MM filter papers.
To test individual plaques, areas on the plates with a confirmed positive signal were excised and placed into 1 ml SM buffer (5.8 g l−1 NaCl, 2 g l−1 MgSO4.7H2O, 5 ml l−1 of 2% gelatine solution in 20 mM TRIS-HCl, pH 7.5) together with one droplet of chloroform for phage diffusion and storage at 4 °C. These were further tested by plaque hybridization with the Musa rbcS probes on plates with higher plaque dilutions (50–100 pfu plate−1) to recognize distinct plaques. λ-DNA preparations were carried out on positive plaques including a proteinase K treatment (Wizard Lambda-DNA Prep Kit; Promega) and confirmed for Musa rbcS by PCR using primers A5.2 and B8.1. A total of 5 μg λ-DNA was cut with XbaI for 2 h at 37 °C and fragments were isolated by gel electrophoresis and subcloned into pBluescript II SK(+) (Stratagene). Sequence analysis was carried out on both strands using a range of purposely designed primers and revealed a new Musa rbcS sequence from ‘Grand Nain’ with large flanking promoter and terminator regions (DQ088099).
Southern blot analyses
For Southern blot analyses, 16 μg of total DNA (‘Williams’) was digested with restriction enzymes PvuII, Asp718, or NcoI in an overnight reaction and subjected to agarose gel electrophoresis. DNA blotting was performed using capillary action on to a nylon membrane according to standard procedures. After UV-crosslinking with 150 mJ, a total of 20 μl of the DIG-labelled probes was used following procedures for hybridization (DIG Easy Hyb buffer) with the mixture of three Musa rbcS probes (based on DQ088100, DQ088101, and DQ088125; see above) and detection (anti-DIG alkaline phosphatase Fab fragment; CSPD substrate) according to the manufacturer's instructions (DIG-labelling and detection system; Roche).
Promoter–reporter–terminator constructs
To test promoter activities by direct gene transfer using biolistics, the promoter–reporter constructs prbcS350sGFP, prbcS250sGFP, prbcS300GUS, and prcbS300exGUST were constructed, which contain promoter sequences from rbcS-Ma5, rbcS-Ma3, and rbcS-Ma1, respectively. The plasmids prbcS350sGFP and prbcS250sGFP contain 1296 bp and 851 bp, respectively, of the promoter region and 5’ UTR (just upstream of the start codon), and were constructed by inserting a BamHI/XbaI- and a BglII/XbaI-cleaved PCR product (amplified from the cloned full-length sequences DQ088100 and DQ088101 using primer pairs 5′-TCTAGACTCTGTCAATGTCAACGCTCT-3′/5′-GCGGATCCTCTTGCTGTTGCTCGTTGACC-3′ and 5′-TTTCTAGATAAGTATGGGATGCGGCAGT-3′/5′-GGAAGCCAGATCTCTTGCTGTCGTTC-3′, respectively) into the BamHI/XbaI-cleaved vector pUbiGFP (Schenk et al., 1999); replacing the maize ubiquitin promoter. The plasmid prbcS300GUS contains 1141 bp of the 5’ untranslated region of rbcS-Ma1 (see above: ‘rbcS-Ma5 respectively’ refers there to prbcS300GUS), the uidA (GUS) reporter gene and the Agrobacterium tumefaciens nopaline synthase (Nos) gene polyadenylation region. It was constructed by cleavage of pUbiGUS (Schenk et al., 1999) with HindIII and BamHI and insertion of a PCR fragment containing 1.1 kb of rbcS-Ma1 upstream region. This PCR fragment was obtained from the subcloned rbcS-Ma1 genomic clone in pBluescript II SK(+) (see Genomic library screening) with primers ProXA (5′-AAGCTTTAAGAGTTCTAACCAATA-3′) and ProXB (5′-GGATCCTTGATTCCTCTTTCTGAC-3′) that were designed with restriction enzyme sites HindIII and BamHI at the 5’ ends (in bold), to replace the maize ubiquitin promoter of pUbiGUS. The plasmid prbcS300exGUST contains 2.5 kb of upstream non-coding (promoter) region (including a 1.3 kb portion of unsequenced DNA at the 5’ end) and 592 bp of downstream non-coding (polyadenylation) region of rbcS-Ma1 (DQ088099). It was constructed from prbcS300GUS by increasing its promoter region to 2.5 kb via insertion (300 bp upstream from the transcriptional start site) of a 2.2 kb ClaI fragment that was directly excised from the genomic library sub-clone containing rbcS-Ma1. Next, the Nos polyadenylation region of this vector was replaced with 592 bp of the 3’ non-coding region of rbcS-Ma1 by cleavage with SacI and EcoRI (in bold) and insertion of a PCR fragment containing 592 bp of rbcS-Ma1 downstream region using primers Rbc3NCRA (5′-GGCGAGCTCACAATTCGGATCCGGCTC-3′) and Rbc3NCRB (5′-CGAATTCGAATGATGCTCGCACGT-3′) that contained the corresponding restriction enzyme sites.
To test the rbcS-Ma1 (‘Grand Nain’) putative terminator region in combination with heterologous promoters and for a direct comparison with the Nos terminator, the plasmids pCvGFPT, pMyGFPT, and pCvGUST were constructed. The plasmids pCvGFPT and pMyGFPT contain 1322 bp and 2105 bp of pregenomic RNA promoter from banana streak Obino l'Ewai virus and banana streak Mysore virus, respectively (isolated from plasmids pCvGFP and pMyGFP, respectively; Schenk et al., 2001) in the XbaI site of pBluescriptII SK+ (Stratagene), together with the sgfp (S65T) reporter gene (Chiu et al., 1996) in the PstI site, and 592 bp of the putative rbcS-Ma1 3’ non-coding (polyadenylation) region in the XhoI site. The plasmid pCvGUST was constructed from pCvGUS (Schenk et al., 2001) by replacing the Nos polyadenylation region (restrictions sites SacI/EcoRI) with 592 bp 3’ non-coding region of the rbcS-Ma1.
Transgene expression studies
Ten-day-old wheat (Triticum aestivum cv. Kennedy) or etiolated maize leaves (sweetcorn ‘Iochief’) were bombarded with plasmid-coated gold particles and transgene expression was visualized as described previously (Schenk et al., 1998, 2001). Quantitative transient assays were carried out in sugarcane (Q117) suspension cells with the plasmid pUbiLuc (Bower et al., 1996) as an internal standard using the luciferase assay as described previously (Remans et al., 2005).
PCR, cloning, and sequencing to obtain rbcS sequences for phylogenetic comparisons
Amplicons were obtained using the Expand High Fidelity PCR system (Roche). Reaction: 5 μl of 10× buffer, 5 μl of MgCl2 (25 mM), 0.75 μl of high fidelity enzyme mix, 0.2 mM of dNTPs (Biotech Australia), 0.4 μM primers of RT35A (5′-ACCTCCCCAGCAACGGTG-3′) and rbc500R (5′-TGTTGTCGAAGCCGATGATG-3′) (Proligo Lismore, Australia), 1 μl of DNA extract and Milli-Q H2O to a final volume of 50 μl. The PCR was cycled at 94 °C for 2 min, 35 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 90 s, and a final extension step at 72 °C for 10 min. After agarose gel electrophoresis, two or three products were observed for each of the cultivars and purified using the UltraClean Gelspin DNA purification kit (Mo Bio Laboratories, USA) according to the manufacturer's directions. Ligations and transformations were performed with the Zero Blunt PCR cloning kit (Invitrogen). Plasmid DNA preparations from recombinant (white) colonies were performed using the Nucleospin-Plasmid purification kit (Macherey-Nagel, Germany). Sequencing was performed using BigDye V3.1 as described by the manufacturer's directions (Applied Biosystems). Forward and reverse sequences were obtained with primers M13F (5′-GTAAAACGACGGCCAG-3′) and M13R (5′-CAGGAAACAGCTATGAC-3′) using an AB3730XL sequencer by the Australian Genomic Research Facility (AGRF, Brisbane).
Phylogenetic analysis
Forward and reverse sequences were aligned and edited using Sequencher 3.0 (Gene Codes Corporation, Ann Arbor, MI, USA). Edited sequences were aligned using MegAlign (DNASTAR, Madison, WI, USA), the alignments were checked and corrected by eye, and then the complete alignment was trimmed so that all sequences started and finished at the same place. Phylogenetic analysis was performed using Paup 4.0b8 (Swofford, 2001). Bootstrap analysis (1000 replicates) was performed using a parsimonious heuristic search with random addition of sequences (1000 replicates), tree bisection–reconnection, branch swapping, and MULPAR effective to give the most parsimonious distance tree with bootstrap values.
Expression studies
For RNA dot blot analyses, DIG-labelled probes were generated by PCR using gene-specific primers (5′-CCCGGCTACTAACTACTCTG-3′ and 5′-GACCGTAGTACATGAGAGTC-3′ for rbcS-Ma3; 5′-GATGGCCACTAGTAGGCAGC-3′ and 5′-AACCTTATTGGATGCTTGGGAAG-3′ for rbcS-Ma5) and total DNA (‘Williams’) according to the manufacturer's instructions (Roche). RNA was isolated from young and old leaves blotted as 2 μl drops (in duplicated dilution series containing between 10 ng and 2 μg of total RNA) onto a nylon filter (GE Healthcare; wetted first with water, then with 1× SSC) and fixed by UV-crosslinking. Hybridization and detection of DIG-labelled probe were carried out as described above.
Quantitative real-time PCR
Quantitative real-time PCR analysis was carried out in optical 96-well plates (Applied Biosystems) using an ABI PRISM© 7700 Sequence Detection System (Applied Biosystems) with the standard thermal profile and dissociation stage (Anderson et al., 2004). Reaction master mix using SYBR Green was made as described previously (Kruse et al., 2005). The PCR primer efficiencies were determined as described by McGrath et al. (2005). Gene copy numbers of rbcS-Ma3 and rbcS-Ma5 relative to single copy gene MWPL1 (encoding banana pectate lyase) were calculated for the DNA of each Musa cultivar using the equation relative ratiogene/MWPL1=[Egene(–Ct gene)]/[EMWPL1(–Ct MWPL1)] (Schenk et al., 2003).
Results
Isolation of rbcS genes from Musa acuminata
Degenerate PCR primers were designed in conserved amino acid regions with low coding redundancy based on the alignment of RbcS sequences from rice, wheat, maize, and Lemna and assuming that G-T binding will occur. Three partial gene sequences of 250–350 bp were initially amplified from banana (Musa acuminata ‘Williams’) by PCR using degenerate primers A5/B8 and A5.2/B8.1. Sequence analysis showed that all three sequences (DQ088100, DQ088101, and DQ088125) encoded amino acid sequences with strong homology to known RuBisCo proteins, which were interrupted by an intronic region. However, the open reading frame of DQ088125 was shifted several times, suggesting that this sequence was derived from a dysfunctional rbcS pseudogene. The other two new M. acuminata rbcS genes are from here onwards referred to as rbcS-Ma3 and rbcS-Ma5. To determine the total copy number of rbcS sequences in M. acuminata, Southern blot analysis was carried out using a mixture of three probes from the above sequences (Fig. 1A). The result suggested that the genome of M. acuminata ‘Williams’ contains three or four unique copies of rbcS genes and it was decided to isolate full-length sequences for these (except for the pseudogene).
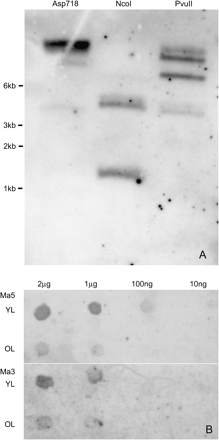
(A) Southern blot analysis of Musa acuminata ‘Williams’ total DNA cleaved with restriction enzymes Asp718 (lane 1), NcoI (lane 2), and PvuII (lane 3), and hybridized with a mixture of three DIG-labelled probes from rbcS-Ma3, rbcS-Ma5, and an rbcS pseudogene. (B) Dot blot analysis for the comparison of expression levels of rbcS-Ma5 and rbcS-Ma3 in young and old leaves. Serial dilutions of RNA were blotted on to two membranes, then probed with gene-specific DIG-labelled probes (rbcS-Ma5, top; rbcS350, bottom) developed from the 3′ non-coding region of each gene.
To isolate the complete coding sequences flanking promoter and terminator sequences from rbcS-Ma3 and rbcS-Ma5, an inverse PCR approach was used. Sequence analyses of the PCR fragments obtained confirmed the presence of the complete rbcS-Ma3 and rbcS-Ma5 genes including upstream and downstream sequences. This was confirmed by independent isolation and sequencing of a coherent fragment from genomic DNA. Totals of 1970 bp and 2350 bp were obtained for rbcS-Ma3 (DQ088100) and rbcS-Ma5 (DQ088101), respectively. Sequence analysis showed that the open reading frame for both genes was interrupted by two introns (exons at positions 854–1033, 1117–1248, and 1340–1570, and 1328–1510, 1600–1731, and 1912–2142 for rbcS-Ma3 and rbcS-Ma5, respectively).
To search for additional rbcS genes, a genomic library from M. acuminata ‘Grand Nain’ was screened by using the same three probes as used previously for the Southern blot analysis. Positive plaque areas were further analysed by subcloning into plasmids and sequencing, and one additional distinct M. acuminata rbcS gene was identified and isolated, which is referred to as rbcS-Ma1 (DQ088099). This sequence contains a total of 2481 bp and, similarly to rbcS-Ma3 and rbcS-Ma5, the open reading frame for rbcS-Ma1 was also interrupted by two introns (exons at positions 1142–1321, 1418–1549, and 1645–1875). Three additional partial rbcS sequences were also found in ‘Grand Nain’ using PCR with conserved primers (see Phylogenetic analyses). In summary, initially it was found that M. acuminata contained at least four distinct rbcS genes.
Musa rbcS sequence analyses
Sequence comparison between the promoter regions of rbcS-Ma1, rbcS-Ma3, and rbcS-Ma5 showed little overall sequence alignment homology (26.2–26.8% in 850 bp and 30.7–37.7% in 300 bp upstream of the start codon), but a number of highly conserved regulatory elements were identified in similar locations. The position of important regulatory elements, namely the TATA-box, I-box, G-box, and the monocot conserved sequence element, are presented in Table 1.
Structure of the Musa acuminata RuBisCo genes rbcS-Ma1, rbcS-Ma3, and rbcS-Ma5 including 5′ untranslated region with positions of putative promoter elements, coding sequence, introns, and 3′ untranslated (polyadenylation) region
| Region | rbcS-Ma1 DQ088099 | Position (bp) | |
| rbcS-Ma3 DQ088100 | rbcS-Ma5 DQ088101 | ||
| I-box (GATAAG) | –223/–218 and –173/–168 | –241/–236 | –182/–177 and –157/–152 |
| G-box [CACGTGGC(AGT)] | CATGTGGC(TCGT) –202/–191 | CACGCGGC(ACGC) –217/–206 | NA |
| Monocot conserved sequence (GAACGTGAGCAATCCCACA) | CAACGTACAAGAT –99/–87 CCACA –83/–79 | GATCGGAGACAC –109/–98 CCACC –94/-90 | GAACGACACCGGC –97/–85 CCTCA –81/−77 |
| TATA-box (TATATATA) | –36/–29 | –34/–27 | –37/–30 |
| 5′ UTR | 1–64 | 1–75 | 1–69 |
| Transit peptide | 65–238 (174) | 76–249 (174) | 70–246 (177) |
| Exon 1 | 239–244 (6) | 250–255 (6) | 247–252 (6) |
| Intron 1 | 245–340 (96) | 256–338 (83) | 253–341 (89) |
| Exon 2 | 341–472 (132) | 339–470 (132) | 342–473 (132) |
| Intron 2 | 473–567 (95) | 471–565 (95) | 474–653 (180) |
| Exon 3 | 568–795 (228) | 566–793 (228) | 654–881 (228) |
| 3′ UTR putative poly(A) site | 983 | 953 or 1112 | 998 |
| Region | rbcS-Ma1 DQ088099 | Position (bp) | |
| rbcS-Ma3 DQ088100 | rbcS-Ma5 DQ088101 | ||
| I-box (GATAAG) | –223/–218 and –173/–168 | –241/–236 | –182/–177 and –157/–152 |
| G-box [CACGTGGC(AGT)] | CATGTGGC(TCGT) –202/–191 | CACGCGGC(ACGC) –217/–206 | NA |
| Monocot conserved sequence (GAACGTGAGCAATCCCACA) | CAACGTACAAGAT –99/–87 CCACA –83/–79 | GATCGGAGACAC –109/–98 CCACC –94/-90 | GAACGACACCGGC –97/–85 CCTCA –81/−77 |
| TATA-box (TATATATA) | –36/–29 | –34/–27 | –37/–30 |
| 5′ UTR | 1–64 | 1–75 | 1–69 |
| Transit peptide | 65–238 (174) | 76–249 (174) | 70–246 (177) |
| Exon 1 | 239–244 (6) | 250–255 (6) | 247–252 (6) |
| Intron 1 | 245–340 (96) | 256–338 (83) | 253–341 (89) |
| Exon 2 | 341–472 (132) | 339–470 (132) | 342–473 (132) |
| Intron 2 | 473–567 (95) | 471–565 (95) | 474–653 (180) |
| Exon 3 | 568–795 (228) | 566–793 (228) | 654–881 (228) |
| 3′ UTR putative poly(A) site | 983 | 953 or 1112 | 998 |
Structure of the Musa acuminata RuBisCo genes rbcS-Ma1, rbcS-Ma3, and rbcS-Ma5 including 5′ untranslated region with positions of putative promoter elements, coding sequence, introns, and 3′ untranslated (polyadenylation) region
| Region | rbcS-Ma1 DQ088099 | Position (bp) | |
| rbcS-Ma3 DQ088100 | rbcS-Ma5 DQ088101 | ||
| I-box (GATAAG) | –223/–218 and –173/–168 | –241/–236 | –182/–177 and –157/–152 |
| G-box [CACGTGGC(AGT)] | CATGTGGC(TCGT) –202/–191 | CACGCGGC(ACGC) –217/–206 | NA |
| Monocot conserved sequence (GAACGTGAGCAATCCCACA) | CAACGTACAAGAT –99/–87 CCACA –83/–79 | GATCGGAGACAC –109/–98 CCACC –94/-90 | GAACGACACCGGC –97/–85 CCTCA –81/−77 |
| TATA-box (TATATATA) | –36/–29 | –34/–27 | –37/–30 |
| 5′ UTR | 1–64 | 1–75 | 1–69 |
| Transit peptide | 65–238 (174) | 76–249 (174) | 70–246 (177) |
| Exon 1 | 239–244 (6) | 250–255 (6) | 247–252 (6) |
| Intron 1 | 245–340 (96) | 256–338 (83) | 253–341 (89) |
| Exon 2 | 341–472 (132) | 339–470 (132) | 342–473 (132) |
| Intron 2 | 473–567 (95) | 471–565 (95) | 474–653 (180) |
| Exon 3 | 568–795 (228) | 566–793 (228) | 654–881 (228) |
| 3′ UTR putative poly(A) site | 983 | 953 or 1112 | 998 |
| Region | rbcS-Ma1 DQ088099 | Position (bp) | |
| rbcS-Ma3 DQ088100 | rbcS-Ma5 DQ088101 | ||
| I-box (GATAAG) | –223/–218 and –173/–168 | –241/–236 | –182/–177 and –157/–152 |
| G-box [CACGTGGC(AGT)] | CATGTGGC(TCGT) –202/–191 | CACGCGGC(ACGC) –217/–206 | NA |
| Monocot conserved sequence (GAACGTGAGCAATCCCACA) | CAACGTACAAGAT –99/–87 CCACA –83/–79 | GATCGGAGACAC –109/–98 CCACC –94/-90 | GAACGACACCGGC –97/–85 CCTCA –81/−77 |
| TATA-box (TATATATA) | –36/–29 | –34/–27 | –37/–30 |
| 5′ UTR | 1–64 | 1–75 | 1–69 |
| Transit peptide | 65–238 (174) | 76–249 (174) | 70–246 (177) |
| Exon 1 | 239–244 (6) | 250–255 (6) | 247–252 (6) |
| Intron 1 | 245–340 (96) | 256–338 (83) | 253–341 (89) |
| Exon 2 | 341–472 (132) | 339–470 (132) | 342–473 (132) |
| Intron 2 | 473–567 (95) | 471–565 (95) | 474–653 (180) |
| Exon 3 | 568–795 (228) | 566–793 (228) | 654–881 (228) |
| 3′ UTR putative poly(A) site | 983 | 953 or 1112 | 998 |
As expected the coding regions of these rbcS transcripts had significant homology to each other, whereas the non-coding regions had very limited similarity. The 5’ untranslated regions of the three rbcS genes were of a similar length but with low homology to each other (30–46%). The transit peptides, which make up almost one-third of the coding region, were almost identical in length (RbcS-Ma5 contains one extra amino acid) and had 86.0–91.4% sequence homology to each other. The rbcS-Ma1, rbcS-Ma3, and rbcS-Ma5 genes were of the same length and with a homology of 89.4–91.9% to each other. There is very little sequence homology between the introns among the rbcS gene families and the majority of sequence variation within each family was based within the introns.
Figure 2A summarizes the amino acid sequence comparison of M. acuminata rbcS-Ma3 and rbcS-Ma5 as sequenced from ‘Williams’ with rbcS-Ma1 from ‘Grand Nain’ and the only previous Musa rbcS sequence submission from GenBank (AF008214). Currently, apart from Musa, the order Zingiberales has only one other rbcS sequence listed in GenBank, DQ418485 Zingiber officinale (ginger). This amino acid sequence, along with four rbcS sequences with the highest homology to the Musa rbcS sequences, was included in the alignment. All sequences were trimmed to the same length (comprising the first two-thirds of the rbcS sequence) and a phylogenetic analysis was performed to show the interspecies relationships (Fig. 2B). The parsimonious results found that the ginger rbcS sequence formed a unique branch between the rbcS-Ma1 and the rbcS-Ma3/rbcS-Ma5 clades. It was most closely aligned to rbcS-Ma1 but the members of the Musa rbcS family were more closely related to each other than to the ginger rbcS. After ginger, the next closest phylogenetic relative to the Musa rbcS sequences was Fritillaria agrestis (a distantly related monocot), followed by Lactuca sativa (a dicot). Lemna gibba (from which the original degenerate primers were designed) and Zantedeschia aethiopica were the two other monocots with the next closest homology to the Musa RbcS protein. Interestingly, both, the transit peptide and the mature protein of all rbcS sequences compared in this study, showed near-equal levels of homology between species, indicating that they have diverged at similar rates.
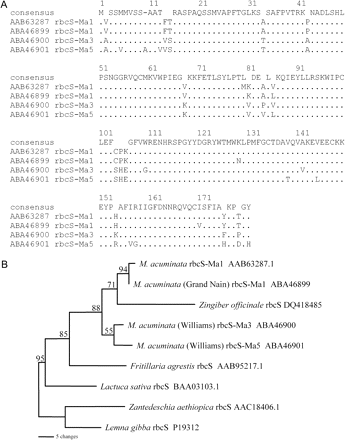
Amino acid sequence comparison of RbcS from Musa and other species. (A) Complete amino acid sequence alignment of RbcS-Ma1, RbcS-Ma3, and RbcS-Ma5, and a previously identified RbcS sequence from Musa together with the closest matches from other species. (B) Maximum parsimony distance tree of the RbcS amino acid sequences from Musa and related species. Tree length=148; number of characters=113; constant characters=45, parsimony uninformative characters=30; parsimony informative characters=38; consistency index=0.872; retention index=0.683. Numbers on branches represent bootstrap percentages (>50%) from 1000 full heuristic replications
Expression analyses, promoter and terminator studies
To determine whether the new M. acuminata rbcS genes were actually expressed and to detect potential differences in transcript abundances, dot blot analyses were carried out with total RNA in a dilution series isolated from young and old leaves of M. acuminata ‘Williams’ using labelled probes specific for rbcS-Ma3 or rbcS-Ma5. The probes showed a detectable signal at a 10-fold higher dilution for rbcS-Ma5 (100 ng) than for rbcS-Ma3 (1 μg) (Fig. 1B) suggesting that rbcS-Ma5 transcripts are more abundant than rbcS-Ma3 transcripts. Similarly, the detection in old leaves was 10-fold lower than in young leaves, suggesting that both genes are highly expressed in young tissues.
Several putative regulatory elements, such as the TATA-box, I-box, G-box, and the monocot conserved sequence element were found in the upstream regions of rbcS-Ma1, rbcS-Ma3, and rbcS-Ma5 (Table 1). To test whether these regions could act as promoters to drive heterologous gene expression, promoter–reporter constructs were produced and tested in transient expression systems. The plasmids prbcS300GUS, rbcS250sGFP, and prbcS350sGFP contain the 5’ upstream sequences from rbcS-Ma1 (1141 bp), rbcS-Ma3 (853 bp), and rbcS-Ma5 (1327 bp), respectively. An additional construct (prbcS300exGUST) with a larger 5’ upstream region (2.5 kb) from rbcS-Ma1 as well as 592 bp of its 3’ downstream region (putative polyadenylation region) was also included. Bombardment on to wheat leaves led to transgene expression for all four constructs (Fig. 3A–D) and maize (data not shown). No significant differences in the intensity or number of detectable transgenic cells could be observed for any of the constructs.
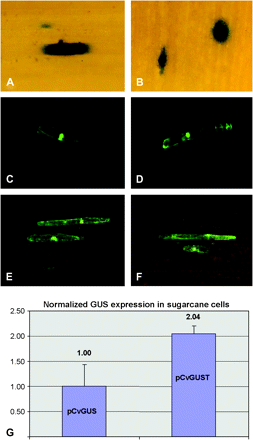
Transient promoter–reporter gene expression assays in wheat leaf cells following bombardment with constructs prbcS300GUS (rbcS-Ma1 promoter, A); prbcS300exGUST (rbcS-Ma1 promoter+polyadenylation region, B); prbcS250sGFP (rbcS-Ma3 promoter, C); prbcS350sGFP (rbcS-Ma5 promoter, D); pMyGFPT (rbcSMa-1 polyadenylation region with promoter from banana streak Mysore virus, E), and pCvGFPT (rbcSMa-1 polyadenylation region with promoter from banana streak Obino l'Ewai virus, F). (G) Comparison of the Nos polyadenylation region (pCvGUS) with the rbcS-Ma1 polyadenylation region (pCvGUST) using a quantitative transient transformation assay in sugarcane suspension cells. Averages and standard deviation are shown from three biological replicates.
To test further whether the polyadenylation region of rbcS-Ma1 can be used for heterologous gene expression independent of its promoter, the constructs pMyGFPT and pCvGFPT were produced. These contain promoters from banana streak virus in combination with 592 bp of the rbcS-Ma1 3’ untranslated region. Both constructs led to transgene expression when bombarded on to wheat leaves, suggesting that the rbcS-Ma1 3’ untranslated region can be used as an efficient terminator (Fig. 3E, F). Furthermore, to directly compare the rbcS-Ma1 polyadenylation region with the commonly used Nos polyadenylation region, the constructs pCvGUS (Nos polyadenylation region) and pCvGUST (rbcS-Ma1 polyadenylation region) were bombarded on to sugarcane suspension cells. Quantification of transient expression, by using an internal luciferase reference construct, showed a significant (P <0.05) 2-fold higher expression of the construct harbouring the rbcS-Ma1 polyadenylation region when compared with the control (Nos polyadenylation region) construct (Fig. 3G).
Phylogenetic analyses within Musa
To analyse the genetic relationship of the commercial cultivars Musa acuminata ‘Williams’ and ‘Grand Nain’ compared with other Musa accessions, a phylogenetic tree was constructed based on rbcS genes. PCR products were generated using the conserved rbcS primers RT35A and rbc500R which had been designed based on sequence data obtained for ‘Williams’ and ‘Grand Nain’ (see above). Genomic DNA from the additional Musa cultivars ‘Goldfinger’, ‘Ladyfinger’, ‘Paka’, and M. balbisiana were used as templates for PCR.
Two different PCR product sizes were observed after gel electrophoresis which were excised, cloned, and sequenced. The larger PCR products (610–660 bp) were found mainly to belong to four rbcS gene subfamilies (1, 2, 5, 6, and occasionally 3; see classification below). The smaller products (520–530 bp) were found to belong to rbcS gene subfamilies 3 and 4. Size differences were mainly due to intron size variation, but some were apparently due to non-specific binding of the forward primer in subfamily 3 as some products were longer or shorter than the average at the start, while all of the 3’ ends stopped at the same point. Six to eight clones of each product size were sequenced in each of the cultivars; many of these proved to be duplicates. Supplementary Table S1 at JXB online shows a list of the 57 distinct sequences submitted to GenBank, and also shows the clone number of each sequence. Many product sizes (up to eight) were observed using different primer combinations; however, these were degenerate primers, which could have resulted in their non-specific binding due to non-stringent reaction conditions in this experiment and thus may not reflect rbcS gene copy number.
All 57 distinct sequences were analysed using a maximum parsimony distance tree (Fig. 4). From the six different Musa accessions, six rbcS gene subfamilies and one pseudogene were identified in the phylogenetic tree. The total sequence divergence between each of the gene subfamilies was 8.1–24.8%. Gene names for the proposed rbcS gene family are rbcS1, rbcS2, rbcS3, rbcS4, rbcS5, and rbcS6. The inclusion of the Musa accessions with mixed A and B genomes (‘Goldfinger’ AAAB and ‘Lady Finger’ AAB) as well as accessions with solely A genomes (‘Williams’ AAA, ‘Grand Nain’ AAA, ‘and Paka’ AA) or just B genomes (M. balbisiana BB) allows the association of Musa rbcS genes to either the A or B genomes. These appear as different clusters on the phylogenetic tree and within each gene subfamily the letters ‘Ma’ or ‘Mb’ were added to the gene name in front of the rbcS subfamily number to show their association with either the A or B genome (e.g. rbcS-Ma1).
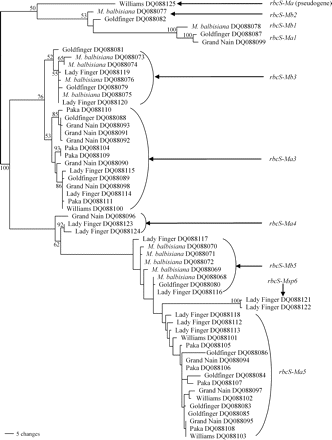
Maximum parsimony distance tree of the rbcS gene family in Musa species. The tree identifies at least six gene subfamilies including one pseudogene for rbcS genes of six different Musa accessions. Tree length=449; number of characters=627; constant characters=381; parsimony uninformative characters=63; parsimony informative characters=183; consistency index=0.655; retention index=0.942. Numbers on branches represent bootstrap percentages (>50%) from 1000 full heuristic replications. Numbers (1–6) after the gene names represent proposed gene subfamilies. Ma=Musa acuminata, Mb=M. balbisiana, Msp=Musa acuminata or M. balbisiana.
Observation of the sequence data showed that two of the genes (rbcS-Ma3 and rbcS-Ma5) were represented by four or five alleles in the diploid accessions ‘Paka’ and M. balbisiana (Fig. 4). The sequences of these alleles are very similar and these genes are likely to have undergone a recent gene duplication event. To determine the copy number of these two genes, quantitative real-time PCR was carried out to measure the relative abundance within each genome (Fig. 5). The sequence and quantitative real-time PCR data showed that rbcS-Ma3 was present once per haploid A genome and twice in the B genome and that the rbcS-Ma5 gene was present twice per haploid A genome and was most likely present four times in the B genome. As expected the Musa×paradisiaca cultivars (mix of A and B genomes) had an intermediate number of alleles, i.e. rbcS-Ma3 ‘Goldfinger’ (AAAB) had 1.23±0.07 copies (1.25 expected) and ‘Lady Finger’ (AAB) had 1.33±0.16 copies (1.33 expected; Fig. 5). These ratios are consistent with the number of A and B genomes of these cultivars. Furthermore, these results suggest that an rbcS gene duplication event occurred in the B genome after Musa A and B genomes diverged (estimated to be around 1–2 million years ago; Lescot et al., 2005).
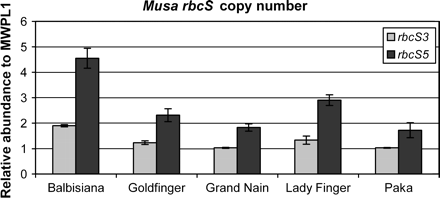
Distribution of proposed copy numbers for the rbcS subfamilies 3 and 5 in five different banana accessions, based on quantitative real-time PCR. Averages and standard deviations are shown from three replicates.
Discussion
The isolation and characterization of the rbcS genes and flanking regions in this study provide a number of interesting results that give us insights into the evolution of rbcS genes in Musa and also provide some starting material for potential applications in plant biotechnology.
Promoters from several rbcS genes have been successfully used to drive high-level transgene expression in stably transformed plants. The present transient gene expression data in wheat and maize demonstrate that the 5’ upstream regions of rbcS-Ma1, rbcS-Ma3, and rbcS-Ma5 contain functional promoters that are able to drive transgene expression in heterologous systems. Furthermore, the terminator of rbcS-Ma1 was functional in different promoter–reporter gene constructs and showed a 2-fold higher expression than the commonly used Nos terminator. To determine fully the potential of Musa rbcS promoters and terminators as biotechnological tools, further studies should follow to measure promoter strengths and specificities in stably transformed transgenic plants.
Interestingly, relative to other characterized species, Musa is highly divergent for the rbcS gene family, more than Arabidopsis, which has four rbcS genes, and wheat, which has five (Sasanuma, 2001). In the present studies, six rbcS genes and one pseudogene were identified in Musa by phylogenetic analyses. There are two distinct clades in three of these gene subfamilies, representing the two different parent lineages, Musa acuminata (A genome) and Musa balbisiana (B genome). Furthermore, based on the close sequence homology of rbcS genes within each subfamily, it can be concluded that at least three of the six gene families were present before Musa A and B genomes separated. As there are multiple alleles in each of the rbcS genes there are potentially 31 or more copies of the rbcS gene in the tetraploid cultivar Goldfinger (AAAB).
Unlike most other monocots analysed to date, the rbcS genes in the Musa genome have two introns compared with one as usual, whereas dicots contain two or more introns (GenBank search of all rbcS genes in Liliopsida including wheat, maize, rice, Lemna gibba, etc). The rbcS intron positions in algae, dicots, and monocots are presented in brief by Yamazaki et al. (2005) where these conserved positions have been designated 1–5. Intron position 1 is conserved in all higher plants (except Lemna gibba) whereas intron position 4, found in the dicots, is missing in most monocot species. The present studies show that the positions of both introns from dicots (positions 1 and 4) are also found in the monocot Musa. Fritillaria agrestis is the only other monocot species known to have two introns in rbcS genes (positions 1 and 4). The rbcS genes of the monocot Lemna gibba contain only one intron at position 4 (Silverthorne et al., 1990; but incorrectly shown at position 5 in Yamazaki et al., 2005), while the typical intron at position 1 present in the rbcS genes of all higher plants is missing.
The present results suggest that an rbcS gene duplication event occurred in the B genome after Musa A and B genomes had diverged. Recent duplication of rbcS genes has been speculated in other plants (Clegg et al., 1997), e.g. wheat (Sasanuma, 2001) and the ice plant (Derocher et al., 1993). While very little sequence information is available for M. balbisiana, the polymorphism of AFLP markers indicates a high degree of speciation (Nwakanma et al., 2003) and a high divergence within this species, with two major groups (Ude et al., 2002). Each of the three Musa rbcS subfamilies were more closely related to each other than to the closest known sequences from other species (Fig. 2B), suggesting that these subfamilies arose from duplication events of a common rbcS ancestor. The duplication event of this particular rbcS gene family is most likely to have occurred in the Zingiberales lineage as the closest rbcS sequence was found in the Zingiberaceae, ginger. To properly resolve the evolution of the rbcS gene family in the Musa genus, additional sequences isolated from species throughout the Zingiberales would be required. Such a study would also yield a genetic resource useful for supporting or improving the phylogeny of Zingiberales.
We wish to thank Gregory May for kindly providing the ‘Grand Nain’ genomic library, Tony Remans for help with the luciferase assays and useful discussions, Julianne Henderson and Julie Pattemore for the MWPL1 primers, Ralf Dietzgen and Haibao Zhang for useful discussions, and the Alexander von Humboldt Foundation for providing a Feodor Lynen Fellowship to Peer Schenk.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments