





Abstract
A formate dehydrogenase gene (fdh) originated from Candida boidinii was co-expressed in E. coli BL21 (DE3) with the cyclohexanone monooxygenase gene (chmo) cloned from Acinetobacter calcoaceticus NCIMB 9871. The co-expression system was then used as a whole-cell biocatalyst to synthesize chiral phenyl methyl sulfoxide (PMSO) from thioanisole (PMS) and the reaction conditions were investigated. When the initial concentration of PMS was 20 mM, the specific productivity of PMSO in this system was 2.07 μmol g−1 cw min−1 (cw: wet cell weight) and the ee value for the R-sulfoxide was 99 %. In contrast, when chmo was the only gene expressed in E. coli, the specific productivity of PMSO was 0.053 μmol g−1 cw min−1 with no exact enantioselectivity. Further determination of NADPH concentration in the whole-cell catalysts suggested that co-expression of fdh with chmo significantly improved NADPH supply. Thus, this whole-cell biocatalyst system is highly advantageous for the synthesis of optically pure R-sulfoxide.
Electronic supplementary material
The online version of this article (doi:10.1007/s10295-013-1288-0) contains supplementary material, which is available to authorized users.
Introduction
The synthesis of chiral sulfoxides has been the focus of synthetic organic chemistry [7, 11, 15]. They can be prepared using chemical methods or biological oxidation. With advances in biotechnology such as DNA sequencing, high-throughput screening, and protein engineering with the assistance of molecular simulation, the ability of biocatalysis to compete with chemical methods has been increasing, and more and more biocatalysts are used in many different chemical transformations [6, 18, 23].
There is an increased interest in employing Baeyer–Villiger monooxygenases (BVMOs) as synthetic tools [8]. BVMOs are able to catalyze Baeyer–Villiger oxidations, sulfoxidations, oxidations of selenium- and boron-containing compounds, epoxidations, and N-oxidations [2]. Several BVMOs such as CHMO, cyclopentanone monooxygenase (CPMO), phenylacetone monooxygenase (PAMO), and hydroxyacetophenone monooxygenase (HAPMO) have been shown to be excellent biocatalysts for the enantioselective oxidation of a large variety of alkyl aryl sulfides, dialkyl sulfides, or dialkyl disulfides [2].
To develop an oxidoreductase-catalyzed process, cofactor regeneration must be considered because of its stoichiometric requirement and high cost. The irreversibility of the reaction and low cost of the substrate has made formate dehydrogenase (FDH) one of the most frequently used enzymes for NADH regeneration [22]. However, wild-type FDHs are highly specific for NAD+ [21], and they have to be engineered if the proteins are to be used in combination with NADPH-dependent oxidoreductases such as cyclohexanone monooxygenase (CHMO). Using engineered FDH from Pseudomonas sp. 101 and CHMO from Acinetobacter calcoaceticus NCIMB 9871, Rissom et al. [20] reported a two-enzyme system for the synthesis of chiral ε-lactones. Recently, Wu et al. [9] reported that an engineered FDH mutant originally from Candida boidinii had substantially higher catalytic efficiency toward NADP+ than that of the FDH mutant from bacterium Pseudomonas sp. 101.
In this study, a whole-cell biocatalyst co-expressing the chmo Acineto and the fdh mutant gene (mfdh) was constructed. The reaction conditions and its application in biooxidation of PMS were investigated. To our knowledge, this is the first study in which FDH was used to save NADPH consumption in biosulfoxidation with BVMO.
Materials and methods
Chemicals, media, and cultivation
NADP+ and NADPH were obtained from Sangon Biotech (Shanghai, China). E. coli DH5α and E. coli BL21 (DE3) were cultivated in Luria Broth (LB) medium routinely and were used as the cloning and expression hosts, respectively. When necessary, antibiotics (100 μg ml−1 ampicillin or 34 μg ml−1 chloromycetin) were supplemented to the medium to maintain the plasmids. Growth of all strains was analyzed by measuring the optical density (OD) at 600 nm.
Protein expression
The expression plasmids were constructed routinely. First, the chmo gene was amplified from the genomic DNA of A. calcoaceticus NCIMB 9871 and subcloned into the vector pET-22b to get the plasmid pMM4 [5]. The Cbo fdh gene from C. boidinii was mutated by point mutation (Cbo mFDH) based on Wu and other’s report [24] using the Stratagene Quikchange protocol and then subcloned into the pACYCDuet-1 (Novagen) vector to generate the new plasmid pAFD. Similar to the result in Wu’s report [24], the mutated enzyme D195Q/Y196R/Q197N showed significant NADP+ specificity and high catalytic efficiencies (data not shown). For a tandem system, the chmo gene was inserted into the pAFD resulting in pAMFD, which was then transformed into E. coli BL21 (DE3). In a two-plasmid system, E. coli BL21 (DE3)-pAMF was constructed harboring both the pMM4 and pAFD plasmids, which contained chmo and mfdh, respectively. E. coli BL21 (DE3) cells harboring the expression plasmids were grown in LB medium containing appropriate antibiotics at 37 °C overnight with shaking at 200 rpm. The culture (5 ml) was transferred to Terrific Broth (TB) medium (500 ml) in a 2-l shake flask and shaken at 200 rpm at 37 °C. IPTG (0.1 mM) was added when the OD600 value reached 0.6–0.8. The cells were further incubated at 20 °C for 16 h with shaking at 150 rpm, harvested by centrifugation (4,000 rpm, 4 °C, 20 min), and the cell pellet was washed with 0.1 M phosphate buffer (pH 7.5). It was then stored at 4 °C (less than 1 week) before being used in the asymmetric oxidation and enzyme activity analysis. SDS-PAGE (12 % polyacrylamide) was carried out as described by Laemmli [16].
Enzyme activity assays
The culture (5 ml) was harvested by centrifugation, and the pellet was resuspended in disruption buffer (20 mM Tris–HCl containing 500 mM NaCl, pH 8.0). After ultrasonic disruption, cell debris was removed by centrifugation at 4 °C for 15 min to obtain a crude enzyme solution for the enzyme assay. The CHMO activity was measured using a spectrophotometric NADPH consumption assay based on the method of Donoghue described in Cheesman et al. [4]. One unit of CHMO activity was defined as the activity of enzyme catalyzing the cyclohexanone-stimulated oxidation of 1 μmol of NADPH per minute. The specific CHMO activity was expressed as units per milligram of cellular protein.
The FDH assay was performed at room temperature with a 1-ml reaction mixture comprising 9 mM phosphate buffer (pH 7.5), 100 μM sodium formate, 0.24 μM NADP+, and the extraction sample containing 70 μl mFDH. The speed of the initial increase in the absorbance at 340 nm was monitored. One unit of FDH activity was defined as the activity of enzyme that catalyzes the formation of one micromole of NADPH per minute. The protein concentration was measured using Bradford’s method [3] with bovine serum albumin as the standard.
Condition optimization for whole-cell biocatalysis
The general procedure for optimization was as the following. Cells were centrifuged, washed with Tris–HCl (pH 8.0), and resuspended in the same buffer at a concentration of 0.04 g ml−1 to make the volume up to 2 ml. PMS, NADP+ and sodium formate were added at concentrations of 10 mM, 317 μM and 50 mM, respectively. The production of PMSO was monitored at 25 °C for 12 h and the PMSO concentration was measured.
The conditions for optimization included the amount of biocatalyst (0.01–0.06 g ml−1 reaction mixture), substrate concentration (10–100 mM), temperature (20–45 °C), pH (7.0–9.4) addition of NADP+ (0–317 μM) and DMSO (0–20 %).
Assay of whole-cell biocatalytic activity
The whole-cell biocatalytic activity was assayed by measuring the increase of PMSO in the reaction mixture. After the reaction was stopped, the cells were removed by centrifugation (12,000 rpm, 1 min), and solid NaCl was added until the water was saturated. The products were then extracted with isopropanol twice. The organic layers were combined and dried over anhydrous calcium chloride, filtered, and concentrated under reduced pressure. The PMSO was detected by absorbance measurements at 245 nm using a Shimadzu LC-20A series HPLC and a Chiralpak OD-H (250 × 4.6 mm) column with a flow rate of 1 ml min−1 (82 % hexane/18 % 2-propanol) for 18 min. The retention times of the (R)-and (S)-enantiomers were 9.429 and 11.297 min, respectively.
One unit of the whole-cell biocatalytic activity was defined as the amount of wet cells that catalyzes the formation of 1.0 μmol of PMSO per minute at 35 °C.
High-performance liquid chromatographic analysis of NADPH in cells
For NADPH quantification, 100 mg of the cells (wet weight) was re-suspended in 1.8 ml 0.1 M phosphate buffer (pH 7.5) and 200 μl sodium formate. After the addition of 2 ml of chloroform, nucleotides were extracted by rotating the sample on a shaker for 0.5–3.5 h at room temperature. After centrifugation for 5 min at 7,000 rpm, the aqueous phase was isolated, filtered, and subjected to HPLC analysis using a Supelcosil LC-18-T column (Supelco, L × ID 25 cm × 4.6 mm). A gradient of buffer A (0.1 M potassium phosphate buffer/4 mM tetrabutylammonium hydrogen sulfate, pH 6.0) and buffer B (70 % buffer A and 30 % methanol, pH 7.5) was applied by mixing 30 % buffer B after 5 min, 60 % buffer B after 10 min, 100 % buffer B after 13 min, and 30 % buffer B again after 17.01 min. The total separation time was 22 min, and the flow rate was 1.5 ml min−1 [13].
Results
Construction of the whole-cell biocatalyst
To construct the whole-cell biocatalyst, two types of expression systems with NADPH-regeneration capacities were examined. In the beginning, a tandem system was used. To construct this system, the chmo gene was inserted into the pAFD to result in pAMFD, which was then transformed into E. coli BL21 (DE3).
A crude extract of induced E. coli BL21 (DE3)-pAMFD showed a CHMO activity of 2.26 × 10−2 U mg−1, but the protein band was not obvious in the SDS-PAGE (data not shown). Further, the extract showed a FDH activity of 3.02 × 10−2 U mg−1 (Table 1) in the presence of sodium formate.
Enzyme activity of whole-cell catalysts containing different plasmids
| Whole-cell catalyst | Plasmid (s) | CHMO activity (U mg−1 protein of crude extract) | FDH activity (U mg−1 protein of crude extract) |
|---|---|---|---|
| BL21(DE3)-pMM4 | pET-22b-chmo | 0.936 | No |
| BL21(DE3)-pAMF | pET-22b-chmo + pACYC-fdh | 1.697 | 3.14 × l0−2 |
| BL21(DE3)-pAMFD | pACYC-chmo-fdh | 2.26 × 10−2 | 3.02 × 10−2 |
| BL21(DE3)-pAFD | pACYC-fdh | No | 2.72 × 10−2 |
| BL21(DE3)-pAET | pET-22b + pACYCDuet-1 | No | No |
| Whole-cell catalyst | Plasmid (s) | CHMO activity (U mg−1 protein of crude extract) | FDH activity (U mg−1 protein of crude extract) |
|---|---|---|---|
| BL21(DE3)-pMM4 | pET-22b-chmo | 0.936 | No |
| BL21(DE3)-pAMF | pET-22b-chmo + pACYC-fdh | 1.697 | 3.14 × l0−2 |
| BL21(DE3)-pAMFD | pACYC-chmo-fdh | 2.26 × 10−2 | 3.02 × 10−2 |
| BL21(DE3)-pAFD | pACYC-fdh | No | 2.72 × 10−2 |
| BL21(DE3)-pAET | pET-22b + pACYCDuet-1 | No | No |
BL21 (Dh3)-pAET was set as control
No no activity was detected
Enzyme activity of whole-cell catalysts containing different plasmids
| Whole-cell catalyst | Plasmid (s) | CHMO activity (U mg−1 protein of crude extract) | FDH activity (U mg−1 protein of crude extract) |
|---|---|---|---|
| BL21(DE3)-pMM4 | pET-22b-chmo | 0.936 | No |
| BL21(DE3)-pAMF | pET-22b-chmo + pACYC-fdh | 1.697 | 3.14 × l0−2 |
| BL21(DE3)-pAMFD | pACYC-chmo-fdh | 2.26 × 10−2 | 3.02 × 10−2 |
| BL21(DE3)-pAFD | pACYC-fdh | No | 2.72 × 10−2 |
| BL21(DE3)-pAET | pET-22b + pACYCDuet-1 | No | No |
| Whole-cell catalyst | Plasmid (s) | CHMO activity (U mg−1 protein of crude extract) | FDH activity (U mg−1 protein of crude extract) |
|---|---|---|---|
| BL21(DE3)-pMM4 | pET-22b-chmo | 0.936 | No |
| BL21(DE3)-pAMF | pET-22b-chmo + pACYC-fdh | 1.697 | 3.14 × l0−2 |
| BL21(DE3)-pAMFD | pACYC-chmo-fdh | 2.26 × 10−2 | 3.02 × 10−2 |
| BL21(DE3)-pAFD | pACYC-fdh | No | 2.72 × 10−2 |
| BL21(DE3)-pAET | pET-22b + pACYCDuet-1 | No | No |
BL21 (Dh3)-pAET was set as control
No no activity was detected
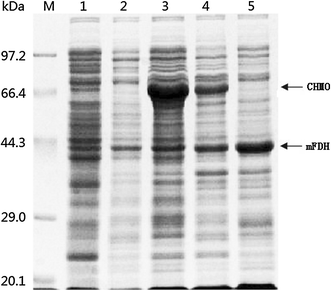
SDS-PAGE analysis of the CHMO and mFDH from different whole-cell catalysts. Lane M marker proteins, lane 1 E. coli BL21 (DE3), lane 2 E. coli BL21 (DE3)-pACYCDuet-1 + pET-22b, lane 3 E. coli BL21 (DE3)-pMM4, lane 4 E. coli BL21 (DE3)-pAMF, lane 5 E. coli BL21 (DE3)-pAFD. The CHMO (ca. 66 kDa) and mFDH (ca. 44 kDa) are indicated by the arrows
Investigation of reaction conditions
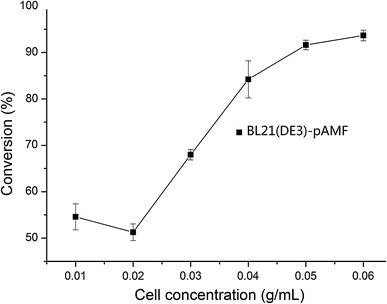
Effect of cell density on reaction conversion. The reaction was performed for 1 h in a 2-ml reaction mixture containing Tris–HCl (pH 8.0), 10 mM of PMS, 317 μM of NADP+ and 50 mM of sodium formate. Cell concentration means weight of wet cell (g) per 1 ml of reaction mixture. Three repeats were performed for each concentration. Error bar mean ± SD
By measuring PMSO concentration sampled at 1, 7, and 14 h, with 20 mM of PMS, the conversion rate was 41.69, 88.90, and 79.03 %, respectively. The turnover rate (based on the product) reached the maximum when the reaction time was 7 h. So the best sampling time was at 7 h when the substrate concentration was 20 mM. When the reaction time was further extended, the conversion rate was decreased. A similar trend was found when the substrate concentration was changed to a different one, although the best sampling time may have some marginal variation (data not shown).
For pH optimization, the biocatalytic oxidation of PMS was carried out in different buffers with various pH values. The maximum reaction rate was obtained with phosphate buffer of pH 7.5. The optimal temperature for this reaction was found to be 35 °C. The relative activity at 40 °C was only 44 % of that at 35 °C. At a higher temperature, the reaction rate began to decrease dramatically, possibly because of the thermal deactivation of the enzymes in the cells. On the basis of these results, pH 7.5 and 35 °C were selected for the subsequent experiments.
The effect of supplementing NADP+ or DMSO in the reaction system was further evaluated. The sulfoxidation rate was increased accordingly with increased addition of the NADP+. Considering high price of NADP+, 317 μM of NADP+ was added when the substrate concentration was 10 mM. When 1 % of DMSO was used, the conversion rate improved significantly and reached the maximum within 45 min. With the addition of more DMSO, the sulfide conversion decreased sharply.
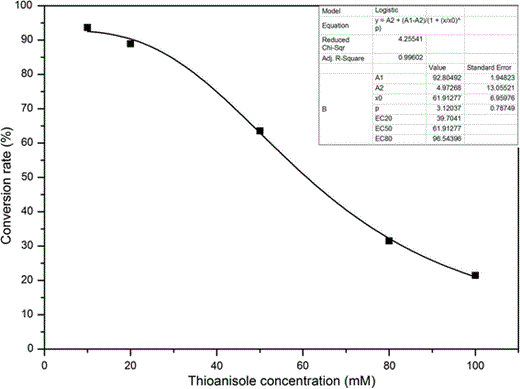
Effects of the substrate concentration on the oxidation process. The conversion of the oxidation reaction by BL21(DE3)-pAMF at different substrate concentration was tested at the different time points, and the maximal conversion was used to make the simulation curve. The reaction was performed in a 2-mL reaction mixture containing Tris–HCl (pH 8.0), 0.08 g cells of E. coli (wet weight), 317 μM of NADP+ and 50 mM of sodium formate. Three repeats for each point
Biotransformation by the co-expressed whole-cell biocatalyst
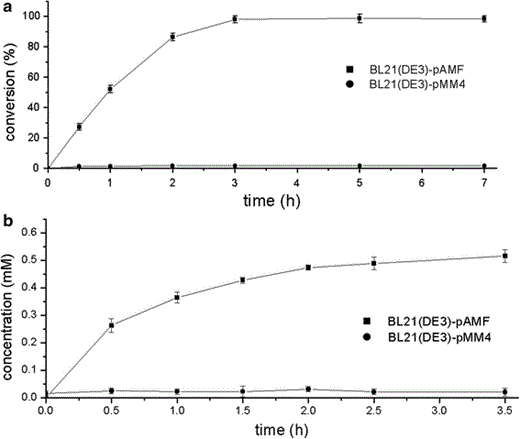
Biotransformation by the co-expressed whole-cell system. The reaction mixture (2 ml) was comprised of 0.10 g of E. coli cells (wet weight), 20 mM of PMS, 317 μM of NADP+ and 50 mM of sodium formate in phosphate buffer (pH 7.5). a Time courses for oxidation of thioanisole with E. coli BL21 (DE3) transformants harboring pAMF (filled square) or pMM4 (filled circle). b The NADPH concentration during the oxidation process in E. coli BL21 (DE3) transformants harboring pAMF (filled square) or pMM4 (filled circle). Three repeats for each point. Error bar mean ± SD
The NADPH concentrations in both kinds of cells were then determined (Fig. 4). In the non-growing E. coli BL21 (DE3)-pAMF cells, an increasing trend was observed, which was similar to that of the PMSO concentration. As a comparison, the NADPH concentration in the resting E. coli BL21 (DE3)-pMM4 cells was very low with no trend of increase (as shown in Fig. 4) which indicated that coexpression of fdh with chmo significantly improved NADPH supply.
Discussion
In this study, pACYCDuet-1 (Novagen) was selected as a co-expression vector because it has two multiple cloning sites, each proceeded by a T7 promoters [10]. However, the chmo gene was expressed at a much higher level when the gene was inserted into plasmid pET-22b (Fig. 1). The plasmids pET-22b and pACYCDuet-1 could coexist in E. coli, expressed independently of each other, and the recombinants could be readily obtained through double-antibiotic screening. Therefore, this two-plasmid system was selected for the further continued experiments in this study.
As reported previously, the reaction time could influence the yield catalyzed by BVMO [12]. This may be due to over-oxidation of the substrate and generation of sulfone from sulfoxide [19]. In all cases, NADP+ was not only the co-substrate of the reaction, it was also crucial to the enzyme structure [1]. In the present experiment, the NADP+ supplement could improve the sulfoxidation reaction rate significantly, which suggests that the intracellular NADP+ concentration was not enough for the reaction and extra NADP+ addition was necessary. DMSO functioned as a co-solvent in the reaction because the substrate PMS was poorly soluble in water, and 1 % supplement increased the productivity significantly (Fig. 2). However, a higher concentration of DMSO might have adverse effects on the enzyme as most organic reagents do. Though many organic solvents such as methanol had influence on the product enantioselectivity [9], there was no evidence that DMSO had a significant effect on the PMSO enantioselectivity.
The whole-cell activity of E. coli BL21 (DE3)-pAMF was 1.04 U ml−1 when it was used in the sulfoxidation reaction, while that of E. coli BL21 (DE3)-pMM4 was only 0.022 U ml−1. The low oxidation efficiency of E. coli BL21 (DE3)-pMM4 strain may indicate that the NADP+ in the cell was slowly reduced to NADPH by the enzyme originating from the wild bacterium itself. As a comparison, the fdh coexpressed with chmo in the cells could be used to regenerate the NADPH cofactor efficiently when sodium formate was provided as a co-substrate, which was confirmed by the determination of NADPH concentration (see Fig. 4). The NADPH concentration in the non-growing E. coli BL21 (DE3)-pAMF cells showed a trend of increase, similar to that of the PMSO concentration. In contrast, the NADPH concentration in the resting E. coli BL21 (DE3)-pMM4 cells was very low and had no trend of increase (Fig. 4). Thus, we confirmed the ability of the mutant FDH to regenerate NADPH. The introduction of this gene into E. coli BL21 (DE3) co-expressing chmo significantly improves the ability of the bacterium to regenerate the cofactor.
In conclusion, this study experimentally demonstrated that an efficient conversion was obtained by using a co-expressed whole-cell system for the enantioselective conversion of an aromatic sulfide, which is the first such study based on our knowledge. However, like other monooxygenases biotransformation [17], the substrate inhibition was still a major drawback for CHMO biosulfoxidation. Further study using a resin-based in situ SFPR strategy may be a good choice to resolve this problem [14].
Acknowledgments
This work was supported by a project (20101131N06) from the Science and Technology Department of Hangzhou, China.

{kind=link}
{kind=link}
{kind=link}
{kind=link}