





Abstract
Chemokine–chemokine receptor (CKR) interactions are traditionally described by a two-step/two-site mechanism that details the major contact points between chemokine ligands and CKRs leading to ligand recognition and receptor activation. Chemokine recognition site 1 (CRS1) encompasses interactions between the CKR N-terminus and the globular chemokine core. Chemokine recognition site 2 (CRS2) includes interactions between the unstructured chemokine N-terminus and the binding pocket of the receptor. The two-step/two-site paradigm has been an adequate framework to study the intricacies of chemokine:CKR interactions, but emerging studies highlight the limitations of this model. Here, we present studies of CRS2 interactions between the chemokine CCL20 and its cognate receptor CCR6 driven by the hypothesis that CCL20 interacts with CCR6 as described by the two-step/two-site model. CCL20 is a chemokine with an unusually short N-terminus of 5 residues (NH2-ASNFD), compared to the average length of 10 residues for chemokine ligands. We have investigated how well CCL20 tolerates manipulation of the N-terminus by monitoring binding affinity of variants and their ability to activate the receptor. We show the CCL20 N-terminus tolerates truncation of up to 3 residues, extension by up to 5 additional residues, and point mutations at 4 of 5 positions with minimal loss of binding affinity and minimal impairment in ability to stimulate calcium mobilization, inositol triphosphate accumulation, chemotaxis, and β-arrestin-2 recruitment. Mutation of the fifth residue, aspartate, to alanine or lysine has a dramatic impact on binding affinity for CCR6 and ligand potency. We postulate CCL20 does not activate CCR6 through the canonical two-step/two-site mechanism of CKR activation.
Systematic structure-function analysis of the CCL20 N-terminus reveals extreme tolerance of N-terminal extension, truncation, or modification to maintain activity at CCR6.
1 INTRODUCTION
Chemokines are small, secreted proteins that interact with cell-surface G protein-coupled receptors (GPCRs) to promote directed cellular migration. Chemokines play a role in orchestrating the complex and specific movement of cells for homeostatic functions like embryonic and lymphoid development and play a crucial role in the immune response. Dysregulation of chemokine signaling has been implicated in many disease states, such as tumor metastasis, HIV infection, and autoimmune disorders.1 Chemokines are promiscuous in nature such that a single chemokine ligand often interacts with multiple chemokine receptors (CKRs) and vice versa.2 The promiscuity of the family is not entirely redundant, because chemokine signaling is regulated by spatial and temporal expression of ligands. Additionally, different ligands have been shown to elicit diverse signaling outcomes at the same receptor.3 Few chemokine/CKR pairs act in a nonpromiscuous fashion, including CCL20 and CCR6.4
CCL20 is an ∼8-kDa chemokine ligand that regulates T cell trafficking to sites of injury or infection via binding and activation of CCR6.1,2 In psoriatic skin, CCL20 expression by keratinocytes and endothelial cells is up-regulated to promote migration of CCR6-expressing immune cells into the epidermis.5,6 Mice deficient in CCR6 were highly resistant to IL-23-induced psoriasis-like inflammation compared to their wild type (WT) counterparts, and a neutralizing anti-CCL20 mAb had a similar protective effect.7,8 The CCL20/CCR6 signaling axis is a promising target as a novel psoriasis therapeutic, but a lack of structure-function information presents a challenge in development of a highly specific CCL20/CCR6 therapy.9
Chemokine–CKR interactions have been well studied for other family members, resulting in the establishment of a paradigm to describe these interactions. This paradigm, referred to as the two-step/two-site model of interaction, separates the chemokine–CKR interaction sites both spatially and functionally. The first site of interaction, chemokine recognition site 1 (CRS1), involves interactions between the chemokine core and the CKR N-terminus; ligand specificity is generally ascribed to these interactions. The second site of interaction occurs between the chemokine N-terminus and the CKR transmembrane (TM) core. This site, chemokine recognition site 2 (CRS2), is a key for activation of the receptor.10
The understanding of chemokine–CKR interactions has grown exponentially with the publication of three crystal structures and one model of distinct chemokine–CKR complexes. The crystal structures of CX3CL1 bound to the viral chemokine receptor US28,11 the viral chemokine vMIP-II bound to CXCR4,12 and the chemokine antagonist [5P7]CCL5 bound to CCR5,13 and the model of the CXCL12:CXCR4 complex14 provide an extraordinarily detailed look at the chemokine–CKR interface, including those contacts defined as CRS2 interactions and buried within the receptor binding pocket. These models, however, in addition to emerging functional studies, challenge the simplicity of the two-step, two-site paradigm.15
The two-step/two-site paradigm still proves useful since chemokine ligands adopt a well-conserved tertiary structure. Despite this conserved structure, chemokine ligands share low sequence identity. In particular, the N-termini of ligands are highly variable in both sequence and length. More than 50% of chemokine ligands have an N-terminus length of 8 residues or greater, with an average length of approximately 10 residues. The CCL20 N-terminus is a unique case as it is just 5 residues long (NH2-ASNFD). A wealth of structure-function studies show chemokine N-termini are crucial for receptor activation in the canonical model of chemokine–CKR interactions. Truncations of the N-terminus have been shown to turn chemokine ligands into potent antagonists,10,16 while chemical modifications of the N-terminus of CCL5 have been thoroughly studied to show many variant-specific outcomes.17
To our knowledge, no systematic structure-function studies have been performed on the CCL20/CCR6 signaling axis. However, two variants of CCL20 that differ in their N-terminus sequence have been reported: CCL20 (1-70) and CCL20 (2-70).18 The shorter form arises from an in-frame deletion of three base pairs from the signal peptide sequence, which affects the location of signal peptide cleavage. One group reported no significant difference in the chemotactic potency of CCL20 (1-70) and CCL20 (2-70), while a second group reported full-length CCL20 to be 2.5-fold more efficacious than the shorter form.18,19 CCL20 also contains an interesting N-terminal DCCL motif, wherein the first two conserved cysteines of CCL20 are flanked by aspartate and leucine (Asp5-Cys6-Cys7-Leu8). This motif is found in 3 other human chemokines—CCL19, CCL21, and CCL25—and has been postulated to be important for receptor activation.20,21
To gain insight into CCL20 interactions with CCR6 at chemokine recognition site 2, we report the investigation of 19 recombinant CCL20 variants designed to probe the importance of the CCL20 N-terminus length, by extension and truncation, and composition, by point mutation. We investigate chemokine activity by calcium mobilization, chemotaxis, inositol triphosphate (IP3) accumulation, and β-arrestin-2 recruitment studies, and chemokine binding by radioligand competition assay, and combine the functional analysis of the CCL20 N-terminus with modeling data to propose key interactions between CCL20 and CCR6. We show that CCL20 tolerates manipulation of its N-terminus with minimum impairment to ligand potency as long as Asp5 is present. Our findings diverge from the two-step/two-site paradigm and support further investigation of the CCL20/CCR6 signaling axis to better understand this unusual interaction.
2 MATERIALS AND METHODS
2.1 CCL20 extension and truncation variants
CCL20 extension (A1, A2, A3, A4, A5) and truncation (Δ1, Δ2, Δ3, Δ3AA) variants were generated from full length CCL20 cDNA with PCR-based cloning to introduce or remove residues. CCL20 point mutation (A1K, A1D, A1G, S2A, N3A, F4A, SNF/AAA, D5A, D5N, D5K) variants were generated by site-directed mutagenesis using complementary primers and the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). Mutations of CCL20 were confirmed by DNA sequencing.
2.2 Recombinant protein expression and purification
CCL20 WT and variant plasmids were expressed and purified as previously described.22 Briefly, Escherichia coli cells were lysed by French Press and centrifuged for 30 min at 18,000 × g. The insoluble fraction was resuspended in a 6 M guanidine buffer and nickel chromatography purified. Eluted protein was refolded drop-wise into a 100 mM Tris pH 8.0 buffer containing 10 mM cysteine and 0.5 mM cystine. The 6HIS-SUMO tag was cleaved by recombinant ULP1 to ensure a native N-terminus, and cation exchange chromatography was used to separate protein from cleaved tag. Protein elution from cation exchange was HPLC purified and lyophilized. Protein purity and molecular weight were confirmed by SDS-PAGE and direct injection MS.
2.3 In vitro mammalian cell culture
Jurkat cells were stably transfected with pORF-hCCR6 plasmid (InvivoGen, San Diego, CA, USA) using Lipofectamine 2000 protocol and reagents from ThermoFisher Scientific (Waltham, MA, USA). Cells expressing hCCR6 were selected by cell sorting using PE anti-mouse CCR6 Ab (BioLegend, San Diego, CA, USA). COS-7 cells were transiently transfected with the pCDNA3.1(+) CCR6 vector by calcium phosphate precipitation approximately 40 h prior to use. Cells were additionally co-transfected with Gqi4myr, a large G protein chimera with the Gαi recognition interface and a Gαq output, for IP3 accumulation assays. The PathHunter U2OS-A2 β-arrestin-2 cell line (DiscoverX, Fremont, CA, USA) was transfected with CCR6 vector using FuGENE® 6 approximately 40 h prior to use in β-arrestin-2 recruitment assays. Cell lines were maintained as previously described.23
2.4 Radioligand displacement
125I-CCL20 WT was produced by oxidative iodination using ChloramineT (PerkinElmer) and purified by reverse-phase HPLC as previously described.24 Radioligand displacement assays were performed as previously described.23 Briefly, CCR6+ COS-7 cells were seeded at a density of 35,000 cells/well in 96-well plates. After 24 h, the cells were washed into a 50 mM HEPES buffer supplemented with 5 g/L BSA, then chilled to 4°C. Unlabeled CCL20 WT and variant ligands were added to the cells shortly before the 125I-CCL20 tracer. The cells were incubated at 4°C for 3 h, and then washed with a 50 mM HEPES buffer containing 0.5% (w/v) BSA and 0.5 M NaCl. Cells were lysed, transferred to shielded vials, and the gamma radiation of the lysate was directly quantitated.
2.5 IP3 accumulation assay
IP3 accumulation assays were performed as previously described.23 Briefly, COS-7 cells cotransfected with CCR6 and Gqi4myr were seeded into 96-well plates at 35,000 cells/well. Cells were maintained for 24 h in growth media supplemented with [3H]myo-inositol (5 μL/mL, 2 μCi/mL). Cells were incubated at 37°C with CCL20 ligands in a 10 mM LiCl solution in HBSS buffer. Cells were lysed with a 10 mM formic acid lysis solution after 90 min. Lysates were transferred to white, opaque 96-well plates, and mixed with a solution of agitated SPA-Ysi beads (80 μL/well, 12.5 mg/mL, PerkinElmer, Waltham, MA, USA). After 30 min shaking, the cells equilibrated for 8 h and scintillation was measured using a Packard Top Count NXT™ counter (PerkinElmer).
2.6 Calcium mobilization
Transfected CCR6+ Jurkat cells were washed twice in HBSS containing 20 mM HEPES (pH 7.4) and 0.2% (w/v) BSA. Cells were plated at a density of 2 × 106 cells/mL into 96-well half-area microplates (Corning, Corning, NY, USA) and incubated with FLIPR Calcium 6 dye (Molecular Devices, San Jose, CA, USA) at room temperature for 20 min, then at 37°C and 5% CO2 for 20 min. Chemokine dilutions were made in HBSS buffer with 20 mM HEPES (pH 7.4). Fluorescence at 515 nm was observed using a Flexstation 3 (Molecular Devices) for a total read time of 80 s, beginning 20 s before addition of chemokine.
2.7 Chemotaxis
Chemotaxis assays were performed as previously described.23 Briefly, chemokine dilutions were added to the lower well of HTS Transwell® 96-well plates with 5 μm pore size (Corning) and transfected CCR6+ Jurkat cells were added to the upper well at a density of 0.375 × 106 cells/mL. Plates were incubated at 37°C, 5% CO2 for 2 h. Migrated cells in the lower well were quantified with a BD™ LSR II flow cytometer and counts were normalized to maximum WT response, with the average of 3 replicates from 2 separate days presented as the mean ± sem.
2.8 β-Arrestin-2 recruitment
The β-arrestin recruitment assay was prepared as previously described.23 U2OS-A2 cells transfected with CCR6-PK1 were seeded into 96-well plates at 35,000 cells/well and incubated at 37°C, 5% CO2 for 24 h. Ligands were added to the cells and the PathHunter™ β-arrestin-2 recruitment assay was carried out according to the manufacturer’s instructions (DiscoverX).
2.9 Homology modeling
Homology modeling of CCR6 was performed using the RosettaCM protocol and the Rosetta protein modeling software suite.25,26 A total of 7 template structures were chosen based on availability of structural information and sequence similarity to human CCR6: human CCR5 (PDB ID: 4MBS), human CXCR4 (PDB ID: 4RWS), human CCR2 (PDB ID: 5T1A), human CCR9 (PDB ID: 5LWE), human Angiotensin II Receptor Type I (PDB ID: 4YAY), human Delta Opioid Receptor (PDB ID: 4RWA), and murine Mu Opioid Receptor (PDB ID: 4DKL). The sequences of the 7 template structures were aligned to human CCR6 (35-333) using Clustal Omega,27 then manually adjusted to eliminate gaps in the TM regions and align the conserved cysteine residue in extracellular loop 2 (ECL2). The human CCR6 sequence was obtained from UniProt (UniProtKB Accession Number: P51684).28 CCR6 fragments were generated by the Robetta full-chain protein structure prediction server29 and the TM topology of CCR6 was predicted using OCTOPUS.30 The RosettaCM method generated 5000 hybridized decoys that were each relaxed to the nearest local energy minimum twice to generate 10,000 total decoys. The 10 decoys with the lowest overall Rosetta energy score were visually inspected and 3 decoys were selected for further analysis based on the presence of structural characteristics known to be conserved in chemokine receptor structures.31 These 3 models were subjected to 300 ns all-atom molecular dynamics (MD) simulations using the Desmond software package.32
2.10 Sequence alignment of chemokine receptors
Structure-based sequence alignment was performed by GPCRdb.33 For key residues in ECL2 and ECL3, distance from the nearest conserved cysteine residue was used for alignment.
2.11 Data analysis and figure generation
Calcium flux, radioligand displacement, IP3 accumulation, and β-arrestin-2 recruitment data were analyzed and plots were generated using GraphPad Prism version 6 (GraphPad Software, La Jolla, CA, USA). EC50 and IC50 values were calculated by fitting a sigmoidal dose-response curve to the data (Table 1). Chemotaxis curves were graphed using the ggplot2 plotting system for R. Simple comparisons of means and standard deviations of data were made using a two-tailed, unpaired Student’s t-test. A P-value less than 0.05 is considered statistically significant. Images of the CCR6 homology model and chemokine–CKR complexes were generated using PyMOL.34
Calculated IC50 values for 125I-CCL20 WT displacement and calculated EC50 values for 3H-IP3 accumulation, calcium mobilization, and β-arrestin-2 recruitment of CCL20 WT and variants
| 125I-CCL20 WT displacement | 3H-IP3 accumulation | Calcium mobilization | β-arrestin-2 recruitment | |||||||||
| IC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | |
| CCL20 WT | 7.58 | –8.12 | 0.296 | –9.46 | 52.7 | –7.28 | 1.91 | –8.68 | ||||
| (0.12) | (0.12) | (0.02) | (0.17) | |||||||||
| (n = 7) | (n = 9) | (n = 6) | (n = 8) | |||||||||
| A5 | 69.4 | –7.16** | 9.2 | 1.98 | –8.70** | 6.7 | 365 | –6.44** | 6.9 | 12.4 | –7.91** | 6.5 |
| (0.04) | (0.14) | (0.07) | (0.22) | |||||||||
| (n = 3) | (n = 5) | (n = 5) | (n = 3) | |||||||||
| Δ3 | 93.2 | –7.03** | 12.3 | 6.75 | –8.17** | 22.8 | 358 | –6.47** | 6.8 | 11.8 | –7.93** | 6.2 |
| (0.07) | (0.17) | (0.07) | (0.26) | |||||||||
| (n = 3) | (n = 5) | (n = 5) | (n = 3) | |||||||||
| Δ3AA | 350 | –6.46** | 46.2 | 58.8 | –7.23** | 199 | 425 | –6.38** | 8.1 | 458 | –6.34** | 240 |
| (0.06) | (0.15) | (0.08) | (0.38) | |||||||||
| (n = 3) | (n = 5) | (n = 3) | (n = 3) | |||||||||
| D5A | 29.3 | –7.53** | 3.9 | 32.7 | –7.49** | 110 | 988 | –6.01** | 18.7 | 105 | –6.98** | 55.1 |
| (0.07) | (0.19) | (0.08) | (0.10) | |||||||||
| (n = 4) | (n = 4) | (n = 4) | (n = 3) | |||||||||
| D5K | 62.6 | –7.20** | 8.3 | 66.4 | –7.18** | 224 | 859 | –6.07** | 16.3 | 390 | –6.41** | |
| (0.05) | (0.17) | (0.05) | (0.11) | |||||||||
| (n = 4) | (n = 4) | (n = 6) | (n = 3) | |||||||||
| D5N | 8.67 | –8.06 | 1.1 | 3.83 | –8.42** | 13.0 | 139 | –6.86** | 2.6 | 1.59 | –8.80 | –1.2 |
| (0.08) | (0.20) | (0.04) | (0.12) | |||||||||
| (n = 4) | (n = 4) | (n = 5) | (n = 3) | |||||||||
| 125I-CCL20 WT displacement | 3H-IP3 accumulation | Calcium mobilization | β-arrestin-2 recruitment | |||||||||
| IC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | |
| CCL20 WT | 7.58 | –8.12 | 0.296 | –9.46 | 52.7 | –7.28 | 1.91 | –8.68 | ||||
| (0.12) | (0.12) | (0.02) | (0.17) | |||||||||
| (n = 7) | (n = 9) | (n = 6) | (n = 8) | |||||||||
| A5 | 69.4 | –7.16** | 9.2 | 1.98 | –8.70** | 6.7 | 365 | –6.44** | 6.9 | 12.4 | –7.91** | 6.5 |
| (0.04) | (0.14) | (0.07) | (0.22) | |||||||||
| (n = 3) | (n = 5) | (n = 5) | (n = 3) | |||||||||
| Δ3 | 93.2 | –7.03** | 12.3 | 6.75 | –8.17** | 22.8 | 358 | –6.47** | 6.8 | 11.8 | –7.93** | 6.2 |
| (0.07) | (0.17) | (0.07) | (0.26) | |||||||||
| (n = 3) | (n = 5) | (n = 5) | (n = 3) | |||||||||
| Δ3AA | 350 | –6.46** | 46.2 | 58.8 | –7.23** | 199 | 425 | –6.38** | 8.1 | 458 | –6.34** | 240 |
| (0.06) | (0.15) | (0.08) | (0.38) | |||||||||
| (n = 3) | (n = 5) | (n = 3) | (n = 3) | |||||||||
| D5A | 29.3 | –7.53** | 3.9 | 32.7 | –7.49** | 110 | 988 | –6.01** | 18.7 | 105 | –6.98** | 55.1 |
| (0.07) | (0.19) | (0.08) | (0.10) | |||||||||
| (n = 4) | (n = 4) | (n = 4) | (n = 3) | |||||||||
| D5K | 62.6 | –7.20** | 8.3 | 66.4 | –7.18** | 224 | 859 | –6.07** | 16.3 | 390 | –6.41** | |
| (0.05) | (0.17) | (0.05) | (0.11) | |||||||||
| (n = 4) | (n = 4) | (n = 6) | (n = 3) | |||||||||
| D5N | 8.67 | –8.06 | 1.1 | 3.83 | –8.42** | 13.0 | 139 | –6.86** | 2.6 | 1.59 | –8.80 | –1.2 |
| (0.08) | (0.20) | (0.04) | (0.12) | |||||||||
| (n = 4) | (n = 4) | (n = 5) | (n = 3) | |||||||||
P < 0.05,
P < 0.01 calculated by two-tailed unpaired Student’s t test.
Calculated IC50 values for 125I-CCL20 WT displacement and calculated EC50 values for 3H-IP3 accumulation, calcium mobilization, and β-arrestin-2 recruitment of CCL20 WT and variants
| 125I-CCL20 WT displacement | 3H-IP3 accumulation | Calcium mobilization | β-arrestin-2 recruitment | |||||||||
| IC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | |
| CCL20 WT | 7.58 | –8.12 | 0.296 | –9.46 | 52.7 | –7.28 | 1.91 | –8.68 | ||||
| (0.12) | (0.12) | (0.02) | (0.17) | |||||||||
| (n = 7) | (n = 9) | (n = 6) | (n = 8) | |||||||||
| A5 | 69.4 | –7.16** | 9.2 | 1.98 | –8.70** | 6.7 | 365 | –6.44** | 6.9 | 12.4 | –7.91** | 6.5 |
| (0.04) | (0.14) | (0.07) | (0.22) | |||||||||
| (n = 3) | (n = 5) | (n = 5) | (n = 3) | |||||||||
| Δ3 | 93.2 | –7.03** | 12.3 | 6.75 | –8.17** | 22.8 | 358 | –6.47** | 6.8 | 11.8 | –7.93** | 6.2 |
| (0.07) | (0.17) | (0.07) | (0.26) | |||||||||
| (n = 3) | (n = 5) | (n = 5) | (n = 3) | |||||||||
| Δ3AA | 350 | –6.46** | 46.2 | 58.8 | –7.23** | 199 | 425 | –6.38** | 8.1 | 458 | –6.34** | 240 |
| (0.06) | (0.15) | (0.08) | (0.38) | |||||||||
| (n = 3) | (n = 5) | (n = 3) | (n = 3) | |||||||||
| D5A | 29.3 | –7.53** | 3.9 | 32.7 | –7.49** | 110 | 988 | –6.01** | 18.7 | 105 | –6.98** | 55.1 |
| (0.07) | (0.19) | (0.08) | (0.10) | |||||||||
| (n = 4) | (n = 4) | (n = 4) | (n = 3) | |||||||||
| D5K | 62.6 | –7.20** | 8.3 | 66.4 | –7.18** | 224 | 859 | –6.07** | 16.3 | 390 | –6.41** | |
| (0.05) | (0.17) | (0.05) | (0.11) | |||||||||
| (n = 4) | (n = 4) | (n = 6) | (n = 3) | |||||||||
| D5N | 8.67 | –8.06 | 1.1 | 3.83 | –8.42** | 13.0 | 139 | –6.86** | 2.6 | 1.59 | –8.80 | –1.2 |
| (0.08) | (0.20) | (0.04) | (0.12) | |||||||||
| (n = 4) | (n = 4) | (n = 5) | (n = 3) | |||||||||
| 125I-CCL20 WT displacement | 3H-IP3 accumulation | Calcium mobilization | β-arrestin-2 recruitment | |||||||||
| IC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | EC50 (nM) | log M (± sem) | Fold Δ | |
| CCL20 WT | 7.58 | –8.12 | 0.296 | –9.46 | 52.7 | –7.28 | 1.91 | –8.68 | ||||
| (0.12) | (0.12) | (0.02) | (0.17) | |||||||||
| (n = 7) | (n = 9) | (n = 6) | (n = 8) | |||||||||
| A5 | 69.4 | –7.16** | 9.2 | 1.98 | –8.70** | 6.7 | 365 | –6.44** | 6.9 | 12.4 | –7.91** | 6.5 |
| (0.04) | (0.14) | (0.07) | (0.22) | |||||||||
| (n = 3) | (n = 5) | (n = 5) | (n = 3) | |||||||||
| Δ3 | 93.2 | –7.03** | 12.3 | 6.75 | –8.17** | 22.8 | 358 | –6.47** | 6.8 | 11.8 | –7.93** | 6.2 |
| (0.07) | (0.17) | (0.07) | (0.26) | |||||||||
| (n = 3) | (n = 5) | (n = 5) | (n = 3) | |||||||||
| Δ3AA | 350 | –6.46** | 46.2 | 58.8 | –7.23** | 199 | 425 | –6.38** | 8.1 | 458 | –6.34** | 240 |
| (0.06) | (0.15) | (0.08) | (0.38) | |||||||||
| (n = 3) | (n = 5) | (n = 3) | (n = 3) | |||||||||
| D5A | 29.3 | –7.53** | 3.9 | 32.7 | –7.49** | 110 | 988 | –6.01** | 18.7 | 105 | –6.98** | 55.1 |
| (0.07) | (0.19) | (0.08) | (0.10) | |||||||||
| (n = 4) | (n = 4) | (n = 4) | (n = 3) | |||||||||
| D5K | 62.6 | –7.20** | 8.3 | 66.4 | –7.18** | 224 | 859 | –6.07** | 16.3 | 390 | –6.41** | |
| (0.05) | (0.17) | (0.05) | (0.11) | |||||||||
| (n = 4) | (n = 4) | (n = 6) | (n = 3) | |||||||||
| D5N | 8.67 | –8.06 | 1.1 | 3.83 | –8.42** | 13.0 | 139 | –6.86** | 2.6 | 1.59 | –8.80 | –1.2 |
| (0.08) | (0.20) | (0.04) | (0.12) | |||||||||
| (n = 4) | (n = 4) | (n = 5) | (n = 3) | |||||||||
P < 0.05,
P < 0.01 calculated by two-tailed unpaired Student’s t test.
3 RESULTS
3.1 CCL20 N-terminus tolerates significant changes in length
Since short truncations of chemokine N-termini have been demonstrated to eliminate activity, the unusually short length of the CCL20 N-terminus raises questions about its role in activation of the receptor.10,35 Perhaps the lack of promiscuity observed in the CCL20/CCR6 system stems from a precise interaction between the short CCL20 N-terminus and a CCR6 orthosteric pocket unable to accommodate the lengthier N-termini of other chemokines. To probe the relevance of the CCL20 N-terminus length, 5 extension variants (introducing up to 5 additional alanine residues to the native N-terminus) and 3 truncation variants (removing up to 3 residues from the native N-terminus) of CCL20 were produced and tested in biochemical assays (Fig. 1). To monitor binding to CCR6, each variant was used to displace 125I-CCL20 WT bound to CCR6 on U2OS-A2 cells. All extension and truncation variants maintained binding to CCR6 with A5 and Δ3 displacing CCL20 WT with IC50 values of 69.4 and 93.2 nM, respectively, compared to WT IC50 of 7.58 nM (Fig. 2A, Table 1). Modification of the length of the CCL20 N-terminus produced appreciable affinity loss compared to WT protein with decreases in affinity occurring in a length-dependent fashion.

Sequences of generated CCL20 N-terminus variants. Conserved cysteine residues shaded grey
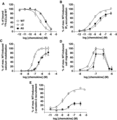
Biochemical characterization of CCL20 extension and truncation variants. CCL20 variant A5 introduced 5 alanine residues to the native N-terminus and variant Δ3 removed 3 residues from the native N-terminus. (A) CCL20 WT and variant protein binding was observed by displacement of 125I-CCL20 WT from CCR6+ transfected COS-7 cells. The IC50 values calculated for displacement by WT, A5, and Δ3 were calculated as 7.58 nM (n = 7), 69.4 nM (n = 3), and 93.2 nM (n = 3), respectively. (B) CCL20 WT, A5, and Δ3 stimulated accumulation of 3H-IP3 in CCR6+ COS-7 cells, with EC50 values calculated as 0.296 nM (n = 9), 1.98 nM (n = 4), and 6.75 nM (n = 4), respectively. (C) CCR6+ Jurkat cells treated with CCL20 WT or variants display a dose-dependent increase in intracellular calcium release. EC50 values were calculated as 52.7 nM (n = 6), 365 nM (n = 3), and 358 nM (n = 5) for WT, A5, and Δ3, respectively. (D) CCR6+ Jurkat cells migrate in a transwell assay in response to CCL20 WT, A5, and Δ3 treatment, with similar chemotactic profiles observed (n = 2). (E) CCL20 WT, A5, and Δ3 promote β-arrestin-2 recruitment to CCR6 in U2OS-A2 cells with calculated EC50 values of 1.91 nM (n = 8), 12.4 nM (n = 3), and 11.8 (n = 3), respectively. The maximum recruitment response of A5 and Δ3 stimulation was ∼30% of maximal WT response, indicating considerable ligand efficacy loss
Wild-type CCL20 activates CCR6 to promote numerous signaling outcomes including 3H-IP3 accumulation, calcium mobilization, β-arrestin-2 recruitment, and chemotaxis. To assess how changes in CCL20 N-terminus length affect downstream G protein signaling, 3H-IP3 accumulation in CCR6+ transfected COS-7 cells and intracellular calcium mobilization of CCR6+ transfected Jurkat cells were stimulated with CCL20 extension and truncation variants. All variants stimulated 3H-IP3 accumulation; A5 and Δ3 had approximately sevenfold (1.98 nM) and ∼23-fold (6.75 nM) increases in EC50, respectively, compared to WT EC50 of 0.296 nM (Fig. 2B, Table 1). Intracellular calcium mobilization was stimulated by all extension and truncation variants. A5 and Δ3 had approximately sevenfold (365 and 358 nM, respectively) increases in EC50 compared to WT EC50 of 52.7 nM (Fig. 2C, Table 1). In both assays, A5 and Δ3 experienced ∼20% reduction in maximum signaling compared to WT response. These data demonstrate extension or truncation of the CCL20 N-terminus was tolerated to maintain downstream G protein signaling through CCR6, but potency was lost in a length-dependent fashion. For all extension variants and the Δ1 and Δ2 truncation variants, the fold change in potency for the tested G protein signaling assays correlated with fold change in binding affinity (Table 1 and Supplementary Table 1), suggesting the observed potency losses were primarily due to loss in binding affinity rather than capacity of the ligand to activate CCR6.
To assess the chemotactic potency of the CCL20 extension and truncation variants, migration of CCR6+ Jurkat cells was assessed in a transwell assay. WT CCL20 stimulated maximum Jurkat cell migration at a concentration of 100 nM. Extension and truncation variants also stimulate CCR6+ Jurkat cell migration with no substantial change in the chemotactic profiles of A5 and Δ3 compared to WT CCL20 (Fig. 2D). At a concentration of 100 nM, A5 and Δ3 promoted migration of ∼20% fewer cells than observed for WT. These results illustrate large changes in N-terminus length do not dramatically alter the chemotactic potency of CCL20 as would have been expected for a ligand behaving according to the two-step/two-site paradigm.
Finally, to test how changes to the native CCL20 N-terminus length affect β-arrestin-2 recruitment, U2OS-A2 cells were stimulated with extension and truncation variants. All variants promoted β-arrestin-2 recruitment to CCR6, with calculated EC50 values of 12.4 and 11.8 nM for A5 and Δ3, respectively. These approximately sixfold increases in A5 and Δ3 EC50 compared to WT (1.91 nM) correlated with the observed fold changes in affinity for CCR6 (Table 1). In addition to potency loss, ligand efficacy was severely depressed for A5 and Δ3. These variants stimulated β-arrestin-2 recruitment to CCR6 at levels ∼70% lower than maximal WT response (Fig. 2E).
The loss of ligand potency observed for all 5 N-terminus extension variants and Δ1 and Δ2 truncation variants as measured by IP3 accumulation, calcium mobilization, and β-arrestin-2 recruitment correlated with the observed loss in ability to displace wild-type CCL20 from CCR6 (Table 1). The ∼23-fold increase in Δ3 EC50 as determined by IP3 accumulation assay was larger than the fold increase in IC50 of displacement (∼12-fold), but changes in EC50 for calcium mobilization and β-arrestin-2 recruitment assays correlated with loss of binding affinity (Table 1 and Supplementary Table 1). These results demonstrate that the high selectivity of the CCL20/CCR6 system is likely not due to a precise interaction between the short CCL20 N-terminus and a smaller than normal CCR6 orthosteric pocket. Extension of the CCL20 N-terminus led to impairment in ligand binding affinity for CCR6, but did not eliminate binding or activity outright. Furthermore, truncation of the CCL20 N-terminus by more than 2 residues had a severe impact on ligand affinity for and activity through CCR6, but removal of 3 out of 5 N-terminal residues was not sufficient to completely eliminate activity in any of the tested assays. These findings suggest the fourth and/or the fifth residue of the CCL20 N-terminus, Phe4 and Asp5, as crucial for CCL20 activity and, perhaps, CCL20/CCR6 specificity.
3.2 Asp5 of CCL20 is crucial for migration of CCR6+ cells
Point mutations within the chemokine N-terminus have been shown to drastically alter ligand activity and convert ligands into potent antagonists, emphasizing the role of the chemokine N-terminus in receptor activation.10,36 To characterize the importance of each individual residue of the CCL20 N-terminus in receptor binding and activation, an alanine scan was performed (A1G, S2A, N3A, F4A, and D5A variants, Fig. 1) and the ability of these variants to stimulate CCR6+ cell migration was assessed. Migration of CCR6+ Jurkat cells was dramatically altered by mutation of Asp5 to alanine (D5A, Fig. 3A). Cell migration stimulated with D5A displayed a maximal chemotactic response at 700 nM (Fig. 3B), while S2A and F4A stimulated maximal migration at 100 nM, similar to WT, and N3A stimulated maximal migration at 300 nM (data not shown). To further study the role of Asp5 in promoting chemotaxis of CCR6-expressing cells, 2 additional CCL20 point variants were generated (D5K and D5N). The loss of chemotactic potency due to D5A mutation was partially rescued with D5N mutation of CCL20. A significant loss of ligand efficacy was observed with D5K mutation, recruiting ∼25% of the number of cells recruited by CCL20 WT (Fig. 3B). These data indicate that Asp5 of CCL20 is crucial for promoting migration of CCR6-expressing cells. The shift in the chemotactic profile for D5N suggests the negative charge of Asp5 plays a key role in CCL20 chemotactic activity whereas the efficacy loss observed with D5K further emphasizes a precise interaction between Asp5 of CCL20 and CCR6 to promote cell migration.
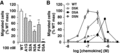
Migration of CCR6+ Jurkat cells in response to CCL20 point variants. (A) CCR6+ transfected Jurkat cells migrated in response to treatment with 100 nM concentration of CCL20 WT, A1G, S2A, N3A, or F4A in a transwell assay. Treatment with 100 nM CCL20 D5A did not promote migration. (B) CCL20 WT, D5A, D5K, and D5N promoted migration of CCR6+ Jurkat cells with maximum migration observed at concentrations of 100, 700, 300, and 300 nM, respectively. D5K treatment recruited ∼20% of cells recruited at maximal WT response. **P < 0.01 versus WT
3.3 Asp5 mutations impair CCL20 signaling through CCR6
The three Asp5 variants (D5A, D5N, and D5K), in addition to 7 other CCL20 point variants were assayed to analyze the role of individual N-terminal residues in CCR6 binding, G protein activation, and β-arrestin-2 recruitment (Fig. 1). First, binding of these variants to CCR6 was studied by displacement of 125I-CCL20 WT from CCR6+ transfected COS-7 cells. All point variants bound CCR6 to displace 125I-CCL20 WT with the largest change in binding affinity observed for mutations at Asp5 (Table 1 and Supplementary Table 1). Asp5 variants displaced 125I-CCL20 WT with IC50 values of 29.3 nM for D5A, 8.67 nM for D5N, and 62.6 nM for D5K (Fig. 4A). D5A and D5K had approximately four- and eightfold increases in IC50, respectively, compared to WT (7.59 nM), while D5N was not significantly different (Table 1).
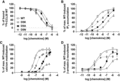
Biochemical characterization of mutations at Asp5 of CCL20. (A) Binding of CCL20 Asp5 point variants was assessed by displacement of 125I-CCL20 WT from CCR6+ transfected COS-7 cells. D5A, D5K, and D5N variants displaced WT CCL20 with calculated IC50 values of 29.3, 62.6, and 8.67 nM (n = 4), respectively. (B) D5A, D5K, and D5N treatment promoted 3H-IP3 accumulation in CCR6+ COS-7 cells with calculated EC50 values of 32.7, 66.4, and 3.83 nM (n = 4), respectively. (C) Asp5 variants stimulated intracellular calcium release of CCR6+ transfected Jurkat cells. Calculated EC50 values for D5A, D5K, and D5N were 988 nM (n = 4), 859 nM (n = 6), and 139 nM (n = 5), respectively. D5K exhibited a significant loss in efficacy, with maximal calcium mobilization signal at ∼40% of maximal WT signal. (D) Treatment with D5A, D5K, and D5N promoted recruitment of β-arrestin-2 to CCR6 in U2OS-A2 cells with calculated EC50 values of 105 nM, 390 nM, and 1.59 nM (n = 3), respectively. D5K and D5N showed significant loss of efficacy and recruited β-arrestin-2 to levels equal to ∼60 and 70% of maximal WT response, respectively
G protein signaling of CCR6 was assessed by stimulation of 3H-IP3 accumulation in CCR6+ transfected COS-7 cells and intracellular calcium mobilization in CCR6+ transfected Jurkat cells with each of the 10 point variants. All point variants promoted 3H-IP3 accumulation and intracellular calcium mobilization with significant impairment in ligand potency observed with D5A and D5K (Table 1 and Supplementary Table 1). In 3H-IP3 accumulation, D5A and D5K had ∼92-fold (32.7 nM) and ∼190-fold (66.4 nM) increases in EC50, respectively, compared to WT EC50 of 0.353 nM. D5N restored substantial activity (EC50 = 3.83 nM) compared to D5A and D5K but was still less potent than WT CCL20 (Fig. 4B, Table 1). This trend held true for intracellular calcium mobilization; D5A and D5K had calculated EC50 values of 988 nM (∼20-fold increase from WT) and 859 nM (∼16-fold increase from WT), respectively. D5K also had a significant loss of ligand efficacy, as evidenced by a maximum response equal to ∼35% of maximum WT response. D5N restored most of the lost potency with an EC50 of 139 nM, compared to WT EC50 of 52.7 nM (Fig. 4C, Table 1).
To determine how β-arrestin-2 recruitment by CCR6 is impacted by point mutations within the CCL20 N-terminus, CCR6+ U2OS-A2 cells were stimulated with the 10 point variants. All variants stimulated β-arrestin-2 recruitment to CCR6 and N3A was significantly more potent than WT CCL20 (calculated EC50 values of 0.299 and 1.91 nM for N3A and WT CCL20, respectively, Supplementary Table 1). D5A and D5K had highly significant ∼95-fold and ∼350-fold increases in EC50, respectively, compared to WT. The calculated EC50 values were 105 nM for D5A and 390 nM for D5K. In contrast, D5N restored considerable ligand potency over D5A and D5K with a calculated EC50 value of 1.59 nM, but promoted a maximum response equal to ∼70% of maximum WT response (Fig. 4D).
In sum, mutation of CCL20 Asp5 to alanine or lysine led to significant decrease in binding affinity for CCR6 and potency in IP3 accumulation, calcium mobilization, and β-arrestin-2 recruitment assays; mutation of Asp5 to asparagine did not significantly impair ligand affinity for CCR6 or β-arrestin-2 recruitment, but did significantly impair downstream G protein signaling. Considered together with all functional data for CCL20 N-terminus point mutations, Asp5 is highlighted as a key residue for potent activation of CCR6.
3.4 The N-terminus of CCL20 is amenable to cumulative changes in length and composition
The chemokine N-terminus is an integral component in chemokine receptor activation, making key contacts within the TM bundle of the receptor to stimulate intracellular signaling. The data presented for CCL20 truncation variants show CCR6 does not require stimulation deep within the TM bundle to elicit activation. The findings from thorough analysis of CCL20 N-terminus point variants underscored Asp5 as a key residue for full ligand activity. None of the tested CCL20 variants completely eliminated ligand binding to CCR6 or ligand activity as measured by chemotaxis, IP3 accumulation, calcium mobilization, and β-arrestin-2 recruitment experiments. To attempt to generate an inactive variant of CCL20, we produced the Δ3AA variant of CCL20. This variant replaced the native CCL20 N-terminus with two alanine residues to test the effect of cumulative truncation and point mutations on CCL20 activity (Fig. 1). Δ3AA maintained binding to CCR6 and displaced 125I-CCL20 bound to CCR6 on COS-7 cells with a calculated IC50 value of 350 nM (∼46-fold increase over WT, Table 1). At the maximum treatment concentration, Δ3AA was unable to displace all 125I-CCL20 WT bound to CCR6 (Fig. 5A). Treatment with Δ3AA stimulated 3H-IP3 accumulation in COS-7 cells with an EC50 of 58.8 nM (Fig. 5B, Table 1) and intracellular calcium mobilization in CCR6+ Jurkat cells with an EC50 of 425 nM (Fig. 5C, Table 1). The calculated EC50 values for 3H-IP3 accumulation and intracellular mobilization are ∼200- and eightfold increases over WT EC50, respectively. In addition to the significant loss of ligand potency as measured by downstream G protein outputs, Δ3AA was deficient in promoting migration of CCR6+ Jurkat cells in a transwell assay. At a protein concentration of 100 nM, Δ3AA stimulated a maximal migratory response equal to ∼20% of cells observed to migrate at maximum WT response (Fig. 5D). Finally, Δ3AA stimulated recruitment of β-arrestin-2 to CCR6 in U2OS-A2 cells with a calculated EC50 value of 458 nM (∼240-fold increase over WT), but the maximum treatment concentration achieved a response equal to ∼30% of maximum WT response (Fig. 5E, Table 1). Despite stripping the CCL20 N-terminus of many identifying features (length reduced by 3 residues and the crucial Asp5 mutated to alanine), Δ3AA maintained the capacity to bind CCR6, promote low levels of CCR6+ cell migration, and stimulate G protein signaling and β-arrestin-2 recruitment, though ligand affinity and activity were significantly impaired compared to WT. Taken together, these data support the importance of CCL20 Asp5 in both affinity for CCR6 and ligand activity at the receptor.
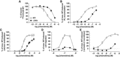
Biochemical characterization of CCL20 Δ3AA variant. The Δ3AA variant removed 3 residues from the native CCL20 N-terminus and replaced the 2 remaining N-terminal residues with alanine residues. (A) Binding of Δ3AA to CCR6 was monitored by displacement of 125I-CCL20 WT from CCR6+ transfected COS-7 cells and had a calculated IC50 value of 350 nM (n = 3). Δ3AA promoted 3H-IP3 accumulation in CCR6+ COS-7 cells with an EC50 value of 6.75 nM (n = 5) (B) and intracellular calcium mobilization in CCR6+ transfected Jurkat cells with an EC50 value of 358 nM (n = 5) (C). Both IP3 accumulation and calcium mobilization showed a ∼20% decrease in response with Δ3AA treatment compared to WT response. (D) CCR6+ Jurkat cells migrated in response to Δ3AA stimulation in a transwell assay to ∼20% of the maximum WT response. (E) Δ3AA promoted β-arrestin-2 recruitment to CCR6 in U2OS-A2 cells with a calculated EC50 value of 11.8 nM (n = 3) and a notably depressed response (∼40% of WT maximum at the highest tested concentration) compared to WT treatment
3.5 Asp5 of CCL20 likely interacts with basic residues of CCR6
The functional data presented herein illustrate the importance of Asp5 of CCL20 in G protein signaling and β-arrestin-2 recruitment at CCR6, as well as stimulation of cell migration. Asp5 tolerates mutation to asparagine with no change in binding affinity. We therefore hypothesized Asp5 of CCL20 interacts with a basic residue of CCR6. To consider specific interacting partners for Asp5, we first generated a CCR6 homology model as described. The final homology model showed stabilization of the backbone RMSD at 2.25 Å over the course of a 300 ns all-atom MD simulation with moderate flexibility in the intracellular and extracellular loop regions and minimal flexibility in the TM regions (data not shown). This homology model has 7 TM helices, 1 amphipathic C-terminal helix, a β-hairpin in ECL2, and 2 disulfide bonds between Cys36/Cys288 and Cys118/Cys197. Inspection of the CCR6 homology model reveals 14 basic residues accessible to a chemokine ligand (Fig. 6A). Asp5 of CCL20 is pinned to the globular chemokine core by 2 adjacent disulfide bonds formed between Cys6/Cys32 and Cys7/Cys48; therefore, Asp5 cannot make contacts deep within the CCR6 TM bundle. Six of the 14 basic residues are positioned favorable for interactions with Asp5 of CCL20 and are found within TM1, TM5, TM7, ECL2, or ECL3 and point into the CCR6 binding pocket (Fig. 6B).
![Analysis of basic residues in CCR6. (A) Homology model of CCR6 viewed parallel to the membrane plane with TM helices denoted with roman numerals. Basic residues near the extracellular binding pocket are shown in blue. (B) Top-down view of the CCR6 binding pocket shows six basic residues positioned near the binding pocket surface with potential to interact with Asp5 of CCL20. (C) Sequence conservation of the 6 CCR6 residues of interest among all CKRs. Residues in extracellular loops were aligned according to the residue’s distance from the nearest cysteine residue. Receptors denoted by “d” exclusively bind DCCL and DCCL-like motif chemokines CCL19, CCL20, CCL21, CCL23, and CCL25. CKRs denoted by “*” bind both DCCL and non-DCCL motif chemokines. (D) Top-down view of the structures of chemokine ligands bound to chemokine receptors shown with emphasis on the chemokine N-terminus. CKRs shown as grey cylinders, CKR residues shown in orange, and ligand N-termini shown in teal. Select interactions between ligand and receptor shown by dotted lines. In the structure of [5P7]CCL5:CCR5, ligand residue Ser9 forms a water-mediated hydrogen bond with receptor residue K22N-term. Ligand residue Lys7 of CX3CL1 forms a hydrogen bond with Y922.63 of the viral receptor US28. Interactions between vMIP-II and CXCR4 include an ionic interaction between ligand residue K10 and receptor residue E311.25. The model of CXCL12 and CXCR4 identifies an ionic interaction between R8 of CXCL12 and E321.26 of CXCR4](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jleukbio/104/2/10.1002_JLB.1VMA0218-049R/1/m_jlb10189-fig-0006-t.gif?Expires=1750219239&Signature=y9Tr1otk4MWtqCPfKYdHftK17rGvRZSISm9OjyJMKvLpPSe~j--aTnIcFWGl9d6vYv-nFtPSp6P~nK3rSRSfszdDAKeJOuAcREKg13Kyy4pNyzHIm1osoQUln7eGdb0nl1PpvLf~M3P5XMgnFDQo30t8rXOq~5onyfHfDLrSQDjL8RJzApgXQYtiele~-JbVZuJypowix9bxNhVARtlL9HDCDI5nAa8EDedB6VJ2XLLDpFFtRv6SaOWL~cRVkOf~yP35G5ZIx1l-~GJZ836ppEg2rBFDL~q~D2ozeNt9VMgJKcU~48W6N3deJ0bT6IEZg9AIm-YmgoTGUHhB2bsP-w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Analysis of basic residues in CCR6. (A) Homology model of CCR6 viewed parallel to the membrane plane with TM helices denoted with roman numerals. Basic residues near the extracellular binding pocket are shown in blue. (B) Top-down view of the CCR6 binding pocket shows six basic residues positioned near the binding pocket surface with potential to interact with Asp5 of CCL20. (C) Sequence conservation of the 6 CCR6 residues of interest among all CKRs. Residues in extracellular loops were aligned according to the residue’s distance from the nearest cysteine residue. Receptors denoted by “d” exclusively bind DCCL and DCCL-like motif chemokines CCL19, CCL20, CCL21, CCL23, and CCL25. CKRs denoted by “*” bind both DCCL and non-DCCL motif chemokines. (D) Top-down view of the structures of chemokine ligands bound to chemokine receptors shown with emphasis on the chemokine N-terminus. CKRs shown as grey cylinders, CKR residues shown in orange, and ligand N-termini shown in teal. Select interactions between ligand and receptor shown by dotted lines. In the structure of [5P7]CCL5:CCR5, ligand residue Ser9 forms a water-mediated hydrogen bond with receptor residue K22N-term. Ligand residue Lys7 of CX3CL1 forms a hydrogen bond with Y922.63 of the viral receptor US28. Interactions between vMIP-II and CXCR4 include an ionic interaction between ligand residue K10 and receptor residue E311.25. The model of CXCL12 and CXCR4 identifies an ionic interaction between R8 of CXCL12 and E321.26 of CXCR4
To further narrow the list of candidate residues of CCR6 that interact with Asp5 of CCL20, we analyzed the conservation of these residues across the CKR family. CCL20 is one of 6 human chemokines containing a DCCL (CCL19, CCL20, CCL21, and CCL25) or DCCL-like (CCL17 and CCL23) motif in the N-terminus; this motif (Asp5-Cys6-Cys7-Leu8 in CCL20) has been postulated to be important for receptor activation due to its position within the chemokine sequence.20,21 With this information, we hypothesized the residue of CCR6 that interacts with Asp5 of CCL20 is most likely conserved in CKRs that respond to DCCL/DCCL-like motif chemokines. These receptors include CCR1, CCR4, CCR6, CCR7, CCR9, and ACKR4. A structure-based sequence alignment was performed using GPCRdb. The GPCRdb numbering scheme takes the sequence-based Ballesteros-Weinstein nomenclature (e.g., 5.35) and couples it with an additional number (e.g., ×36) to correct for helical bulges and constrictions. Conservation analysis revealed a few interesting trends. At position 1.28 × 28 (CCR6 Arg42), most CC-motif CKRs have a basic residue, while most CXC-motif CKRs have a basic residue at position 1.32 × 32 (CCR6 Arg46). Moderate conservation of a basic residue at position 5.35 × 36 (CCR6 Lys211) is observed across the CKR family. Among the receptors that respond to DCCL/DCCL-like motif chemokines, highest conservation of a basic residue is found at positions 1.28 × 28, 5.35 × 36, and 7.29 × 28 (Fig. 6C). From the structure-based sequence alignment, we predicted Asp5 of CCL20 interacts with Arg42(1.28 × 28) or Lys292(7.29 × 28) of CCR6.
Next, we considered the structures of 3 chemokine–CKR complexes ([5P7]CCL5 bound to CCR5, CX3CL1 bound to US28, and vMIP-II bound to CXCR4) and the model of CXCL12 bound to CXCR4 to look for commonalities in the interactions between ligand N-terminus and receptor to guide our prediction of interactions between CCL20 Asp5 and CCR6.11–14 We specifically looked at direct or indirect interactions between the ligand residue immediately preceding the first conserved cysteine (i.e., the ligand residue analogous to Asp5 of CCL20) and the receptor. In the [5P7]CCL5:CCR5 structure, ligand residue S9 and receptor residue K22N-term interact through a water-mediated hydrogen bond (Fig. 6D, top left). A direct hydrogen bond interaction is observed between CX3CL1 residue K7 and US28 residue Y922.63, found at the top of TM2 (Fig. 6D, top right). CXCR4 residues E311.25 and E321.26 at the top of TM1 participate in direct ionic interactions with vMIP-II residue K10 and CXCL12 residue R8, respectively (Fig. 6D, bottom panels). Taken together, these structures and the CXCL12:CXCR4 model show key interactions between the most proximal residue of the ligand N-terminus and receptor residues in the N-terminus, TM1, or TM2. We therefore predict Arg42(1.28 × 28) of CCR6 is the most likely direct interaction partner of Asp5 of CCL20. Future computational and experimental analysis of CCL20 and CCR6 will be designed to test this hypothesis.
4 DISCUSSION
The current understanding of chemokine–CKR interactions is ever expanding from the simplistic two-step/two-site model. While the canonical model of interaction has served its purpose as a framework for generating hypotheses, advanced structural studies of the chemokine–CKR interaction show an interface that extends beyond the two-site model. Here, we present a systematic mutational study of the CCL20 N-terminus that demonstrates a tolerance of CCL20 to extreme changes in length and point mutations at most positions to maintain ligand signaling through CCR6. With the exception of variants that manipulated Asp5 of the CCL20 N-terminus (D5A, D5N, D5K, and Δ3AA), ligand potency loss due to N-terminus manipulation generally stemmed from a simultaneous loss of binding affinity. These data illustrate a departure from the two-step/two-site paradigm as the full CCL20 N-terminus is not required for receptor activation. Given the monogamous nature of the CCL20/CCR6 signaling axis, an uncommon property of chemokine ligands and receptors, we were not entirely surprised to find a divergence from the established two-step/two-site paradigm.
Our findings show increasing the length of the native CCL20 N-terminus by up to 5 residues or removing up to 3 residues from the native N-terminus does not eliminate binding to CCR6. However, the most extreme extension and truncation variants, A5 and Δ3, had 9.2-fold and 12.3-fold increases in calculated IC50 of displacement of CCL20 WT, respectively, indicating significant loss of ligand affinity for CCR6. These observed affinity losses for A5 and Δ3 translated to reduced ability of the ligand to stimulate downstream G protein signaling events, β-arrestin-2 recruitment, and chemotaxis through CCR6. Additionally, A5 and Δ3 showed substantial reduction in efficacy of β-arrestin-2 recruitment. These data suggest extreme manipulation of the CCL20 N-terminus length converts variant proteins into partial agonists of CCR6.
Mutations at most locations within the CCL20 N-terminus—Ala1, Ser2, Asn3, or Phe4—were well tolerated and in some cases, mutation at these positions enhanced ligand potency. Mutation of Asp5 to alanine or lysine, however, significantly impaired binding to CCR6 and impaired ligand ability to recruit β-arrestin-2, promote cell migration, or stimulate intracellular calcium mobilization. Asp5 is crucial for promoting these key signaling outputs through CCR6 and makes key contributions to binding to CCR6. The effect of cumulative Asp5 mutation and N-terminus truncation on CCL20 affinity and activity was further tested with analysis of a Δ3AA variant of CCL20. This variant had the greatest impairment in binding to CCR6 when compared to all other tested variants and a significantly reduced ability to promote downstream G protein signaling events, β−arrestin-2 recruitment, and CCR6+ cell migration. Δ3AA fared far worse than the Δ3 variant of CCL20, indicating the presence of Phe4 and/or Asp5 play a key role in CCL20 activity at CCR6. The N-terminus of CCL20 is not entirely dispensable, but the overall tolerance of changes in length and composition does not agree with the canonical chemokine–CKR interaction model as other well-studied ligands became completely inactive upon truncation of 1 or 2 residues or mutation of a single residue.
From this extensive structure–function analysis of the CCL20 N-terminus, we concluded the CCL20/CCR6 system retains activity after extensive manipulation to residues within CRS2 provided Asp5 of CCL20 is present. The length and sequence of the CCL20 N-terminus is highly conserved in orthologous CCL20 proteins for many different species, including mouse (NH2-ASNYD) and rat (NH2-ASNFD), despite overall sequence identities to human CCL20 of 66 and 61% for mouse and rat proteins, respectively.37,38 It is possible the CCL20 N-terminus plays a significant role in maintaining receptor specificity and our CCL20 variant proteins may have introduced promiscuity into this highly monogamous system. This study did not directly address this hypothesis, but future work will evaluate receptor specificity of CCL20 variants.
Although mutation of Asp5 to lysine or alanine led to significant decreases in binding affinity and ligand potency, mutation to asparagine did not provoke a change in binding to CCR6 and had a marginal effect of ligand activity. A significant loss of potency in G protein signaling assays was observed for D5N, while no significant difference was found in β-arrestin-2 recruitment. We hypothesized Asp5 likely interacts with a basic residue of CCR6 and identified several candidate residues near the opening of the CCR6 binding pocket. Based upon the conservation of these residues across the chemokine receptor family and scrutiny of available structures of chemokine ligands bound to receptors, we ultimately proposed Arg42(1.28) of CCR6 as the top candidate for a direct interaction with Asp5 of CCL20. Future structure-function studies of CCR6 will be designed to confirm the crucial receptor residues for CCR6 activation by CCL20.
In summary, these functional data indicate Asp5 of CCL20 is crucial for receptor activation and CCL20 does not require the full N-terminus to activate the receptor. Considering the delicate sensitivity of other chemokine/CKR systems to modifications to the chemokine N-terminus, our data suggests the CCL20/CCR6 signaling axis diverges from the two-step/two-site paradigm and these proteins interact in a noncanonical fashion. Future analysis of the CCL20/CCR6 interaction through modeling efforts and structure-function studies of CCR6 will continue to shape our understanding of this unusual monogamous interaction.
AUTHORSHIP
S.J.R., A.E.G., M.M.R., S.T.H., and B.F.V. designed research. S.J.R. and O.L. performed research. All authors contributed to data analysis. S.J.R. wrote the first draft of the manuscript. All authors commented on and contributed to the final draft of the manuscript.
ACKNOWLEDGMENTS
We sincerely thank M. A. Thomas for assistance in homology model development and Dr. H. Fan for evaluation of the model. This work was supported in part by a National Psoriasis Foundation translational grant and National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant 1R01AR063091-01A1 (to S.T.H.). This work was supported in part by National Institutes of Health Grants R01 AI058072 and R01 GM097381 (to B.F.V.).
DISCLOSURES
B.F.V. has ownership interests in Protein Foundry, LLC. The other authors state no conflict of interest.
Abbreviations
- CKR
chemokine receptor
- CRS1/CRS2
chemokine recognition site 1/2
- ECL2/ECL3
extracellular loop 2/3
- GPCR
G protein-coupled receptor
- IP3
inositol triphosphate
- MD
molecular dynamics
- TM
transmembrane
- ULP1
ubiquitin-like-specific protease 1
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}