





Abstract
The genus Nepenthes represents carnivorous plants with pitcher traps capable of efficient prey capture and digestion. The possible involvement of plant chitinases in this process was studied in Nepenthes khasiana. Two different types of endochitinases were identified in the liquid of closed traps exhibiting substrate specificity for either long chitin polymers or N-acetylglucosamine (GlcNAc) oligomers. Injection of chitin into such closed sterile pitchers induced the appearance of additional endochitinase isoenzymes, with substrate specificity only for long chitin polymers. No significant exochitinase (N-acetyl-β-glucosaminidase) or chitobiosidase activity could be detected in the non-induced or induced trap liquid. Four genes representing two subgroups of basic chitinases, denoted as Nkchit1b and Nkchit2b, were isolated from the secretory region of N. khasiana pitchers. The main differences between the two subgroups are the presence of a proline-rich hinge region only in NkCHIT1b and a C-terminal putative vacuole targeting extension only in NkCHIT2b, indicating different compartmentalization of the two enzymes. Reverse transcription–polymerase chain reaction (RT–PCR) evaluation of mRNA levels showed that the Nkchit2b genes are constitutively expressed in the secretory cells while transcription of Nkchit1b genes is induced by chitin injection. These results show for the first time the involvement of genes encoding chitinases in prey–trap interaction and their differential expression and activity during prey trapping.
Abbreviations
- CaMV
cauliflower mosaic virus
- ChiA
Serratia marcescens chitinase
- GlcNAc
N-acetylglucosamine
- 4MU
4-methylumbelliferyl
- Nkchit1b
Nepenthes khasiana basic chitinase 1
- pNP
p-nitrophenyl
- RT–PCR
reverse transcription–polymerase chain reaction
Introduction
Carnivorous plants grown in barren regions with high humidity and acidic soil have developed special organs to capture insects, digest them, and absorb the nutrients. The tropical carnivorous pitcher plant Nepenthes, whose name originated from the poisonous drug used by Helen in Homer's Odyssey, encompasses ∼80 species, with the largest diversity in the islands of Borneo and Sumatra. Additional scattered populations are found in northern India (N. khasiana), Madagascar (N. distillatoria), and the Seychelles (N. perveleii). Pitcher-shaped leaves, that capture insects, characterize all the species. The prey is attracted to the pitcher by its colour and nectar secretion. The upper part of the pitcher has a wax-coated inner epidermis that forms a slippery surface promoting the sliding down of the prey into the lower part of the pitcher, which has >6000 digestive cellular glands per cm2. These glands function in the perception of chemical stimuli, secretion of digestive enzymes, and nutrient absorption (Owen et al., 1999; Schulze et al., 1999).
The presence of proteolytic enzymes in carnivorous plants had already been reported by Darwin (1875). The acidic trap liquid of Nepenthes was shown to have several hydrolytic enzymes such as proteases and phosphatases (Heslop-Harrison, 1975). Five cDNA clones of aspartic proteinase (AP) were isolated from Nepenthes alata and their expression was localized to different parts of the pitcher (An et al., 2002). A plasma membrane H+-ATPase and transporters of ammonium, amino acids, and peptides, involved in acidification of the pitcher fluid and nutrient absorption, respectively, were also identified in N. alata secretory glands (Schulze et al., 1999; An et al., 2001). The presence of proteases, RNases, nucleases, and phosphatases during trap development was studied in Sarracenia purpurea, representing another family of pitcher-possessing plants (Gallie and Chang, 1997). The expression of these hydrolytic enzymes was shown to be developmentally regulated during the pitcher opening stages. Later on, in mature opened pitchers, these enzymes are induced by an, as yet undefined, signal transduction mechanism in response to nucleic acids, proteins, and reduced nitrogen (Gallie and Chang, 1997). Digestion of colloidal chitin by the secreted liquid of the Nepenthes pitcher and by an aqueous extract of Drosera peltata indicated the possible involvement of chitinolytic enzymes in the digestion of insects by carnivorous plants (Amagase et al., 1972). Recently, chitinase activity was detected in the carnivorous genus Utricularia (Sirova et al., 2003) and in Drosera rotundifolia L. (Matusikova et al., 2005). However, as yet, no full-length sequences of chitinase genes or cDNAs have been isolated from carnivorous plants.
Plant chitinases are to be considered pathogen-related (PR) proteins, since their activity is induced by fungal, bacterial, and viral infections, as well as by more general signals of stress such as wounding, salicylic acid, ethylene, and heavy metal salts (Collinge et al., 1993; Graham and Sticklen, 1994). Moreover, chitinases also have important non-defensive functions in growth and development processes (Kasprzewska, 2003). Chitinases are involved in the regulation of nodulation signal molecules (Goormachtig et al., 1998), they affect embryogenesis (De Jong et al., 1992; Helleboid et al., 2000), and they might participate in programmed cell death (Passarinho et al., 2001). Chitin and chitosan are major components of cell walls in insects and fungi. Since insects make up the bulk of the prey found in pitcher traps, chitinases appear to play a double role in carnivorous plants—defence and carnivory. The isolation and characterization of four N. khasiana chitinase genes and their corresponding cDNA sequences is described here. To gain a better understanding of the role of chitinases in pitchers, exogenous chitin was applied to the pitchers as a means to mimic prey capture, and the expression pattern of chitinases was characterized.
Materials and methods
Plant growth conditions
Nepenthes khasiana plants were grown in the Botanical Gardens of Tel Aviv University in a greenhouse under a 16 h/8 h light/dark regime at 25 °C. The plants were watered twice a week with double-distilled water. Closed pitchers were used for all analyses described herein.
Trap liquid sampling and induction of chitinase activity by chitin injection
Trap liquid was collected from closed young pitchers by a sterile syringe, in order to preserve sterility, and stored at −70 °C in aliquots. Approximately 1 mg of colloidal chitin (Vessey and Pegg, 1973), pH 5.0, was injected with a sterile syringe into the side of a closed pitcher that was first surface-sterilized with 70% ethanol. Aliquots of the trap liquid were collected from the closed pitcher at increasing intervals after injection.
Chitinase assays using various substrates
Endochitinase activity was assayed by first separating sterile trap liquid or callus extracts on native 12–15% acrylamide gels using 25 mM TRIS–HCl, pH 8.3, 192 mM glycine as running buffer (Davis, 1964) and then by overlaying an additional acrylamide gel containing glycol-chitin (0.01% w/v) as a substrate (Trudel and Asselin, 1989). After 20 h incubation at 37 °C, the gels were stained for 5 min with 0.01% (w/v) Calcofluor white M2R. Chitinase activity was visualized as dark spots on the glycol-chitin gel by UV light (260 nm).
Chitinolytic activity was also characterized in gels by using 4-methylumbelliferyl (4MU) β-D-N,N′,N″-triacetylchitotrioside as a substrate. After electrophoresis, the gel was washed twice with 100 mM sodium citrate buffer, pH 5.5, and then overlaid with 200 μM substrate solubilized in 1% plant agar. The released 4MU was detected by UV light.
The activity of N-acetyl-β-glucosaminidase (exochitinase), chitobiosidase, and endochitinase in pitcher liquid was determined in microtitre plates by using as substrates the dimer 4MU-N-acetyl-β-D-glucosaminide [4MU-(GlcNAc)1; Sigma], trimer 4MU-β-D-N,N′-diacetylchitobioside [4MU-(GlcNAc)2, Sigma], or tetramer 4MU-β-D-N,N′,N″-triacetylchitotrioside [4MU-(GlcNAc)3, Sigma], respectively. Chitinase specificity was estimated by the cleavage of the β-1,4-bond that releases 4MU from the different oligomers (Kuranda and Robbins, 1991). The chitinase reaction contained 20 μl of trap liquid mixed with 30 μl of 300 μM substrate in 0.1 M sodium citrate buffer, pH 3.0 and incubated at 37 °C for 1 h. The reaction was stopped by addition of 150 μl of 0.64 M glycine-NaOH buffer, pH 10.4. The production of 4MU was measured by a fluorescence spectrophotometer (excitation at 360 nm, emission at 460 nm). Activity units were defined as nM 4MU released min−1.
Kinetic properties of endochitinases were determined according to Tronsmo and Harman (1993) in microtitre plates using p-nitrophenyl (pNP)-β-(GlcNac)3 as the substrate. Reactions (80 μl) containing increasing substrate concentrations ranging from 0 to 250 μM in 50 mM potassium phosphate buffer, pH 6.7, and 30 μl of 5.6-fold concentrated pitcher liquid or 0.0005 U of commercial Serratia marcescens chitinase (Sigma), were incubated for 30 min at 50 °C. Reactions were terminated by adding 50 μl of 0.4 M sodium carbonate, which also enhanced the colour of p-nitrophenol formed by the enzymatic cleavage of the substrate. Absorbance at 405 nm was measured by a Spectromax 190 microplate reader.
Isolation of genomic DNA
Leaf tissue (1 g) was homogenized in 5 ml of buffer consisting of 1 vol. of DNA extraction buffer (0.35 M sorbitol, 0.1 M TRIS–HCl, pH 7.5, 5 mM EDTA, and 0.02 M Na-bisulphite), 1 vol. of nuclei lysis buffer (0.2 M TRIS–HCl, pH 7.5, 50 mM EDTA, 2 M NaCl, 2% CTAB), and 0.4 vol. of 5% sarkosyl. The homogenate was incubated for 20 min at 65 °C and then extracted twice with 1 vol. of chloroform:isoamyl alcohol (24:1 v:v). Three volumes of 6 M NaI were added to the aqueous phase and the genomic DNA was further purified by using a ‘High Pure’ filter tube (High Pure Plasmid Isolation Kit, Boehringer Mannheim).
Isolation of chitinase genes
Conserved amino acid sequences of plant chitinases were used to design degenerate primers for polymerase chain reaction (PCR) amplification of Nepenthes chitinase genes. The following degenerate primers were used: 5′-TTIGGICAA/GACIT/AG/CICAC/TGAG/AAC-3′(direct primer), and 5′-GAG/TICCICCA/GTTIATIATA/GTTT/A/C/GGT-3′ (reverse primer). PCR was carried out using N. khasiana genomic DNA as a template at an annealing temperature of 50 °C. Based on the isolated sequences, sets of marginal primers with reverse orientation were synthesized to isolate the rest of the gene sequences further by an inverse PCR strategy. In brief, genomic DNA (5 μg) digested with HindIII, BamHI, or NdeI was self-ligated to form circles. About 100 ng of this circular DNA was used as a template for PCR amplification at annealing temperatures between 60 °C and 65 °C using TaKaRa Ex Taq and the specifically designed primers.
Isolation of total RNA
Total RNA was isolated from the lower part of the pitcher by a special hot borate/proteinase K method (Schulze et al., 1999).
Isolation of mRNA and cDNA synthesis
To identify differential expression of the defined chitinase genes, mRNA was purified from total RNA by using oligo(dT) conjugated to magnetic DynaBeads (Dynal, Norway) according to the manufacturer's instructions. Thereafter total cDNA was synthesized by M-MLV reverse transcriptase (Promega) using oligo(dT)12–18 as a primer. The primer was annealed at 22 °C for 10 min and the reverse transcription reaction was performed at 40 °C for 60 min. A 1 μl aliquot of the reverse transcription reaction (50 μl) was taken to PCR amplification. The following gene-specific primers were used for the identification of the transcribed genes in the trap secretory cells: Nkchit1b direct primer 5′-CGACGGTCCATATGCATGGGGATACTGTTTCAAG-3′, reverse primer 5′-CAAATGGCCACTGGGTGTAGCAGTCCAAGTTATC-3′; Nkchit2b direct primer 5′-CGTGGGGATATTGCTATCTCAG-3′, and reverse primer 5′-CTACTCGGTGG CCCACAAAAG-3′.
Constructs and plant transformation
Nkchit1b was synthesized by PCR amplification using genomic DNA template, Taq polymerase (Ex.Taq.,TaKaRa), and specific primers corresponding to the 5′ and 3′ ends of the coding sequence. Nkchit2b cDNA was synthesized by a reverse transcription (RT)–PCR strategy. Briefly, mRNA isolated from trap tissue was reverse transcribed by M-MLV reverse transcriptase (Promega). The resulting cDNA was PCR amplified using specific primers designed according to the 5′ and 3′ end of the coding sequence. Nkchit1b and Nkchit2b PCR products were cloned downstream of the constitutive cauliflower mosaic virus (CaMV) 35S promoter in the pPCV702 binary vector, which was used for Agrobacterium-mediated transformation of tobacco (Nicotiana tabacum var. Samsun NN) leaves (Koncz et al., 1989).
Callus generation from transgenic tobacco leaves and estimation of chitinase activity
Leaf discs were excised from sterile young tobacco plants and grown on Murashige and Skoog (MS) medium containing 3% sucrose, 0.8% agar, 5 μg ml−1 α-naphthalene acetic acid (NAA) and 1 μg ml−1 benzylaminopurine (BA). The calli were grown at 25 °C in the dark. Crude extracts of transgenic calli were prepared by grinding the sterile calli in a glass homogenizer without addition of buffer. The homogenates were centrifuged for 5 min at 14 000 rpm in an Eppendorf minifuge and the supernatants were used for the enzyme activity assay on gels.
Results
Pitcher structure
A longitudinal section of N. khasiana closed mature pitcher (Fig. 1A) shows the glandular bottom part of the pitcher and the secretory glands (Fig. 1B, C). The glands are situated within defined depressions of the epidermis (Fig. 1D) partially covered by small epidermal ridges (Fig. 1E). Numerous secretory cells are visualized in each gland (Fig. 1D). The glandular secretory zone extends only to about half of the pitcher length, and in the upper half it is exchanged for a waxy epidermal zone with a distinct network of red pigmented cells (Fig. 1F).
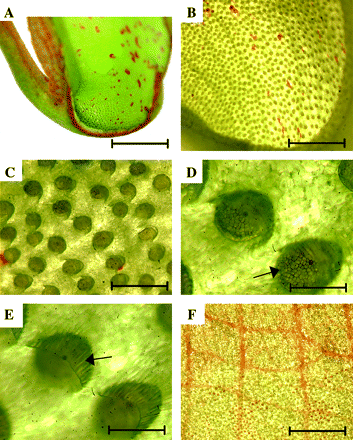
Pitcher structure of Nepenthes khasiana. (A) Longitudinal section of the lower part of a pitcher. Bar=7.5 mm. (B) Bottom part of pitcher visualized under low magnification. Bar=2.5 mm. (C) Magnification of secretory glands. Bar=300 μm. (D) Glands full of secretory granules (arrow). Bar=100 μm. (E) Epidermal ridges (arrow) covering every gland. Bar=100 μm. (F) Waxy epidermal zone covering the inner surface of the upper part of a pitcher. Bar=2 mm.
Induction of chitinase activity in trap liquid
Plants respond to pathogen attack by induced accumulation of lytic enzymes such as chitinases and β-1,3-glucanases (Punja, 2001). To follow the response of the pitcher to a mimicked pathogen attack and prey sensing, changes in protein pattern (Fig. 2A) and chitinase activity (Fig. 2B) in the liquid of a closed trap were analysed before and after chitin injection. Trap liquid samples were collected prior to and after the injection of soluble colloidal chitin (1 mg per pitcher, pH 5.0) into closed pitchers. Total protein concentration in the collected samples was very low, below detectable levels of Coomassie staining when determined on gels or Whatman paper (Marder et al., 1986) or by measuring absorbance at 280 nm. Consequently, samples were separated on a 12% SDS–polyacrylamide gel and visualized by silver staining (Fig. 2A). Chitin injection induced the synthesis of at least four new proteins and enhanced the production of two proteins that were also present before induction (Fig. 2A, arrows and dots, respectively). All the major induced bands were isolated and subjected to tandem mass spectrometry (MS/MS) analysis (Q-STAR, Applied Biosystems), but no oligopeptides showing resemblance to known chitinase sequences were identified. When chitinase activity was estimated using glycol-chitin as a substrate following separation on native gels, at least three new chitinases (Fig. 2B, lanes 2 and 3 indicated by arrows) appeared after 20 h of chitin induction showing different migration rates from the non-induced chitinase (lane 1). Injection of an equal volume of sterile water into the trap also induced chitinase activity but to a lesser extent (data not shown). Hence, chitin injection into the trap induced secretion of new proteins including chitinases.
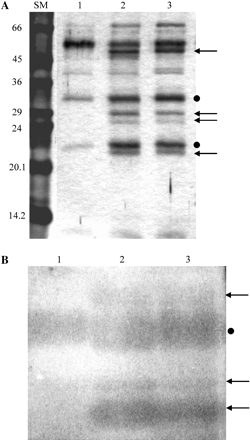
Chitin injection into closed pitchers induced changes in protein pattern and chitinase activity of pitcher liquid. Colloidal chitin (1 mg) was injected into closed young traps. (A) General protein pattern of the trap liquid: liquid samples (100 μl) taken prior to injection (lane 1) and 4 d (lane 2) or 8 d (lane 3) after injection were separated on a 12% SDS–polyacrylamide gel and subsequently stained with a silver stain. Arrows indicate chitin-induced bands, dots indicate chitin-enhanced bands. (B) Chitinase activity estimated on a native gel: samples (30 μl) taken prior to injection (lane 1) and 20 h (lane 2) or 5 d (lane 3) after injection. Following electrophoresis, the gel was overlaid with an additional gel containing 0.01% (w/v) glycol-chitin and assayed for chitinase activity. Arrows show induced chitinase bands and the dot shows the chitin-enhanced band.
Characterization of the trap liquid chitinase activity
To verify endo- and exochitinase activities, differentiating GlcNAc oligomeric substrates were used. The release of 4MU from 4MU-(GlcNAc)1 (dimer), 4MU-(GlcNAc)2 (trimer), or 4MU-(GlcNAc)3 (tetramer) indicates the presence of N-acetyl-β-glucosaminidase (exochitinase), chitobiosidase, or endochitinase activity, respectively. No exochitinase activity was detected in either the non-induced or induced trap liquid (Fig. 3A) since there was no release of fluorescent 4MU from 4MU-(GlcNAc)1. Similarly, the lack of ability to release 4MU from 4MU-(GlcNAc)2 indicated no chitobiosidase activity. However, a significant release of 4MU from 4MU-(GlcNAc)3 was identified only in non-induced trap liquid (Fig. 3A). This endochitinase activity of the non-induced liquid exhibited sigmoid kinetics as compared with S. marcescens endochitinase (commercial preparation), used as a control (Fig. 3B). Sigmoid enzyme kinetics may indicate positive substrate co-operativity, thus only allowing estimation of enzyme affinity towards the substrate, i.e. K0.5 instead of Km. The estimated K0.5 value for 4MU-(GlcNAc)3 was 130 μM. Only a trace amount of endochitinase activity, as evident by 4MU-(GlcNAc)3 hydrolysis, was observed in the induced liquid (Fig. 3A). This seemed to be in contrast to the results shown in Fig. 2B where induction of additional endochitinase activities was observed after chitin injection when glycol-chitin was used as the substrate. To clarify this point, equal amounts of non-induced and induced trap liquid were separated on native gels which were subsequently overlaid with either 4MU-(GlcNAc)3- or glycol-chitin-containing gels as substrates for the chitinase assay. Comparison of the location and intensity of the activity spots in the two gels (Fig. 3C) re-confirmed that the induced endochitinases could only hydrolyse longer chitin polymers (lane 4), but not the tetrameric substrate (lane 2). Thus, a specific chitinase capable of releasing trimeric GlcNAc oligomers from 4MU-(GlcNAc)3 is only present in the non-induced closed trap. Together, these enzymatic assays showed that at least two distinct endochitinases, differing in substrate specificity (Fig. 3A, C, lanes 1 and 3), exist in the non-induced liquid, whereas additional endochitinases, that can only hydrolyse chitin polymers, appeared in the traps after chitin induction (Fig. 3C, lane 4).
![Identification of differential endo- and exochitinase activity, before and after induction, by assaying N-acetyl-β-glucosaminidase, chitibiosidase, and endochitinase activity in trap liquid. (A) Chitinolytic activity of non-induced (N, filled symbols) and induced (I, open symbols) trap liquid samples determined by using 4MU covalently (β-1,4) linked to GlcNAc oligomers (dimer = one, trimer = two, and tetramer = three GlcNAc residues) as substrates. (B) Kinetic analysis of the non-induced trap liquid endochitinase, using 4MU-(GlcNAc)3 as the substrate. Chitinase activity (v), expressed in p-nitrophenol (pNP) absorbance units, was determined using 170 μl (<0.3 ng μl−1 total proteins) of trap liquid (filled squares) or 0.0005 U of Serratia marcescens chitinase (open circles) as enzyme, and the tetrameric pNP-β-(GlcNAc)3 as a substrate ([S]). The sigmoid-type curve was obtained in three independent experiments. (C) In-gel chitinolytic activity of different endochitinases present in trap liquid. Chitinase activity was determined after separating 70 μl of non-induced (N, lanes 1 and 3) and induced (I, lanes 2 and 4) trap liquid on a 10% native gel. The overlaid gels contained either fluorescent tetramer (left gel) or glycol-chitin (right gel) as substrates to differentiate between the two types of endochitinases capable of hydrolysing chitin oligomers or polymers, respectively.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jxb/57/11/10.1093/jxb/erl048/2/m_jexboterl048f03_ht.gif?Expires=1750346132&Signature=jlN-BESLfPMI4aAi0sDUEbRgHzaY5wcRhXVIU4RtXn8U4GyFmM~Myuusymf-mg9~LgY6pMQoJZ0mQuN55bRMzAxlpw0SDT4TTWDhnRBbZw5bP4XIllXJulPLheVHxMWy~BdhGRCCatV4d~kFZgP7kos8c5fFd7M9NR75lIjrVS5CKtiigqlWl-XqFYraSuwuE~pbHIhz6xX789ttow3XASJVD5L3Y4TATvgCqUbiA7~7~OGv2b9i3T-GVaMUNkCF0Ct5StDyL7FVDW-EdZgCt73RLD4SnDcFJajpLZiVMyAfFMTWYRkzk7I3KfC0TLFmf4OXHwjosykXWhufjXxeFA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Identification of differential endo- and exochitinase activity, before and after induction, by assaying N-acetyl-β-glucosaminidase, chitibiosidase, and endochitinase activity in trap liquid. (A) Chitinolytic activity of non-induced (N, filled symbols) and induced (I, open symbols) trap liquid samples determined by using 4MU covalently (β-1,4) linked to GlcNAc oligomers (dimer = one, trimer = two, and tetramer = three GlcNAc residues) as substrates. (B) Kinetic analysis of the non-induced trap liquid endochitinase, using 4MU-(GlcNAc)3 as the substrate. Chitinase activity (v), expressed in p-nitrophenol (pNP) absorbance units, was determined using 170 μl (<0.3 ng μl−1 total proteins) of trap liquid (filled squares) or 0.0005 U of Serratia marcescens chitinase (open circles) as enzyme, and the tetrameric pNP-β-(GlcNAc)3 as a substrate ([S]). The sigmoid-type curve was obtained in three independent experiments. (C) In-gel chitinolytic activity of different endochitinases present in trap liquid. Chitinase activity was determined after separating 70 μl of non-induced (N, lanes 1 and 3) and induced (I, lanes 2 and 4) trap liquid on a 10% native gel. The overlaid gels contained either fluorescent tetramer (left gel) or glycol-chitin (right gel) as substrates to differentiate between the two types of endochitinases capable of hydrolysing chitin oligomers or polymers, respectively.
Isolation of Nepenthes chitinase genes
Preliminary separation of trap liquid on a Mono Q ion exchange column followed by chitinase assays on activity gels revealed that the major part of trap liquid chitinases were eluted at low (∼0.2 M NaCl) salt, indicating a high PI value (data not shown). Since this is a characteristic for basic plant chitinases (Graham and Sticklen, 1994; Meins et al., 1994), we designed the strategy of isolating the corresponding genes accordingly. Four chitinase genes were isolated by PCR amplification using Nepenthes genomic DNA as a template. Degenerate primers, designed according to conserved amino acid sequences of known plant basic chitinases (NCBI GenBank), were used for the initial amplification of the conserved genomic region and then, by using an inverse PCR strategy, the full-length sequences of the genes were obtained and cloned. The genes, denoted as Nkchit1b-1, Nkchit1b-2, Nkchit2b-1, and Nkchit2b-2 (accession numbers AY618881, AY618883, AY618885, and AY618887, respectively), showed a high amino acid homology to plant class I basic chitinases as determined by a multidomain structure analysis and basic PI predictions (data not shown). cDNA clones (accession numbers AY618882, AY618884, AY618886, and AY618888) corresponding to these chitinase genes were isolated using RNA extracted from chitin-induced pitcher secretory tissue as a template for RT–PCR amplification with gene-specific primers. Genomic and cDNA sequence comparisons confirmed the predicted exon–intron structure of the two gene types. Chitinase 1 (Nkchit1b-1 and 2) and chitinase 2 (Nkchit2b-1 and 2) genes are composed of three exons and two introns. The coding regions of the two Nkchit1b genes are identical, while the intron sequences are different. On the other hand, the two introns of chitinase 2 genes (Nkchit2b-1 and 2) are identical, while the exons have sequence differences resulting in six amino acid substitutions (L17/V17, V34/A34, Y44/F44, G55/E55, F64/S64, and A237/E237).
Alignment of the deduced amino acid sequences of the chitinase 1 (Nkchit1b-1 and 2) and one of the chitinase 2 (Nkchit2b-1) genes revealed 66% identity and 74% similarity (NCBI BLAST) (Fig. 4). Both types of basic chitinases contain an N-terminal signal peptide differing in length and amino acid composition, a cysteine-rich domain, and a catalytic domain (Fig. 4). This domain structure resembles that of plant class I basic chitinases that are usually composed of four or five domains: an N-terminal signal peptide, cysteine-rich domain, proline-rich hinge region, catalytic domain, and a C-terminal extension when the chitinase is targeted to the vacuole (Neuhaus et al., 1991; Collinge et al., 1993). However, a proline-rich hinge region is only present in NkCHIT1b. A part of this region (T82–P88) is unique to NkCHIT1b and does not appear in other chitinase sequences reported thus far (BLAST comparisons). NetOGlyc prediction (ExPaSy Molecular Server), which identifies putative post-translational modifications, shows nine putative glycosylation sites in NkCHIT1b (Fig. 4) while none was predicted for NkCHIT2b. Eight of the nine sites are located in the proline-rich hinge region. Comparison of the C-terminal parts of the two types of chitinases revealed the presence of a hydrophobic putative extension only in NkCHIT2b (Fig. 4). A BLAST search for amino acid sequence identity of NkCHIT1b and NkCHIT2b showed highest (66%) identity to Poa pratensis (AF000966) chitinase and to a basic endochitinase precursor (79%) of Vitis vinifera (P51613), respectively. Conserved amino acid residues, suggested to be of functional importance in all plant chitinases, were identified in both chitinases and are indicated by different symbols in Fig. 4. The eight cysteine residues (C), present at identical positions in the chitin-binding region of most basic chitinases, are involved in the formation of disulphide bridges allowing the correct folding of domains into a compact active conformation. The threonine (T) and glutamine (Q) residues, that play a role in the active site geometry, as well as the glutamic acid (E) and asparagine (N) that are important for catalysis (Graham and Sticklen, 1994; Hamel et al., 1997), are all present in both Nepenthes chitinases. On the other hand, the conserved tyrosine (Y), thought to affect substrate binding in the catalytic cleft (Verburg et al., 1993), is altered to phenylalanine (F) in NkCHIT2b. To date, the significance of the Y to F substitution in Nepenthes remains to be determined.

Amino acid sequence alignment of the two types of N. khasiana basic endochitinases, NkCHIT1b-1 and NkCHIT2b-1. Identical residues are shown in black. The putative leader peptide is marked by a continuous line above it, the cysteine-rich domain by a broken line above it, the proline-rich region by asterisks, and the C-terminal extension by crosses. Vertical arrows pointing upward mark the six amino acid residues differing between NkCHIT2b-1 and NkCHIT2b-2. Predicted glycosylation sites (ExPASy—Biochemical Pathways) are marked by open triangles. Residues suggested to be of functional importance are marked above the sequence by the following symbols: cysteines—involved in disulphide bridges (open circles), glutamic acid and asparagine—important for enzymatic catalysis (filled circles), threonine and glutamine—involved in maintenance of active site geometry (open squares), tyrosine—suggested to affect substrate binding in the catalytic cleft (filled square).
Identification of chitinase(s) present in pitcher secretory cells
Figure 2B shows that chitin injection into closed traps induced the appearance of at least three new trap liquid chitinases. Therefore, the presence of Nkchit1b and Nkchit2b transcripts in the trap secretory region, before and after chitin injection, was estimated. Following RNA extraction from the pitcher secretory region, RT–PCR was performed with Nkchit1b- and Nkchit2b-specific primers. Figure 5 clearly demonstrates that the Nkchit2b transcript is present in the secretory region of both non-induced (Fig. 5A) and induced (Fig. 5B) pitchers while the Nkchit1b transcript is only present in the secretory region after induction. Both primer sets yielded longer products when genomic DNA, instead of cDNA, was used as a template, due to the presence of introns in the amplified region. Thus, according to these results, it might be assumed that either the uppermost or lowest endochitinase activity spot shown in Fig. 3C, lane 4, represents Nkchit1b while the activity spots present in both lanes 3 and 4 (middle band) (Fig. 3C) correspond to Nkchit2b, which was shown to be transcribed in the secretory region of both non-induced and induced pitchers. To confirm this conclusion, the Nkchit1b genomic clone and Nkchit2b cDNA, each driven by the 35S promoter, were separately introduced into transgenic tobacco plants. It was assumed that in tobacco plants the alien chitinases would undergo similar post-translational modifications. Figure 6 demonstrates chitinase activity in calli derived from the transgenic tobacco plants. The introduced NkCHIT1b migrated slowly, as did the induced upper band in Fig. 3C, lane 4, while NkCHIT2b co-migrates with the endogenous tobacco chitinase and resembles the migration of the middle band in Fig. 3C, lanes 3 and 4. The slow migration of NkCHIT1b on native gels in comparison with NkCHIT2b could be due to possible glycosylation of the proline-rich region (Fig. 4).
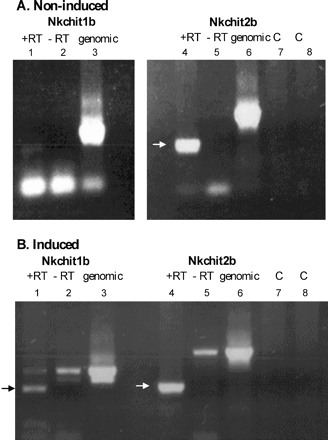
Differential presence of Nkchit1b and Nkchit2b transcripts in the pitcher secretory region before and after induction. RT–PCR analysis was performed on mRNA isolated from closed pitchers either before (A) or 4 d after (B) chitin injection. Lanes 1–3 represent Nkchit1b amplification, and lanes 4–8 represent Nkchit2b amplification with gene-specific primers. Lanes 1 and 4, reactions with reverse transcriptase (+RT); lanes 2 and 5, reactions without reverse transcriptase (–RT); lanes 3 and 6, amplification with genomic DNA as a template; lane 7, Nkchit2b 5′ primer control; and lane 8, Nkchit2b 3′ primer control.
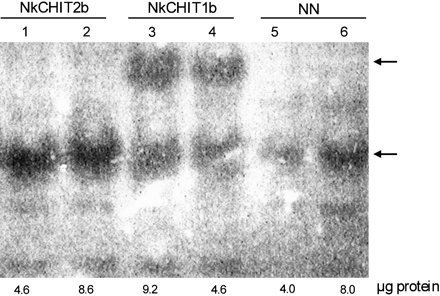
In-gel activity assay of NkCHIT1b and NkCHIT2b expressed in calli derived from transgenic tobacco plants. Crude extracts of transgenic calli constitutively expressing NkCHIT2b (lanes 1 and 2) or NkCHIT1b (lanes 3 and 4) and wild-type NN (lanes 5 and 6) were loaded onto a 10% native gel. The amounts of loaded protein are indicated below the lanes. After electrophoresis, the gel was overlaid with an additional gel containing 0.01% (w/v) glycol-chitin and incubated for 2 h at 37 °C. Chitinase activity was detected by UV light after staining with 0.01% (w/v) Calcofluor white M2R for 5 min.
Discussion
Nepenthes pitchers are highly specialized organs that capture and digest insects for an extra supply of organic precursors when grown in harsh environments. The trapped insects are digested in the pitcher fluid and then the resulting nutrients are absorbed via activated transporters. Prey digestion has been shown to involve the secretion of proteases, RNases, and phosphatases (Matthews, 1960; Amagase, 1972; Amagase et al., 1972). The lack of complete digestion of ants by purified proteases from Nepenthes traps (Amagase et al., 1972) had raised the possibility that additional exoskeleton-digesting enzymes, such as chitinases, are present in the traps. Incubation of concentrated Nepenthes trap liquid with colloidal chitin resulted in considerable digestion of chitin (Amagase et al., 1972). However, this observed activity was partially attributed to the presence of chitinase-producing symbiotic micro-organisms in the open traps. The present study clearly defines the involvement of plant chitinases by using closed Nepenthes pitchers that allowed the estimation of net plant chitinase activity in sterile pitcher liquid prior to any prey intervention and the appearance of secondary micro-organisms. To mimic signals induced by insect capture, colloidal chitin was injected into the closed pitchers under sterile conditions and the liquid was then analysed at different time intervals.
The presence of at least two different chitinases in the N. khasiana pitcher liquid was shown under non-induced conditions. These chitinases differed in their migration rate on native gels and in substrate specificity estimated by the ability to release (GlcNAc)3 from the tetramer 4MU-(GlcNAc)3 or to hydrolyse glycol-chitin (Fig. 3A, C, lanes 1 and 3). The sigmoid velocity curve, observed when increasing tetramer concentrations were used as the substrate (Fig. 3B), indicated enzyme–substrate co-operativity and the requirement for a certain threshold substrate concentration in the trap to increase the activity rate of this chitinase significantly. The second endochitinase, identified in the pitcher liquid under non-induced conditions, could only hydrolyse long chitin polymers (Fig. 3C, lane 3).
Injection of colloidal chitin into the closed pitchers mimicked prey trapping and induced the secretion of at least three additional active chitinases (Fig. 2B). Such an induced reaction resembles the plant response to pathogen attack when chitinases and other PR proteins are induced by different pathogens and specific elicitors (Punja, 2001). Chitin and chitosan were shown to elicit chitinase activity in bacteria (e.g. Vibrio harveyi), fungi (e.g. Trichoderma harzianum), plant suspension cultures, and leaves of the carnivorous plant Drosera rotundifolia (Haran et al., 1995; Svitil et al., 1997; Wu et al., 1997; Matusikova et al., 2005). Mechanical wounding was also shown to up-regulate chitinase isoenzymes differentially in tomato and wheat tissues (Botha et al., 1998; Wu and Bradford, 2003). In Nepenthes pitchers, the chitin-induced chitinases were only able to hydrolyse glycol-chitin (Fig. 2B, Fig. 3C lane 4), but not 4MU-(GlcNAc)3 or shorter oligomers (Fig. 3A, C, lane 2). Similarly, when hydrolysis rates of S. marcescens chitinases A and B were estimated using 4MU-chito-oligosaccharides as substrates, they did not fully correlate with the whole chitinolytic activity measured by using chitin or GlcNAc polymers as substrates (Brurberg et al., 1996, 2001), indicating the presence of endochitinases with specificities for long polymers in addition to those that can hydrolyse short oligomers. The occurrence in the trap liquid of multiple forms of basic endochitinases, exhibiting different substrate specificities, emphasizes the important role that chitinases play in the trap response to prey capturing. It is possible that in Nepenthes the combined action of the two endochitinases, initially present in the closed sterile trap liquid, first cleaves the long polymers and then releases GlcNAc oligomers from already produced ends of the chitin exoskeleton of the intruding prey. The small oligomers, in turn, induce the synthesis of a repertoire of new chitinases possessing the required endochitinase specificities for further efficient digestion of the prey chitin skeleton whose action probably allows the penetration and interaction of the inducible nucleases and proteases with their substrates (Gallie and Chang, 1997).
In tobacco, 10 clearly defined endo-type chitinases with different substrate specificities, that act synergistically, appeared at the initial stages of the defence response (Brunner et al., 1998). Such a platform of different classes of chitinases can liberate hydrolysed products with various lengths, which may possess variable abilities to elicit defence reactions (Roby et al., 1987; Brunner et al., 1998).
Until this study, no Nepenthes chitinase genes have been characterized. Here the isolation of four Nepenthes chitinase genes (Nkchit1b-1 and 2, and Nkchit2b-1 and 2) belonging to two subgroups of basic chitinases, according to amino acid sequence homology and domain structure, is described. Both Nkchit1b and Nkchit2b genes are composed of three exons and two introns and contain a putative leader peptide sequence at the 5′ end of the coding region. The Nkchit1b group contains two genes sharing identical exons and different intron sequences. The two Nkchit2b genes code for proteins differing from each other in six amino acid residues but having identical introns. Since only one of the Nkchit1b promoters has been isolated, it is difficult to estimate why the organism has retained two different genes encoding an identical enzyme. Gene duplication could have occurred long ago, since the intron sequences differ significantly. On the other hand, the exon preservation suggests an important function for the NkCHIT1b enzyme. It is also interesting to note that NkCHIT1b and NkCHIT2b share 66% amino acid homology, while a higher identity exists between NkCHIT2b and several non-carnivorous plant chitinases, for example, chitinases from V. vinifera (79%), Ulmus pumila (74%), Persea americana (75%), and Medicago truncatula (73%).
NkCHIT2b can be considered as a constitutively expressed housekeeping chitinase produced in both non-induced and induced secretory tissue (Fig. 5A, B). According to the sequence of its C-terminal end (NGLLWATE), NkCHIT2b is probably targeted to vacuoles (Chrispeels and Raikhel, 1992). NkCHIT1b, on the other hand, which is synthesized under induced conditions, lacks the C-terminal targeting signal and is secreted into the pitcher fluid. It also contains a proline-rich domain with eight putative glycosylation sites.
This study has unravelled the role of chitinases in inducing the trap digesting machinery and also defined the involvement of genes encoding basic chitinases in this mechanism. The differential expression of the newly characterized chitinases, before and after chitin induction, and the identification of chitinases with different substrate specificities suggest that the insect trap of carnivorous plants is an advanced and efficient modification of the plant defence mechanism. In these traps, traces of GlcNAc oligomers, specifically produced by the sequential action of long polymer- and short oligomer-cleaving enzymes, induce a massive production of endochitinases that digest the trapped insect's inner and outer skeleton to provide carbon and nitrogen sources.
Since some Nepenthes pitchers can contain >3.0 l of secreted liquid, chitin induction might be utilized as a tool for inducing the synthesis of large quantities of soluble endochitinases. Moreover, GlcNAc oligomers were found to play a role in various medical treatments, such as wound healing (Cole et al., 1999; Schwaitzberg et al., 2004), and their selective production by plant-derived enzymes in the closed non-induced pitchers devoid of any animal viruses or bacterial contamination may offer a great biotechnological advantage.
This study was supported by Biotech M.A.H. (Makhteshim-Agan-Hazera) Brurim M.P. Shikmim 79837, Israel. We thank Shoshan Haran for helpful discussions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments