





Abstract
Pneumonia caused by Streptococcus pneumoniae is a leading cause of death worldwide. A growing body of evidence indicates that the successful treatment of bacterial infections results from synergy between antibiotic-mediated direct antibacterial activity and the host's immune defenses. However, the mechanisms underlying the protective immune responses induced by amoxicillin, a β-lactam antibiotic used as the first-line treatment of S. pneumoniae infections, have not been characterized. A better understanding of amoxicillin's effects on host-pathogen interactions might facilitate the development of other treatment options. Given the crucial role of neutrophils in the control of S. pneumoniae infections, we decided to investigate amoxicillin's impact on neutrophil development in a mouse model of pneumococcal superinfection. A single therapeutic dose of amoxicillin almost completely eradicated the bacteria and prevented local and systemic inflammatory responses. Interestingly, in this context, amoxicillin treatment did not impair the emergency granulopoiesis triggered in the bone marrow by S. pneumoniae. Importantly, treatment of pneumonia with amoxicillin was associated with a greater mature neutrophil count in the bone marrow; these neutrophils had specific transcriptomic and proteomic profiles. Furthermore, amoxicillin-conditioned, mature neutrophils in the bone marrow had a less activated phenotype and might be rapidly mobilized in peripheral tissues in response to systemic inflammation. Thus, by revealing a novel effect of amoxicillin on the development and functions of bone marrow neutrophils during S. pneumoniae pneumonia, our findings provide new insights into the impact of amoxicillin treatment on host immune responses.
1. Introduction
Respiratory infections constitute a leading cause of morbidity and mortality worldwide.1Streptococcus pneumoniae is a major cause of community-acquired pneumonia and may also damage the lung barrier, leading to further dissemination of the bacteria into the bloodstream and thus bacteremia and meningitis.2S. pneumoniae can pose even greater threats to global public health upon influenza A virus (IAV) co-infection.3,4 Although vaccines and antibiotics have proven extremely useful in respectively the prevention and treatment of pneumococcal pneumonia, S. pneumoniae has been listed by the World Health Organization as a priority pathogen that requires the development of novel therapeutic strategies.5
The activity of antibiotics relies primarily on the direct killing or growth inhibition of bacteria. However, the literature data indicate that host immunity contributes to the effectiveness of antibiotics.6–8 Thus, antibiotics are often less effective in immunocompromised patients. Furthermore, macrolides and fluoroquinolones show anti-inflammatory effects in vivo in addition to their direct antimicrobial action.7–10 The first-line antibiotic used to treat S. pneumoniae pneumonia is the β-lactam amoxicillin (AMX).11 Penicillins like AMX exert their bactericidal activity by targeting penicillin-binding proteins and thus by interfering with bacterial peptidoglycan synthesis and cell wall integrity. The current general hypothesis is that β-lactam’s action can directly regulate innate immune responses against bacteria. Upon treatment with a β-lactam, dying and dead bacteria release various compounds, including pathogen-associated molecular patterns that might modulate local and/or systemic antimicrobial immunity.12 However, mechanistic evidence of β-lactam–induced protective immune responses in the host is currently lacking.
The innate immune response to a S. pneumoniae lung infection is critical for pathogen clearance and disease control.13 The recognition of S. pneumoniae by epithelial and myeloid cells expressing pattern recognition receptors (e.g. Toll-like receptors) results in the production of inflammatory mediators, the recruitment of immune cells, and changes in adaptive immunity. In this context, neutrophils have a significant role in the control of a S. pneumoniae infection via their ability to rapidly engulf bacteria in phagosomes.14,15 The phagosomes then mature further upon fusion with preformed granules. Together with reactive oxygen species (ROS) generated by NADPH oxidase, the granule contents (proteases, defensins, and antimicrobial peptides) directly interfere with the pathogen's survival in the phagosomal vacuole.16
Acute microbial infections induce a host phenomenon known as emergency granulopoiesis, which maintains the supply of neutrophils to the infected tissue.17 Neutrophils are derived from progenitor cells in the bone marrow (BM). Studies in the mouse have provided a description of the successive steps leading to the generation of mature neutrophils.18,19 The granulocyte/monocyte progenitors (GMPs) develop into granulocyte progenitors (GPs), which in turn further differentiate locally into immature and then mature neutrophils. Mature neutrophils subsequently leave the BM, enter the bloodstream, and circulate as quiescent cells at homeostasis. Upon infection, circulating neutrophils migrate to the infected tissue, where they encounter bacteria and become fully activated. Emergency granulopoiesis is a complex and tightly regulated process involving interleukins, chemokines, transcription factors, and differentiation factors (primarily granulocyte colony-stimulating factor [G-CSF]).20
Given the major role of neutrophils in S. pneumoniae clearance, we looked at whether AMX treatment alters neutrophil development and function during pneumonia. We established a murine model of IAV-S. pneumoniae (IAV/Sp) superinfection and found that AMX treatment was associated with a greater number of mature neutrophils in the BM. These neutrophils had a distinct transcriptomic profile and distinct functional responses.
2. Methods
2.1 Bacterial strains and cultures
Serotype 2 S. pneumoniae (D39V strain) was obtained from J.W. Veening Laboratory (University of Lausanne, Switzerland).21 Fresh colonies grown on blood agar plates were incubated in Todd Hewitt Yeast Broth (Merck) at 37 °C until the OD600nm reached 0.7 to 0.9 units. Cultures were stored at −80 °C in Todd Hewitt Yeast Broth medium containing 12% glycerol (vol./vol.) for up to 3 mo. For intranasal infection, working stocks were thawed, washed twice in phosphate-buffered saline (PBS) (Thermo Fisher Scientific), and diluted to adjust the concentration.
2.2 Mouse model of infection and AMX treatment
Seven- to 10-week-old male wild-type C57BL/6J were purchased from Janvier Labs. CX3CR1gfp/gfp homozygous mice (The Jackson Laboratory) were crossed with C57BL/6J mice to generate heterozygous CX3CR1gfp/+ progeny. CX3CR1gfp/+ were used for an optimal characterization by flow cytometry of myeloid cell populations and their progenitors in the BM. Mice were maintained in ventilated cages in a biosafety level 2 facility in the Animal Resource Center at the Institut Pasteur de Lille. Housing and experiments were carried out according to institutional regulations and ethical guidelines (E59-350 009, Institut Pasteur de Lille; Protocol APAFIS 16966-20180531141769v3). A primary infection with the murine-adapted H3N2 IAV Scotland/20/74 was performed to sensitize the mice to S. pneumoniae infection. Briefly, mice were anesthetized by intraperitoneal injection of 1.25 mg of ketamine and 0.25 mg of xylazine in 250 μL of PBS and then intranasally infected with 30 μL of PBS containing 70 plaque-forming units of IAV. Seven days later, mice were infected (or not) intranasally with 5 × 104 colony-forming units (CFUs) of S. pneumoniae in 30 μL of PBS. Twelve hours postinfection, mice were intragastrically treated with 150 μg of AMX (Merck) in 200 μL water or left untreated. To evaluate bacterial counts in lung and spleen, mice were sacrificed at selected times using intraperitoneal injection of 11.4 mg of sodium pentobarbital in 200 μL of PBS. Tissues were collected in sterile PBS and homogenized with an UltraTurrax homogenizer (IKA-Werke). Viable bacteria were determined by plating serial dilutions onto blood agar plates and incubating them at 37 °C for 24 h. Survival and body weight were monitored daily after infections.
2.3 In vivo model of inflammation
Ultrapure lipopolysaccharide (LPS) from Escherichia coli 0111: B4 strain (5 μg/mouse; Invitrogen) was administered by the intraperitoneal route (200 μL) 12 h post-AMX treatment. Five hours later, femurs, blood, lungs, and peritoneal fluid were collected and cell suspensions were prepared for flow cytometry analysis.
2.4 Real-time polymerase chain reaction assays
Total RNA from lungs or BM neutrophils were extracted with the Nucleospin RNA Plus kit (Macherey-Nagel) and reverse-transcribed with the High-Capacity cDNA Archive kit (Applied Biosystems). The complementary DNA was amplified using SYBR green–based real-time polymerase chain reaction (PCR) on a QuantStudio 12 K PCR system (Applied Biosystems). The primers used were designed using National Center for Biotechnology Information and are listed in Table 1. Bm2 and Actb were used as housekeeping genes. The relative messenger RNA (mRNA) level was determined (1) by normalizing the cycle thresholds (Cts) for the gene of interest to the housekeeping genes (Bm2 and Actb) and (2) the values of group of interest were compared with the values of control group (=2−ΔΔCt). Genes with Ct values >35 cycles were considered nondetectable.
Sequences of the primers used for quantitative PCR assays.
| Target gene | Forward primer (F) | Reverse primer (R) |
|---|---|---|
| B2m | TGGTCTTTCTGGTGCTTGTC | GGGTGGCGTGAGTATACTTGAA |
| Actb | CGTCATCCATGGCGAACTG | GCTTCTTTGCAGCTCCTTCGT |
| Il6 | GTTCTCTGGGAAATCGTGGAAA | AAGTGCATCATCGTTGTTCATACA |
| Il1b | AATCTATACCTGTCCTGTGTA | TGGGTATTGCTTGGGATCCA |
| Tnf | CATCTTCTCAAAATTCGAGTGACAA | CCTCCACTTGGTGGTTTGCT |
| S100a9 | CACCCTGAGCAAGAAGGAAT | TGTCATTTATGAGGGCTTCATTT |
| Cxcl2 | CCCTCAACGGAAGAACCAAA | CACATCAGGTACGATCCAGGC |
| Csf3 | CCTGGAGCAAGTGAGGAAGA | CAGCTTGTAGGTGGCACACA |
| Lcn2 | CCATCTATGAGCTACAAGAGAACAAT | TCTGATCCAGTAGCGACAGC |
| S100a8 | ATCCTTTGTCAGCTCCGTCTT | TCTCCAGTTCAGACGGCATT |
| Mmp8 | AACGGGAAGACATACTTCTTCATAA | GGGTCCATGGATCTTCTTTG |
| Target gene | Forward primer (F) | Reverse primer (R) |
|---|---|---|
| B2m | TGGTCTTTCTGGTGCTTGTC | GGGTGGCGTGAGTATACTTGAA |
| Actb | CGTCATCCATGGCGAACTG | GCTTCTTTGCAGCTCCTTCGT |
| Il6 | GTTCTCTGGGAAATCGTGGAAA | AAGTGCATCATCGTTGTTCATACA |
| Il1b | AATCTATACCTGTCCTGTGTA | TGGGTATTGCTTGGGATCCA |
| Tnf | CATCTTCTCAAAATTCGAGTGACAA | CCTCCACTTGGTGGTTTGCT |
| S100a9 | CACCCTGAGCAAGAAGGAAT | TGTCATTTATGAGGGCTTCATTT |
| Cxcl2 | CCCTCAACGGAAGAACCAAA | CACATCAGGTACGATCCAGGC |
| Csf3 | CCTGGAGCAAGTGAGGAAGA | CAGCTTGTAGGTGGCACACA |
| Lcn2 | CCATCTATGAGCTACAAGAGAACAAT | TCTGATCCAGTAGCGACAGC |
| S100a8 | ATCCTTTGTCAGCTCCGTCTT | TCTCCAGTTCAGACGGCATT |
| Mmp8 | AACGGGAAGACATACTTCTTCATAA | GGGTCCATGGATCTTCTTTG |
Sequences of the primers used for quantitative PCR assays.
| Target gene | Forward primer (F) | Reverse primer (R) |
|---|---|---|
| B2m | TGGTCTTTCTGGTGCTTGTC | GGGTGGCGTGAGTATACTTGAA |
| Actb | CGTCATCCATGGCGAACTG | GCTTCTTTGCAGCTCCTTCGT |
| Il6 | GTTCTCTGGGAAATCGTGGAAA | AAGTGCATCATCGTTGTTCATACA |
| Il1b | AATCTATACCTGTCCTGTGTA | TGGGTATTGCTTGGGATCCA |
| Tnf | CATCTTCTCAAAATTCGAGTGACAA | CCTCCACTTGGTGGTTTGCT |
| S100a9 | CACCCTGAGCAAGAAGGAAT | TGTCATTTATGAGGGCTTCATTT |
| Cxcl2 | CCCTCAACGGAAGAACCAAA | CACATCAGGTACGATCCAGGC |
| Csf3 | CCTGGAGCAAGTGAGGAAGA | CAGCTTGTAGGTGGCACACA |
| Lcn2 | CCATCTATGAGCTACAAGAGAACAAT | TCTGATCCAGTAGCGACAGC |
| S100a8 | ATCCTTTGTCAGCTCCGTCTT | TCTCCAGTTCAGACGGCATT |
| Mmp8 | AACGGGAAGACATACTTCTTCATAA | GGGTCCATGGATCTTCTTTG |
| Target gene | Forward primer (F) | Reverse primer (R) |
|---|---|---|
| B2m | TGGTCTTTCTGGTGCTTGTC | GGGTGGCGTGAGTATACTTGAA |
| Actb | CGTCATCCATGGCGAACTG | GCTTCTTTGCAGCTCCTTCGT |
| Il6 | GTTCTCTGGGAAATCGTGGAAA | AAGTGCATCATCGTTGTTCATACA |
| Il1b | AATCTATACCTGTCCTGTGTA | TGGGTATTGCTTGGGATCCA |
| Tnf | CATCTTCTCAAAATTCGAGTGACAA | CCTCCACTTGGTGGTTTGCT |
| S100a9 | CACCCTGAGCAAGAAGGAAT | TGTCATTTATGAGGGCTTCATTT |
| Cxcl2 | CCCTCAACGGAAGAACCAAA | CACATCAGGTACGATCCAGGC |
| Csf3 | CCTGGAGCAAGTGAGGAAGA | CAGCTTGTAGGTGGCACACA |
| Lcn2 | CCATCTATGAGCTACAAGAGAACAAT | TCTGATCCAGTAGCGACAGC |
| S100a8 | ATCCTTTGTCAGCTCCGTCTT | TCTCCAGTTCAGACGGCATT |
| Mmp8 | AACGGGAAGACATACTTCTTCATAA | GGGTCCATGGATCTTCTTTG |
2.5 Cytokine and growth factor quantification
Levels of interleukin (IL)-6 and G-CSF were quantified in sera using standardized sandwich enzyme-linked immunosorbent assay kits (BD) according to the manufacturer's recommendations.
2.6 Cell preparations
White blood cells were prepared from whole-blood samples obtained by retro-orbital puncture. Briefly, blood was collected in 1.5 mL tube containing heparin and 150 μL of sample was next processed. Red blood cells were removed using a lysis buffer (Pharmlyse; BD Biosciences). Lungs were perfused with PBS, excised, and finely minced, followed by enzymatic digestion for 20 min at 37 °C in PBS containing 1 mg/mL type VIII collagenase (Sigma-Aldrich) and 1 μg/mL DNase type I (Sigma-Aldrich). After red blood cell lysis, lung cell homogenates were suspended in a 20% Percoll and centrifuged at 2000 rpm without brake at room temperature for 12 min. Cell pellets were washed with PBS supplemented with 2% Fetal Calf Serum (FCS), penicillin (10 units/mL), and streptomycin (100 ug/mL) (penicillin/streptomycin) and then filtrated. BM cells were isolated using a centrifugation-based method as previously described.22 Then, red blood cells were lysed and cell suspensions were washed and filtered. Peritoneal cells were obtained by injecting 5 mL of ice-cold sterile PBS into the peritoneal cavity. After abdominal massage, the cell suspension was collected and centrifuged for 10 min at 1400 rpm. The cell suspensions were finally resuspended in PBS/2% FCS/penicillin/streptomycin/0.01% NaN3 before antibody (Ab) labelling. The total number of cells was determined by trypan blue exclusion with a Corning Cell Counter (Life Sciences).
2.7 Reagents and Abs for flow cytometry analysis
Monoclonal Abs (mAbs) against mouse CD3 (biotin conjugated), NK1.1 (biotin conjugated), Ter119 (biotin conjugated), CD45R/B220 (biotin conjugated), CD117 (Brilliant Violet [BV] 711 conjugated), Sca1 (APC conjugated), CD34 (PE conjugated), CD16/CD32 (PE-Cy7 conjugated), Ly6G (BV711 or APC conjugated), CD11b (BV785 conjugated), Ly6C or Gr1 (Percp/Cy5.5 conjugated), Siglec-F (PE/CF594 or APC conjugated), CD45 (BV510, PE, APC, APC-Cy7 or Alexa Fluor 700 conjugated), CD115 (BV421 conjugated), CXCR2 (BV510 or PE conjugated), CXCR4 (APC conjugated), CD101 (PE conjugated), CD62L (PE-conjugated), Ki-67 (APC conjugated), and BV605-conjugated streptavidin were purchased from BioLegend, BD Biosciences, or eBioscience (Table 2). Lineage markers (Lin) include CD3, NK1.1, Ter119, CD11b, CD45R/B220, and Ly6G. Propidium iodide was purchased from BioLegend.
List of monoclonal antibodies used for flow cytometry.
| Antibody (fluorochrome[s]) | Clone | Supplier |
|---|---|---|
| Anti-mouse CD3 (biotin) | 145-2C11 | BioLegend |
| Anti-mouse NK1.1 (biotin) | PK136 | BioLegend |
| Anti-mouse Ter119 (biotin) | TER-119 | BioLegend |
| Anti-mouse CD45R/B220 (biotin) | RA3-6B2 | BioLegend |
| Anti-mouse CD117 (BV711) | 2B8 | BioLegend |
| Anti-mouse Sca1/Ly-6A/E (APC) | D7 | BioLegend |
| Anti-mouse CD34 (PE) | RAM34 | BD Biosciences |
| Anti-mouse CD16/CD32 (PE-Cy7) | 2.4G2 | BD Biosciences |
| Anti-mouse Ly6G (BV711 or APC) | 1A8 | BioLegend |
| Anti-mouse CD11b (BV785) | M1/70 | BioLegend |
| Anti-mouse Gr1 (Percp-Cy5.5) | RB6-8C5 | BioLegend |
| Anti-mouse Ly6C (Percp-Cy5.5) | HK1.4 | BioLegend |
| Anti-mouse Siglec F (PE-CF594) | E50-2440 | BD Biosciences |
| Anti-mouse Siglec F (APC) | S17007I | BioLegend |
| Anti-mouse CD45 (BV510, PE, APC, APC-Cy7, or AF700) | 30-F11 | BioLegend |
| Anti-mouse CD115 (BV421) | T38-320 | BioLegend |
| Anti-mouse CXCR2 (BV510) | V48-2310 | BD Biosciences |
| Anti-mouse CXCR2 (PE) | SA044G4 | BioLegend |
| Anti-mouse CXCR4 (APC) | 2B11 | BD Biosciences |
| Anti-mouse CD101 (PE) | Moushi101 | eBioscience |
| Anti-mouse CD62L (PE) | MEL-14 | eBioscience |
| Anti-mouse Ki-67 (APC) | 11-F6 | BioLegend |
| Antibody (fluorochrome[s]) | Clone | Supplier |
|---|---|---|
| Anti-mouse CD3 (biotin) | 145-2C11 | BioLegend |
| Anti-mouse NK1.1 (biotin) | PK136 | BioLegend |
| Anti-mouse Ter119 (biotin) | TER-119 | BioLegend |
| Anti-mouse CD45R/B220 (biotin) | RA3-6B2 | BioLegend |
| Anti-mouse CD117 (BV711) | 2B8 | BioLegend |
| Anti-mouse Sca1/Ly-6A/E (APC) | D7 | BioLegend |
| Anti-mouse CD34 (PE) | RAM34 | BD Biosciences |
| Anti-mouse CD16/CD32 (PE-Cy7) | 2.4G2 | BD Biosciences |
| Anti-mouse Ly6G (BV711 or APC) | 1A8 | BioLegend |
| Anti-mouse CD11b (BV785) | M1/70 | BioLegend |
| Anti-mouse Gr1 (Percp-Cy5.5) | RB6-8C5 | BioLegend |
| Anti-mouse Ly6C (Percp-Cy5.5) | HK1.4 | BioLegend |
| Anti-mouse Siglec F (PE-CF594) | E50-2440 | BD Biosciences |
| Anti-mouse Siglec F (APC) | S17007I | BioLegend |
| Anti-mouse CD45 (BV510, PE, APC, APC-Cy7, or AF700) | 30-F11 | BioLegend |
| Anti-mouse CD115 (BV421) | T38-320 | BioLegend |
| Anti-mouse CXCR2 (BV510) | V48-2310 | BD Biosciences |
| Anti-mouse CXCR2 (PE) | SA044G4 | BioLegend |
| Anti-mouse CXCR4 (APC) | 2B11 | BD Biosciences |
| Anti-mouse CD101 (PE) | Moushi101 | eBioscience |
| Anti-mouse CD62L (PE) | MEL-14 | eBioscience |
| Anti-mouse Ki-67 (APC) | 11-F6 | BioLegend |
List of monoclonal antibodies used for flow cytometry.
| Antibody (fluorochrome[s]) | Clone | Supplier |
|---|---|---|
| Anti-mouse CD3 (biotin) | 145-2C11 | BioLegend |
| Anti-mouse NK1.1 (biotin) | PK136 | BioLegend |
| Anti-mouse Ter119 (biotin) | TER-119 | BioLegend |
| Anti-mouse CD45R/B220 (biotin) | RA3-6B2 | BioLegend |
| Anti-mouse CD117 (BV711) | 2B8 | BioLegend |
| Anti-mouse Sca1/Ly-6A/E (APC) | D7 | BioLegend |
| Anti-mouse CD34 (PE) | RAM34 | BD Biosciences |
| Anti-mouse CD16/CD32 (PE-Cy7) | 2.4G2 | BD Biosciences |
| Anti-mouse Ly6G (BV711 or APC) | 1A8 | BioLegend |
| Anti-mouse CD11b (BV785) | M1/70 | BioLegend |
| Anti-mouse Gr1 (Percp-Cy5.5) | RB6-8C5 | BioLegend |
| Anti-mouse Ly6C (Percp-Cy5.5) | HK1.4 | BioLegend |
| Anti-mouse Siglec F (PE-CF594) | E50-2440 | BD Biosciences |
| Anti-mouse Siglec F (APC) | S17007I | BioLegend |
| Anti-mouse CD45 (BV510, PE, APC, APC-Cy7, or AF700) | 30-F11 | BioLegend |
| Anti-mouse CD115 (BV421) | T38-320 | BioLegend |
| Anti-mouse CXCR2 (BV510) | V48-2310 | BD Biosciences |
| Anti-mouse CXCR2 (PE) | SA044G4 | BioLegend |
| Anti-mouse CXCR4 (APC) | 2B11 | BD Biosciences |
| Anti-mouse CD101 (PE) | Moushi101 | eBioscience |
| Anti-mouse CD62L (PE) | MEL-14 | eBioscience |
| Anti-mouse Ki-67 (APC) | 11-F6 | BioLegend |
| Antibody (fluorochrome[s]) | Clone | Supplier |
|---|---|---|
| Anti-mouse CD3 (biotin) | 145-2C11 | BioLegend |
| Anti-mouse NK1.1 (biotin) | PK136 | BioLegend |
| Anti-mouse Ter119 (biotin) | TER-119 | BioLegend |
| Anti-mouse CD45R/B220 (biotin) | RA3-6B2 | BioLegend |
| Anti-mouse CD117 (BV711) | 2B8 | BioLegend |
| Anti-mouse Sca1/Ly-6A/E (APC) | D7 | BioLegend |
| Anti-mouse CD34 (PE) | RAM34 | BD Biosciences |
| Anti-mouse CD16/CD32 (PE-Cy7) | 2.4G2 | BD Biosciences |
| Anti-mouse Ly6G (BV711 or APC) | 1A8 | BioLegend |
| Anti-mouse CD11b (BV785) | M1/70 | BioLegend |
| Anti-mouse Gr1 (Percp-Cy5.5) | RB6-8C5 | BioLegend |
| Anti-mouse Ly6C (Percp-Cy5.5) | HK1.4 | BioLegend |
| Anti-mouse Siglec F (PE-CF594) | E50-2440 | BD Biosciences |
| Anti-mouse Siglec F (APC) | S17007I | BioLegend |
| Anti-mouse CD45 (BV510, PE, APC, APC-Cy7, or AF700) | 30-F11 | BioLegend |
| Anti-mouse CD115 (BV421) | T38-320 | BioLegend |
| Anti-mouse CXCR2 (BV510) | V48-2310 | BD Biosciences |
| Anti-mouse CXCR2 (PE) | SA044G4 | BioLegend |
| Anti-mouse CXCR4 (APC) | 2B11 | BD Biosciences |
| Anti-mouse CD101 (PE) | Moushi101 | eBioscience |
| Anti-mouse CD62L (PE) | MEL-14 | eBioscience |
| Anti-mouse Ki-67 (APC) | 11-F6 | BioLegend |
2.8 Flow cytometry and cell sorting
Briefly, 3 to 5 × 106 cells were plated in 96-well plates, resuspended in 50 µL of the appropriate combination of Abs and incubated on ice for 20 min. For Ki-67 labeling, after extracellular staining, cells were fixed and permeabilized using the Foxp3 Fixation/Permeabilization kit (Invitrogen) and incubated for 20 min at room temperature with anti-Ki67 mAb. After the last wash, cell populations (CD45+ cells) were analyzed with a BD LSR Fortessa (BD Biosciences) and the FlowJo software v10 (TreeStar). Cell subsets were identified as follows: for GMPs, Lin−ckit+sca1−/+CD34+CD16/CD32highCX3CR1−Ly6C−CD115−; for GPs, Lin−ckit+sca1−/+CD34+CD16/CD32highCX3CR1−Ly6C+CD115− and for total neutrophils, Ly6G+CD11b+CX3CR1− (Siglec-F− for the lung) composed in the BM of immature (CXCR2lowCD101−) and mature (CXCR2highCD101+) neutrophils. In some experiments, dead cells were excluded by propidium iodide staining. BM mature neutrophils (Ly6G+CD11b+CX3CR1−CXCR2high) were sorted using a FACSAria III cell sorter (BD Biosciences), and this protocol yielded >98% cell purity as evaluated by fluorescence-activated cell sorting.
2.9 Intravascular labelling of lung neutrophils
Mice were injected with 5 μg of APC-labeled rat anti-CD45 mAb via the retro-orbital venous plexus. The mAb was allowed to circulate for 5 min in order to label all leukocytes in the vascular space including circulating and marginated leukocytes.23 After perfusion, lungs were removed and leukocytes were prepared as previously described. Cell suspensions were then labeled with appropriate mAbs for neutrophil detection and BV510-labeled anti-CD45 mAb to stain all leukocytes. Marginated neutrophils were defined as double positive for CD45-APC and CD45-BV510.
2.10 Phagocytosis and ROS production
Production of ROS was assessed using the CellROX Deep Red Reagent (Invitrogen) and flow cytometry. Briefly, BM cells (5 × 105/mL) were resuspended in RPMI 1640 supplemented with 2% FCS and incubated or not with TBHP (400 μM) at 37 °C for 15 min. Then, CellROX (500 nM) was added and cells incubated at 37 °C for 30 min. After washes, cells were labelled with appropriate Abs to identify mature neutrophils. SYTOX blue reagent was added to exclude dead cells. As negative control, BM cells were incubated with NAC (1 mM) for 30 min at 37 °C before treatment with TBHP. Analysis of the Median Fluorescence Intensity (MFI) provides a semiquantitative assessment of ROS production. To assess phagocytosis capacity, BM cells (2 × 105/mL) were plated in 96-well plates and resuspended with the pHrodo Red bioparticles (killed E. coli) (Invitrogen) for 1 h at 37 °C 5%CO2. As a control, some BM cells were incubated with bioparticles at 4 °C. After washes, BM cells were labeled with appropriate Abs for mature neutrophil identification. The percentages of mature BM neutrophils positive for E. coli-pHrodo as well as the MFI were quantified by flow cytometry.
2.11 RNA sequencing analysis
Total RNA from 5 × 106 mature neutrophils was extracted and RNA quality was evaluated by spectrophotometry (Nanodrop) and RIN with the TapeStation 4200 (Agilent Technologies). Libraries were prepared with the QIAseq stranded mRNA library kit (Qiagen), according to the manufacturer's protocol. All libraries were controlled by TapeStation 4200 and quantified by quantitative PCR (KAPA Library Quantification Kit for Illumina platforms; KapaBiosystems). Libraries were normalized and pooled in an equimolar way, before sequencing on a NovaSeq sequencer (Illumina) in 2 × 150 bp. RNA sequencing (RNA-seq) data in the form of FASTQ files were subsequently mapped to the mouse genome build (GRCm39) using the STAR (STAR-2.7.10a) alignment software. The mapped reads were then annotated using GENCODE M30 gtf file. Raw counts were used to perform a differentially expressed gene (DEG) analysis using DESeq2 (R version 4.2.1; R Foundation for Statistical Computing). Genes with a false discovery rate <0.05 were considered as differentially regulated and used to generate volcano plots. Overrepresentation analysis through Gene Ontology (GO) enrichment (GO Biological Process 2021 and GO Cellular Component 2021) of DEGs was performed using Enrichr.24 The data have been deposited in National Center for Biotechnology Information’s Gene Expression Omnibus 53 and are accessible through GEO Series accession number GSE232689 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE232689).
2.12 Mass spectrometry proteomic analysis
Mature BM neutrophils (5 × 105) were collected for the proteomic analysis. The sorted cells were washed twice in PBS at 200 g for 3 min. Pellets were resuspended in 20 μL of TruPAGE loading buffer containing dithiothreitol (Sigma-Aldrich). Samples were stored at −20 °C up until use. Liquid chromatography tandem mass spectrometry was performed using UltiMate 3000 RSLCnano System (Thermo Fisher Scientific) for peptide elution, as previously described.25 Peptides were identified using Proteome Discoverer Software (v.1.4) (Thermo Fisher Scientific). Mass spectra were searched against UniProtKB database (taxonomy 10090, April 2022, 88,037 entries) restricted to Mus musculus using the Mascot search engine (version 2.4.0; Matrix Science). Mass tolerance was set to 0.02 Da for precursor ions and 10 ppm for fragment ions. One tryptic miscleavage was considered as well as chemical modifications of cysteine (carbamidomethylation and propionamidation), methionine (oxidation), and protein N-terminal acetylation. The identification results were validated using ProLine software v2.0. Peptide spectrum >9 residues and ion scores >10 were retained. Only peptides with a false discovery rate <1% were considered for analysis (using Mascot Modified Mudpit score). Spectra counts were quantified with Proline 2.0. The data have been deposited to the ProteomeXchange Consortium via the PRIDE26 partner repository with the dataset identifier PXD042496 and 10.6019/PXD042496.
2.13 Statistical analysis
All statistical analysis were performed using GraphPad Prism software (GraphPad Software v9). Unpaired t test or 1-way analysis of variance followed by Bonferroni's post hoc t test were used for statistical analysis when 2 groups or multiple groups were analyzed, respectively. When data did not follow a Gaussian distribution, the statistical significance was evaluated using nonparametric Mann-Whitney or Kruskal-Wallis (followed by a Dunn’s posttest). We used 2-way analysis of variance when multiple parameters were studied. The survival of mice was analyzed using the Kaplan-Meier method and a log-rank test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
3. Results
3.1 Administration of a single dose of AMX protects against S. pneumoniae superinfection
In order to test the efficacy of a single, intragastric dose of AMX, we set up a murine model of pneumococcal superinfection in C57BL/6 and CX3CR1gfp/+ mice, used in flow cytometry to track myeloid lineage cell subpopulations. Animals were infected with a sublethal dose of IAV, followed 7 d later by 5 × 104 CFUs of S. pneumoniae (Fig. 1A). In untreated mice, the bacterial load increased gradually over time and reached 1.8 × 107 CFUs per lung 24 h after the pneumococcal infection (Fig. 1B and C). High numbers of bacteria were also detected in the spleen 24 h after inoculation with S. pneumoniae, suggesting that bacteria disseminate from the lungs into the blood (Fig. 1C). In the antibiotic-treated group, animals were administered with a single, intragastric dose of AMX (150 μg per mouse, 7.5 mg/kg) 12 h after the bacterial infection. This time point was defined as the t0 for all analyses (Fig. 1A). With this regimen, the serum AMX concentration peaked at 2.88 μg/mL 30 min after administration, fell rapidly, and became undetectable at 8 h (Supplementary Fig. 1). Treatment with AMX gradually decreased the bacterial loads in the lung and spleen, and the bacteria were cleared almost completely 12 h after the start of treatment (Fig. 1B and C). Treatment with AMX was associated with a significant higher survival rate, relative to infected, untreated mice (Fig. 1D). Taken as a whole, these data indicate that both strains of mice developed similar pneumococcal superinfections and the selected AMX dose of treatment was effective in terms of reducing the bacterial load and promoting survival.
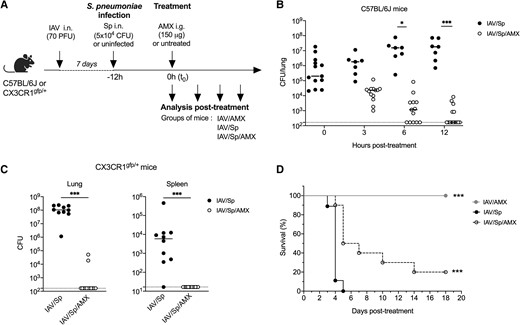
A single dose of AMX protects against Streptococcus pneumoniae (Sp) superinfection. (A) C57BL/6J or CX3CR1gfp/+ mice were infected intranasally (i.n.). with IAV and 7 days later with 5 × 104 CFUs of S. pneumoniae (Sp). Mice were treated intragastrically (i.g.) with AMX 12 h after Sp infection (defined as the t0) (IAV/Sp/AMX) or left untreated (IAV/Sp). As a control group, mice only infected with IAV were also treated with AMX (IAV/AMX). Lungs (B, C) and spleen (C) were collected at different times posttreatment in C57Bl/6 mice (B) or at 12 h posttreatment in CX3CR1gfp/+ mice (C), homogenized, and plated in serial dilutions onto blood agar plates to measure the bacterial load. Counts for individual mouse are shown as CFUs per tissue. The solid line represents the median value and the dashed line represents the detection threshold. Data depict pooled results from 2 independent experiments, with each symbol representing an individual mouse (n = 6–14). (B) * P < 0.05 and *** P < 0.001 (2-way analysis of variance test). (C) *** P < 0.001 (Mann-Whitney). (D) The survival rate of C57Bl/6J mice infected with IAV only or IAV/Sp treated or not with AMX was monitored daily for 18 d. Data were compared in a log-rank test (***P < 0.001). PFU = plaque forming units.
3.2 Administration of AMX reduces the inflammatory response during S. pneumoniae infection
We next evaluated the effect of AMX treatment on the production of various innate immunity–related factors, including the inflammatory cytokines IL-6, IL-1β, and tumor necrosis factor (TNF); the chemokine CXCL2; the growth factor G-CSF; and the antimicrobial peptide S100A9. To this end, lungs and sera of superinfected animals treated (or not) with AMX were collected at various posttreatment time points. Levels of mRNA in the lungs and levels of cytokine/growth factor proteins in the blood were quantified and normalized against those measured in animals infected with IAV only and treated with AMX. We found that in the absence of AMX treatment, transcription of the Il6, Il1b, Tnf, S100a9, Cxcl2, and Csf3 genes increased over time after infection with S. pneumoniae (Fig. 2A). The serum concentrations of IL-6 and G-CSF were also higher at the 18-h time point (Fig. 2B). At t0 (12 h postinfection), IAV/Sp and IAV-only mice did not differ significantly with regard to mRNA levels of innate immune system components or the serum IL-6 level (Supplementary Fig. 2A and B). The serum G-CSF concentration was slightly higher in IAV/Sp animals than in IAV-only animals (Supplementary Fig. 2B). AMX treatment of infected animals (i.e. IAV/Sp/AMX) abrogated the expression of inflammatory gene transcripts and systemic IL-6 and G-CSF production at all posttreatment timepoints (Fig. 2A and B); this finding shows that AMX treatment efficiently killed S. pneumoniae and prevented local and systemic inflammatory responses.
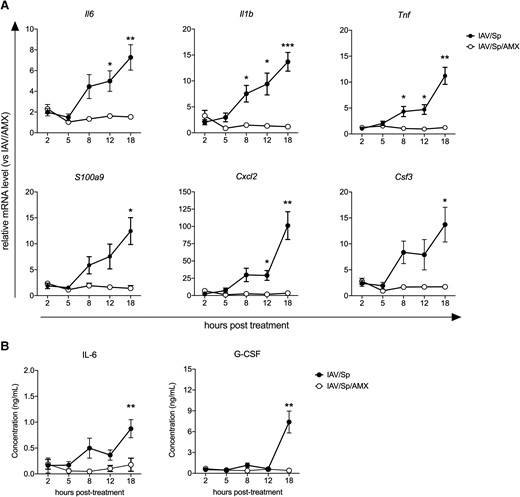
AMX treatment of infected animals reduces the inflammatory response. C57BL/6J mice were infected with Sp 7 days post-IAV infection and treated with AMX 12 h after Sp infection (IAV/Sp/AMX) or left untreated (IAV/Sp). Lungs and blood were collected at different time points posttreatment. (A) Relative expression of selected genes in the lungs was assessed by real-time quantitative PCR. For each gene, mRNA level is normalized to the reference genes Actb and B2m, and then to the control group IAV/AMX. (B) IL-6 and G-CSF concentrations in the sera were measured by enzyme-linked immunosorbent assay. Data represent pooled results from 2 independent experiments (n = 10) and are expressed as mean ± SEM (*P < 0.05, **P < 0.01 and ***P < 0.001; 2-way analysis of variance test).
3.3 AMX treatment promotes the maturation of BM neutrophils
The host's control of S. pneumoniae pneumonia depends on the rapid recruitment of neutrophils to the lungs (i.e. a process regulated [at least in part] by inflammatory molecules like IL-1β, TNF, CXCL1/2, and S100A9).14,27–29 In order to establish whether AMX-associated inhibition of local and systemic inflammation affects neutrophil mobilization, we therefore determined the numbers of lung and blood neutrophils in animals treated with AMX. At t0 (12 h postinfection), the neutrophil count in the lungs (but not in the blood) was slightly higher in superinfected animals than in animals infected with IAV only (Supplementary Fig. 2C). Twelve hours later, significant increases in lung and blood neutrophil counts were observed in IAV/Sp animals (Fig. 3A). Treatment with AMX fully abolished neutrophil recruitment to the lungs but not in the blood, compared with untreated animals (Fig. 3A). We found that fewer than half of the lung neutrophils were located in the vasculature after S. pneumoniae infection and that AMX treatment did not influence their localization (Fig. 3B).
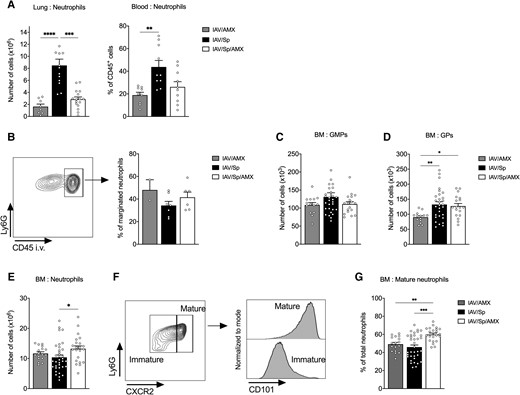
AMX treatment of infected animals promotes BM neutrophil maturation. CX3CR1gfp/+ mice were infected with Sp 7 days post-IAV infection and treated with AMX 12 h after Sp infection (IAV/Sp/AMX) or left untreated (IAV/Sp). As a control, mice only infected with IAV were treated with AMX (IAV/AMX). (A) Absolute number of lung neutrophils and frequency of blood neutrophils were assessed by flow cytometry 12 h posttreatment. Mean ± SEM of 3 experiments are shown (n = 9–16). (B) A representative fluorescence-activated cell sorting dot plot of CD45 in vivo labeling of lung neutrophils (left). Mean ± SEM of the percentage of CD45+ lung neutrophils (marginated pool) are shown (n = 6–7). Absolute number of BM GMPs (C) and GPs (D) were quantified by flow cytometry. (E) Absolute number of BM neutrophils. (F) Gating strategy for mature neutrophils in the BM according to Ly6G, CXCR2, and CD101 expression and (G) frequency of mature BM neutrophils among total neutrophils. Mean ± SEM of biological replicates from at least 3 independent experiments are represented (n = 14–35) (*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; Kruskal-Wallis test).
Acute mobilization of neutrophils in infected tissues requires the rapid production of these cells in the BM. We therefore investigated the potential effects of secondary pneumococcal infection and AMX treatment on the de novo generation of BM neutrophils. Of note, S. pneumoniae was not detected in BM cells of superinfected animals treated or not with AMX (Supplementary Fig. 3A). Using the gating strategy depicted in Supplementary Fig. 3B, we quantified the numbers of neutrophil progenitors (i.e. GMPs and GPs), immature neutrophils, and mature neutrophils in the BM of untreated and AMX-treated superinfected animals (Fig. 3C and D and Supplementary Fig. 2D). Before the administration of AMX (t0), the IAV/Sp and IAV groups were similar in terms of GMP, GP, total neutrophil, and mature neutrophil counts (Supplementary Fig. 2D). The absolute GMP count did not significantly change upon bacterial infection and AMX treatment (Fig. 3C). In contrast, the GP count significantly increased upon infection with S. pneumoniae (Fig. 3D). Interestingly, AMX treatment did not modify the GP count relative to untreated superinfected animals and remained high relative to animals infected with IAV only. Whereas the total neutrophil count did not change significantly upon infection with S. pneumoniae, AMX treatment was associated with an increase in this variable (Fig. 3E). It is noteworthy that as reported elsewhere, the proportion of proliferating neutrophils was much lower than the proportion of neutrophil progenitors (Supplementary Fig. 3C).18,30 Furthermore, AMX treatment did not influence the proportion of Ki-67+ proliferating GMPs, GPs, and neutrophils (Supplementary Fig. 3C). Mature neutrophils were then identified based on the expression of CXCR2 and CD101; the latter marker was recently found to be specific for this cell population (Fig. 3F).18 Interestingly, treatment with AMX significantly enhanced the proportion of mature neutrophils in BM, relative to untreated superinfected animals or IAV-infected animals (Fig. 3G). Taken as a whole, these data indicate that the AMX treatment of secondary pneumococcal infection influences the maturation of neutrophils in the BM.
3.4 Mature BM neutrophils display specific transcriptional and proteomic profiles during the AMX treatment of superinfection
To gain further insights into the biology of mature BM neutrophils, we conducted RNA-seq analysis of Ly6G- and CXCR2-expressing cells from superinfected animals treated (or not) with AMX for 12 h (Fig. 4). A total of 617 DEGs were identified in BM neutrophils from AMX-treated animals: 206 were upregulated and 411 were downregulated, relative to BM neutrophils from untreated animals (Fig. 4A). To further elucidate the relation between DEGs and function, we used Enrichr to perform a GO enrichment analysis. Analysis of the biological processes revealed that AMX treatment significantly enriched genes related to neutrophil activation and degranulation and the production/regulation of cytokines (e.g. Il6) (Fig. 4B and Supplementary Table 1). DEGs were also significantly enriched in cellular components related to granules and phagocytic or secretory vesicles (Fig. 4C and Supplementary Table 2). AMX treatment was associated with lower expression of genes associated with pathogen recognition (Tlr4, Tlr7, Myd88, and Cd14), vesicle transport (Scamp1 and Atp11a), inflammation (Mmp8, S100a8, Adam8, Lrg1, and Lcn2), oxidative stress (Nos2 and Qsox1), and immune regulation (Cd47 and Crispld2) (Fig. 4D–F and Supplementary Fig. 4A). Some of the downregulated genes were further selected for confirmation by real-time quantitative PCR (Supplementary Fig. 4B). With regard to cellular components, AMX treatment was associated with downregulation of Clec4e (coding for a phagocytic C-type lectin) and the RAS-related Rab genes Rab23, Rab20, and Rab31 (known to be crucial for phagosome maturation) (Fig. 4F).
![AMX treatment alters the transcriptional profile of mature BM neutrophils. CX3CR1gfp/+ mice were infected with Sp 7 days post-IAV infection and treated with AMX 12 h after Sp infection (IAV/Sp/AMX) or left untreated (IAV/Sp). Mice were sacrificed 12 h posttreatment. Mature BM neutrophils were sorted on the basis of Ly6G and CXCR2 expression, and RNA was purified for subsequent RNA-seq analysis. (A) Volcano plot showing –log10 (adjusted P value) and log2 (fold change) for DEGs in AMX-treated animals compared with untreated mice. Each data point represents a gene. The genes with a significant log2 (fold change) and an adjusted P value <0.05 are located above the dashed horizontal line with down-regulated genes on the left and up-regulated genes on the right. (B, C) Enrichment analysis of GO on identified DEGs using Enrichr. The top 10 significantly enriched GO terms of the target genes in the biological processes and cellular components are shown. Heat maps showing row-scaled expression (Log2 ([old change]) of the highest DEGs associated with the biological process “neutrophil activation involved in the immune response” (D), all DEG related to the biological process “regulation of IL-6 production” (E), and the cellular components “phagocytic vesicle” (F). Data represent biological triplicates, each replicate being a pool of 3 mice per group.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jleukbio/115/3/10.1093_jleuko_qiad125/1/m_qiad125f4.jpeg?Expires=1750186646&Signature=ajoWLyPq1TW~CYlRNDlNfrGueptvfLfSJ1-qbu~84EE4m-TnY22Z0lvFWGrrss5jOfG807EWWa8XPpCU~hZdKXrTwqryy27R0dQrVvN5FFEkUbtr0~KCXZzj3RVXXuJohuoYd2wLbiUmRv3iJQn~nCWJLzRr5zZFdLeMEY~gIO~-p5O3HL2BgsSFQ0wTe6JOG9JEJ0c-sivEn0kqnv6Fjy8o4~Imo8RvWsZ1UX1eGKQXShcA2P~2aaTDkCupx4qjhPIAl~h1XZo6nfiyBujVY0ZlV4Ay54XFmNdtbapVsYg92OBlyKntv2QFRImm9fV4lPF5158d0wsthFNaEc4owA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AMX treatment alters the transcriptional profile of mature BM neutrophils. CX3CR1gfp/+ mice were infected with Sp 7 days post-IAV infection and treated with AMX 12 h after Sp infection (IAV/Sp/AMX) or left untreated (IAV/Sp). Mice were sacrificed 12 h posttreatment. Mature BM neutrophils were sorted on the basis of Ly6G and CXCR2 expression, and RNA was purified for subsequent RNA-seq analysis. (A) Volcano plot showing –log10 (adjusted P value) and log2 (fold change) for DEGs in AMX-treated animals compared with untreated mice. Each data point represents a gene. The genes with a significant log2 (fold change) and an adjusted P value <0.05 are located above the dashed horizontal line with down-regulated genes on the left and up-regulated genes on the right. (B, C) Enrichment analysis of GO on identified DEGs using Enrichr. The top 10 significantly enriched GO terms of the target genes in the biological processes and cellular components are shown. Heat maps showing row-scaled expression (Log2 ([old change]) of the highest DEGs associated with the biological process “neutrophil activation involved in the immune response” (D), all DEG related to the biological process “regulation of IL-6 production” (E), and the cellular components “phagocytic vesicle” (F). Data represent biological triplicates, each replicate being a pool of 3 mice per group.
Interestingly, one of the upregulated genes (Itgax, encoding the integrin X chain protein CD11c) was recently identified as being essential for the regulation of neutrophil maturation in the BM (Fig. 4A and D).31 AMX treatment was associated with greater expression of genes related to innate immunity (Unc93b1, Camp, Ltf, and Mpeg1), endocytosis/phagocytosis (Creg1, Tcirg1, and Hvcn1), and the regulation of inflammation (Cd300a, Fgl2, Trpm2, Zc3h12a, and Ptpn22) (Fig. 4D–F). A semiquantitative proteomics analysis of mature BM neutrophils identified a number of proteins modulated by AMX treatment (Supplementary Fig. 4C). Some of the downregulated proteins were related to cytoskeleton remodeling (WAS protein, RHOA, and VAV) or neutrophil activity (NCF4 [an NADPH oxidase subunit crucial for ROS production], galectin-9, and Rab31); Rab31 was also identified in the RNA-seq analysis. Some of the proteins upregulated by AMX treatment were linked with the complement system and the coagulation cascade. Our results show that during a superinfection, AMX mainly downregulates activation-related genes, but also upregulates phagocytosis/endocytosis- and regulation-associated genes in mature BM neutrophils.
3.5 AMX treatment impacts the activation and functions of mature BM neutrophils
We next sought to establish whether or not AMX treatment of superinfected animals influenced the activation and effector functions of mature BM neutrophils. To this end, we used flow cytometry to measure the expression of markers associated with cell activation, phagocytosis and ROS production in mature neutrophils among a sample of total BM cells harvested from superinfected animals treated (or not) with AMX (Fig. 5). Treatment with AMX enhanced the expression of CXCR2 and CD62L on mature BM neutrophils, whereas CD11b expression was lower than in untreated animals; this indicated a less activated/primed phenotype (Fig. 5A). Using E. coli bioparticles conjugated to pHrodo, we showed that AMX treatment did not change the proportion of mature BM neutrophils capable of phagocytosing particles in vitro but did significantly enhance the MFI of pHrodo in mature BM neutrophils from AMX-treated animals (Fig. 5B and Supplementary Fig. 5A). It is noteworthy that incubation of neutrophils with E. coli particles at 4 °C, when the phagocytosis is blocked, drastically decreased the percentage of pHrodo positive cells and the MFI of pHrodo to the same extent in the 2 groups (Supplementary Fig. 5A). This finding suggests that mature BM neutrophils from AMX-treated animals have a higher phagocytosis capacity. Basal levels of ROS production were similar in mature BM neutrophils from superinfected mice treated with AMX and those not treated with AMX (Fig. 5C). In contrast, mature BM neutrophils from AMX-treated animals that had been activated ex vivo with tert-butyl hydroperoxide released less ROS than untreated animals did (Fig. 5C and Supplementary Fig. 5B). Overall, our data confirmed that AMX treatment significantly impacted the phenotype and function of mature BM neutrophils.
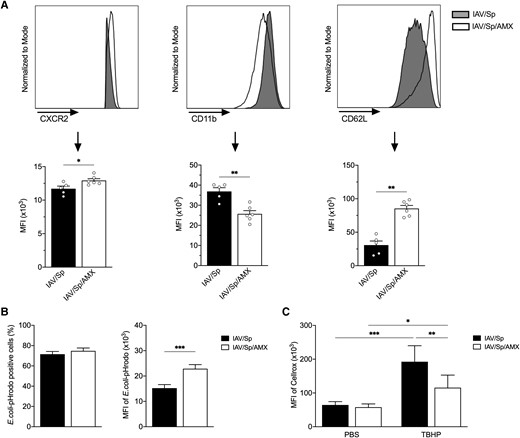
AMX treatment impacts the activation state and functions of mature BM neutrophils. CX3CR1gfp/+ mice were infected with Sp 7 days post-IAV infection and treated with AMX 12 h after Sp infection (IAV/Sp/AMX) or left untreated (IAV/Sp). Mice were sacrificed 12 h post-AMX treatment. (A) The MFIs of CXCR2, CD11b, and CD62L expression on mature BM neutrophils were analyzed by flow cytometry. Representative histograms (top) and mean ± SEM of 1 experiment representative of 3 (lower) are shown (n = 5–6). (B) Total BM cells were incubated with pHrodo E. coli bioparticles and the percentage of mature BM neutrophils positive for E. coli pHrodo (left) as well as the MFI of E.coli pHrodo (right) were quantified by flow cytometry. Data represent pooled results from 3 independent experiments (n = 13) (***P < 0.001; Mann-Whitney). (C) Total BM cells were treated with TBHP (or PBS) and then incubated with CellROX Deep Red Reagent. Results are expressed as mean ± SEM of CellROX MFI of 1 experiment representative of 2 (n = 5–6) (*P < 0.05, **P < 0.01, ***P < 0.001; 2-way analysis of variance).
3.6 Mature BM neutrophils conditioned by AMX treatment are rapidly mobilized in response to systemic inflammation
Because AMX treatment modified the mature BM neutrophils’ transcriptional program and phenotypic and functional characteristics, we next looked at whether or not the antibiotic influenced neutrophil mobilization during acute inflammation. To this end, LPS was administered intraperitoneally into superinfected animals treated (or not) with AMX (Fig. 6A). A rapid drop in the proportion of total BM neutrophils and the complete disappearance of mature neutrophils was observed in both groups 5 h after LPS stimulation—suggesting the egress of neutrophils from the BM (Fig. 6B and C). In both groups of mice, the fall in the BM neutrophil count was associated with the rapid accumulation of neutrophils in the peritoneum (Fig. 6D). It has been reported previously that the intraperitoneal administration of LPS in naive animals promotes the rapid accumulation of neutrophils in the lungs.32,33 Interestingly, LPS challenge was associated with a higher percentage of lung neutrophils in AMX-treated animals and a significantly lower percentage in untreated animals (Fig. 6E). Thus, AMX-conditioned BM neutrophils were rapidly mobilized to peripheral tissues in response to the systemic administration of LPS. A phenotypic analysis of neutrophils that had migrated into the peritoneum after the LPS challenge indicated that LPS promoted the activation of neutrophils in both AMX-treated and untreated superinfected animals, as demonstrated by elevated surface levels of CD11b and lower levels of CD62L and CXCR2 expression (Fig. 6F).34 In both groups of mice, LPS also enhanced the expression of CXCR4, a marker of aged neutrophils.35,36 Importantly, surface CXCR4 expression was higher on neutrophils from AMX-treated animals. Our results suggest that AMX treatment influences neutrophil aging in a context of systemic inflammation.
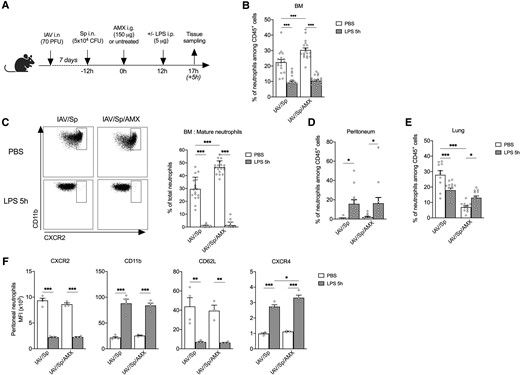
AMX treatment promotes neutrophil recruitment in tissues following administration of LPS in vivo. (A) CX3CR1gfp/+ mice were infected with Sp 7 days post-IAV infection and treated with AMX 12 h after S. pneumoniae infection (IAV/Sp/AMX) or left untreated (IAV/Sp). Then, in each group, animals were injected intraperitoneally (or not) with 5 μg LPS. Mice were sacrificed 5 h later. (B) The percentage of total BM neutrophils among CD45+ population was quantified by flow cytometry. (C) Mature BM neutrophils (CXCR2high) were quantified among total neutrophils in the BM. Neutrophils were quantified in the peritoneum (D) and the lung (E). Mean ± SEM of biological replicates from 3 independent experiments are represented (n = 11–20). (F) The phenotype of neutrophils in the peritoneum was evaluated by flow cytometry. Mean ± SEM of biological replicates of 1 experiment representative of 3 are shown (n = 4) (*P < 0.05, ** P < 0.01, *** P < 0.001; 2-way analysis of variance). PFU = plaque forming units.
4. Discussion
Characterization of the complex relationship between β-lactams and host immunity during the treatment of infection might help to improve treatments. Given the neutrophils’ dominant role in clearing S. pneumoniae infection, we looked at whether or not AMX treatment of pneumococcal pneumonia in vivo influenced neutrophil development and function. Our study is the first to have shown that AMX treatment of pneumonia influenced emergency granulopoiesis in the BM and led to the generation of mature neutrophils with distinct antimicrobial functions. Importantly, these AMX-conditioned mature neutrophils were mobilized rapidly in response to systemic inflammation.
Previous research had shown that neutrophil depletion or delayed neutrophil recruitment during S. pneumoniae pneumonia was associated with higher bacterial loads and a higher mortality rate—highlighting the neutrophils’ critical role in the elimination of bacteria.14 By using a murine model of pneumococcal postinfluenza superinfection, we showed that (1) neutrophils accumulated in the lungs and blood and (2) S. pneumoniae triggered significant innate defense mechanisms in the highly inflammatory context of viral infection.37,38 The pool of neutrophils in the BM did not change in superinfected animals. However, a concomitant enhancement of emergency granulopoiesis, characterized by a greater number of proliferating (Ki-67+) GPs, was observed. This finding is in line with the previously reported emergency granulopoiesis during S. pneumoniae infection, which restores the neutrophil reservoir in the BM.29
We next hypothesized that curative AMX treatment of ongoing pneumonia affects neutrophils by modulating their development, migration, and/or functions. The reduced bacterial burden upon AMX treatment was associated with a lower pulmonary pro-inflammatory response and lung neutrophil count, a hallmark most likely due to the direct effect of AMX on S. pneumoniae. In contrast, AMX treatment did not affect emergency granulopoiesis in the BM: the GP count was the same as in untreated animals. More importantly, the number of mature BM neutrophils was higher upon AMX treatment. Our results suggest that AMX treatment does not interfere with emergency granulopoiesis caused by S. pneumoniae but does reduce the mobilization of mature neutrophils from the BM into the lungs. It is not clear how AMX treatment maintains emergency granulopoiesis. Because no S. pneumoniae could be detected in the BM of animals treated (or not) with AMX, an indirect mode of action for AMX could be postulated. Indeed, these effects were visible when AMX was no longer detectable in the bloodstream (≥8 h). Earlier in vitro and in vivo studies had shown that when exposed to β-lactams, S. pneumoniae can rapidly release the cell wall components teichoic and lipoteichoic acids and the virulence factor pneumolysin.39–41 Further studies will be needed to establish whether AMX-exposed bacteria in the lungs of superinfected animals release bacterial compounds that might trigger BM hematopoietic cells and skew differentiation toward the myeloid lineage.42 We cannot completely rule out a local effect of proinflammatory cytokines (e.g. IL-6 or IFN-γ) or differentiation factors (e.g. G-CSF) upon AMX treatment.43–45
A large body of evidence shows that neutrophils are phenotypically heterogeneous and functionally versatile.46 In response to infection, neutrophils switch from a resting state to a primed or activated state—an essential requirement for the cells’ functions. Our analysis of the mature BM neutrophils’ phenotype highlighted the presence of a less fully activated/primed population in AMX-treated animals. These results are in line with the weak ability of AMX-conditioned mature BM neutrophils to produce ROS upon stimulation. The downregulation of genes related to pathogen recognition (Tlr4, Tlr7, and Myd88), inflammatory responses (Mmp8, S100a8, and Lcn2), and ROS production (Nos2 and Qsox1), and the low observed protein level of the NADPH oxidase subunit NCF4,47 further support our conclusion. However, we were surprised to observe an increase in the ability of AMX-conditioned mature BM neutrophils to phagocyte bacterial particles. This finding was in line with the upregulation of genes related to the phagocytic/endocytic pathway (Creg1, Tcirg, and Hvcn1) and proteins of the complement and coagulation systems.48 On one hand, this is suggestive of greater expression of phagocytosis-related receptors/molecules on AMX-conditioned mature BM neutrophils. On the other hand, the downregulation of genes and proteins related to cytoskeleton remodeling, vesicle transport, and phagosome maturation suggest the impairment of cytoplasmic granule transport and fusion with phagosomes—a prerequisite for neutrophil killing activity49,50—in AMX-conditioned mature BM neutrophils. This apparent discrepancy might be due to a lower rate of phagosomal content digestion in AMX-treated mature BM neutrophils, which would increase the number of bacteria in the phagosomes. The cells’ less primed/activated state is in line with this hypothesis. However, it remains to be determined whether AMX treatment affects the initial steps in phagocytosis and/or phagosome maturation in mature BM neutrophils.
AMX-treated mature BM neutrophils are also endowed with distinct migratory capabilities. In response to systemic administration of LPS, mature neutrophils from AMX-treated animals exited the BM and migrated toward the peritoneum and the lungs. This finding is in line with the rapid mobilization of neutrophils in peripheral tissues upon LPS administration previously reported in naive animals.32,33 Mature BM neutrophils from untreated animals only migrated toward the peritoneum. The low neutrophil count in the lungs might have been due to either local apoptosis of neutrophils in superinfected animals or, as has been described previously, reverse migration from the lungs to the peritoneum upon LPS activation.51 It remains to be established whether or not the lung functions as a reservoir of primed/activated neutrophils in superinfected animals (i.e. a reservoir that can be mobilized to distant tissues on demand). Endotoxemia also induced a significant elevation in the number of neutrophils with an aged phenotype, characterized by enhanced expression of CD11b and CXCR4, in the peritoneum of mice, regardless of treatment with AMX.35,36 Aged neutrophils are rapidly mobilized under inflammatory conditions and have stronger effector functions (e.g. phagocytosis and neutrophil extracellular trap formation) than young neutrophils.36,52 Importantly, the higher expression of CXCR4 on peritoneal neutrophils upon LPS treatment suggests that AMX enhances retention of these cells at the site of inflammation, which would render aged neutrophils more efficient in situ. After completing their functions, the CXCR4high neutrophils traffic back to the BM and are cleared; this physiological phenomenon is important for maintaining immunological homeostasis.53,54
To conclude, we showed that the AMX treatment of a bacterial lung infection can rapidly influence in vivo neutrophil development and functions within the BM. We now intend to elucidate the mechanisms by which AMX reprograms neutrophil progenitors and/or mature neutrophils in the BM and determine how these changes are associated with a therapeutic effect during bacterial pneumonia. This knowledge should facilitate the development of novel antimicrobial drugs and new treatment strategies that harness the host-pathogen interface.
Acknowledgments
We thank Drs. Anne Rogel and Laurye Van Maele for critical reading of the manuscript. We thank the UMS 2014—US 41—PLBS, Lille, and more specifically the BioImaging Center Lille (BICeL) for access to equipment, the animal core facilities for technical assistance and the platforms GO@L and PAGés-P3M for transcriptomic and proteomic analysis, respectively. MM was a fellow of the Innovation Pharmaceutique et Recherche program.
Author contributions
Conceptualization, C.F., J.-C.S., and C.C.; Investigation, M.M., D.S., C.F., B.H., D.B., and J.-M.S.; Data curation: M.M., Y.Z., C.F., and J.-M.S.; Formal analysis, M.M., Y.Z., B.H., C.F., and J.-M.S.; Resources, J.-C.S., C.C., and C.F.; Validation, M.M., C.F., Y.Z., and J.-M.S.; Supervision, C.F.; Writing—original draft: M.M. and C.F.; Writing—review & editing: M.M., C.F., J.-C.S., and C.C.
Supplementary material
Supplementary materials are available at Journal of Leukocyte Biology online.
Funding
C.C. was funded by Université de Lille. Y.Z., J.-C.S., and C.F. were funded by INSERM. B.H. was funded by Lille University Hospital. D.S., J.-M.S., and D.B. were funded by the Institut Pasteur de Lille. The work is supported by the INSERM, Institut Pasteur de Lille, Université de Lille, and the project FAIR that received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 847786, and the project BIP from the Agence Nationale de la Recherche (ANR-19-CE18-0030-02).
References
Author notes
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}