





Abstract
A novel thermostable isoamylase, IAM, was purified to homogeneity from the newly isolated thermophilic bacterium Bacillus sp. CICIM 304. The purified monomeric protein with an estimated molecular mass of 100 kDa displayed its optimal temperature and pH at 70 °C and 6.0, respectively, with excellent thermostability between 30 and 70 °C and pH values from 5.5 to 9.0. Under the conditions of temperature 50 °C and pH 6.0, the K m and V max on glycogen were 0.403 ± 0.018 mg/mg and 0.018 ± 0.001 mg/(min mg), respectively. Gene encoding IAM, BsIam was identified from genomic DNA sequence with inverse PCRs. The open reading frame of the BsIam gene was 2,655 base pairs long and encoded a polypeptide of 885 amino acids with a calculated molecular mass of 101,155 Da. The deduced amino acid sequence of IAM shared less than 40 % homology with that of microbial isoamylase ever reported, which indicated it was a novel isoamylase. This enzyme showed its obvious superiority in the industrial starch conversion process.
Electronic supplementary material
The online version of this article (doi:10.1007/s10295-013-1249-7) contains supplementary material, which is available to authorized users.
Introduction
Isoamylase (EC 3.2.1.68, glycogen-6-glucanogydrolase) catalyzes hydrolysis of α-d-(1,6)-glucosidic branch linkages in amylopectin and glycogen to release amylose and linear malto oligosaccharides [9]. In starch processing industry, application of debranching enzymes such as pullulanase and isoamylase could dramatically improve the efficiency and yield of conversion from starch to fermentable sugar. Isoamylase has three advantages over pullulanase, namely, both endo- and exo-cleavage activity, higher efficient cleavage on α-d-(1,6)-glucoside linkage and less inhibition by its main product maltose, but very limited information relevant to isoamylase is available in contrast with huge amounts of investigations concerning pullulanase of various origins, probably largely due to the insurmountable difficulty in measuring isoamylase activity under strong interference from that of other amylolytic enzymes, and due to the instability of isoamylase itself. To date, Bacillus amyloliquefaciens [29], Flavobacterium odoratum [2], Escherichia coli [8, 16, 25, 36], and Pseudomonas amyloderamosa [3, 7, 13] have been isolated with the capability of producing isoamylase, of which only Pseudomonas amyloderamosa is known to produce industrially useful isoamylase. As high efficient enzymatic liquefaction and saccharification are usually performed at elevated temperature (up to 117 °C) in starch processing, thermoactive and thermostable amylolytic enzymes will show obvious superiority in production of glucose syrup, maltose syrup and maltodextrin. Thermostable amylase from B. acidocaldarius (80 °C) or B. licheniformis (95 °C) has been widely used in current liquefaction of starch [32], while as one of the most important enzymes in starch processing industries, most known microbial isoamylases are thermolabile. Isoamylase was first detected in the intracellular form in yeast [20] and the first bacterial isoamylase was characterized from Pseudomonas amyloderamosa [13]. Although more than four decades have passed, P. amyloderamosa is still the only origin of industrial isoamylase production. Even if it has some advantages, which can be utilized in the starch processing industry, it is not convenient for the P. amyloderamosa isoamylase to be used simultaneously with various other amylolytic enzymes due to the large differences in the optimal temperature range when they were cooperatively used.
As an auxiliary enzyme of glucoamylase or β-amylase in starch processing industry, isoamylase is expected to perform efficient saccharification under the condition of high temperature [17]. But until now, the thermostable isoamylases (optimal temperature above 70 °C or more, stable above 70 °C or more) still remain to be industrially applied. A thermophilic archaeon Sulfolobus solfataricus ATCC 35092 was reported to produce an isoamylase with an optimal temperature of 75 °C and optimal pH of 5 [9], while this enzyme lost most of its activity at pH 7 or above, which restricted its use in collaboration with other amylolytic enzymes. Current starch processing usually requires more than two enzymes to act cooperatively in the same condition, and development of isoamylase with different optimal temperatures and pH will provide researchers with more options to improve the technique.
This study aims to purify and characterize the novel isoamylase from Bacillus sp. strain CICIM 304 so as to investigate its catalytic properties. At the same time, we expect to identify and clone the gene encoding the isoamylase to obtain some insight into the mechanism of the debranching activity.
Materials and methods
Bacterial isolation, identification, and isoamylase production
The bacteria strains used in this study were isolated by screening a soil sample collected from a hot spring of Tengchong volcano in Yunnan, China. The soil sample (1 g) was suspended in 10 ml of sterile water and serial dilutions were made. Then aliquots of appropriate dilutions were plated on Luria–Bertani (LB) agar containing 0.5 % maize amylopectin from Sigma-Aldrich (Deisenhofen, Germany). Debranching enzyme producing strains were identified by purple halo around the colony after iodine staining as described previously [30]. The 16S rRNA gene sequence analysis was applied in order to characterize the newly isolated strain. PCR product was generated with the primers B27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and U1492R (5′-GGTTACCTTGTTACGACTT-3′) targeting the 16S rRNA gene as previously described [24]. The 1,581-bp 16S rRNA gene sequence thus obtained was performed with nucleotide BLAST (http://blast.ncbi.nlm.nih.gov/) at NCBI to identify the nearest neighbor sequences.
For shaking cultivation, organisms were cultivated in a modified Luria–Bertani (MLB) medium containing amylopectin, 2 g/l; tryptone, 5 g/l; Na2HPO4, 0.1 g/l; KH2PO4, 0.15 g/l; NaCl, 1 g/l and MgSO4·7H2O, 0.5 g/l, which was adjusted to pH 7.0 with 6 M HCl. Cultures were incubated by shaking at 250 rpm and 37 °C on shakers. For incubation in the fermentor, a 10 % inoculum derived from a 72-h culture was used to initiate the cultivation in New Brunswick Scientific BioFlo 110 fermentor (Edison, NJ, USA). Culture conditions in the fermentor were controlled as follows: invariable temperature at 37 °C and air flow of 2 l/min. The control of dissolved oxygen (DO) level was achieved by mixing the inlet air with oxygen or nitrogen; the accuracy of the DO control was within 2 % air saturation. Agitation was performed using a gate impeller with a maximum speed of 500 rpm. At the end of cultivation, the cells and debris were removed from the culture by centrifugation at 8,000×g for 20 min at 4 °C. Then the supernatant was collected and stored at 4 °C for enzyme purification.
Purification of isoamylase
The supernatant of culture was applied to microfiltration with ceramic membrane (Hua Zhuang, Wuxi, China, with a nominal pore size of 0.2 μm), followed by ultrafiltration using Spin-Columns (Millipore, MA, USA; cut off 10 kDa). The cell-free and concentrated crude enzyme was fractionally precipitated by ammonium sulfate (30 % saturation, 2 h to remove impurities and 80 % saturation, 12 h to collect fraction with isoamylase activity). The active fraction pellet was collected by centrifugation at 10,000g, 4 °C for 30 min and dissolved in 10 ml of 50 mM Tris–HCl buffer (pH 6.4) and successively passed through a Sephacryl S-100 HR (GE, Uppsala, Sweden) column pre-equilibrated with the same buffer containing 0.15 M NaCl. The fraction with activity was applied to HiPrep 16/10 DEAE FF Column (GE) equilibrated with the same buffer adding 0.2 M NaCl. The column was washed with two column volumes of the equilibration buffer, followed by elution of the bound proteins with the buffer of a 0.15–1 M NaCl liner gradient. The fraction with activity was desalted and concentrated by Labconco Freezone 12 l freeze dry system (Kansas City, MO, USA). The active fraction dissolved in 0.15 M NaCl was finally subjected to MONO Q 4.6/100 PE column (GE) equilibrated and eluted with the same condition as that of HiPrep 16/10 DEAE FF Column. The active fraction was collected and then desalted with a dialysis bag (cut off 7 kDa, Union Carbide Co., Houston, Texas, USA) against water for 48 h at 4 °C, refreshing the water every 4 h.
Isoamylase activity assay
IAM activity was assayed with oyster glycogen (Sigma-Aldrich, Seelze, Germany) as substrate by using the method described previously [1, 4] with modification. An aliquot (1 ml) of glycogen substrate solution (2 % w/v) was pre-incubated at 50 °C for 5 min. An aliquot (1 ml) of pre-incubated isoamylase (0.1 M sodium acetate buffer, pH 6.0) was added, stirred on a vortex mixer and incubated at 50 °C. After exactly 30 min at 50 °C, an aliquot (0.2 ml) of the reacted mixture was removed, added to 5 ml of 0.1 % (w/v) iodine solution and stirred vigorously on a vortex mixer. The absorbance at 600 nm was measured immediately (within a minute). Blanks were prepared by mixing 5 ml of 2 % glycogen with 5 ml of sodium acetate buffer (0.1 M, pH 6.0), and an aliquot (0.2 ml) was removed to mix with iodine solution (5 ml). One unit of IAM activity was defined as the amount of enzyme that increased absorbance of the reaction mixture by 0.01 in 1 h under the conditions of the assay.
Other analytical procedures
The protein concentration was determined by Bradford's method [6] using the Bio-Rad (Richmond, CA, USA) protein assay kit, with bovine serum albumin as the standard. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed on a gel composed of 3 and 12 % polyacrylamide for concentration and separation, respectively. Protein bands were visualized in gels by being stained with Coomassie Brilliant Blue R (CBR; Serva, Heidelberg, Germany) as described [13]. The protein standards (NEB, MA, USA) used for estimation of subunit molecular masses were MBP-β-galactosidase (175 kDa), MBP-truncated-β-galactosidase (80 kDa), MBP-CBD (58 kDa), CBD-Mxe intein-2CBD (46 kDa), CBD-Mxe intein (30 kDa) and CBD-E. coli Par (25 kDa).
Biochemical characterization of isoamylase
The effect of temperature on the activity of isoamylase was determined at temperatures between 30 and 80 °C. Furthermore, the effect of temperature on the stability of the enzyme was carried out by incubating 0.1 ml (1.1 μg) isoamylase in 0.05 M sodium phosphate buffer pH 6.0 at 30–80 °C for 1 h without substrates. The residual enzyme activity was measured as described above.
The optimum pH values for the purified enzyme was obtained by incubating the enzyme at room temperature (20 °C) for 5 min with the following buffers: 0.05 M HAc- NaAc (pH 4.0–5.8), 0.05 M sodium phosphate (pH 5.8–7.8) and 0.05 M Tris–HCl (pH 7.8–9.0), respectively, and measuring the activities at 50 °C. The effect of pH on the stability of purified isoamylase was determined by incubating 0.1 ml enzyme without substrates in buffer solutions (pH 4.0–9.0) at room temperature for 1 h. The residual activities were measured as described above.
To determine the kinetic parameters of IAM, reaction system containing 0.05 M sodium phosphate (pH 6) and glycogen, of which the concentration varies from 2 to 10 mg/ml, were incubated at 50 °C for 30 min, then reducing sugar released were quantified using Benedict’s test [5], and the kinetic parameters (K m and V max) were calculated following general procedures.
To investigate the effect of potential activators or inhibitors on activity of purified isoamylase, the enzyme was mixed with various concentrations of chemical agents and incubated with glycogen for 30 min at 50 °C, followed by measuring its activity accordingly. Metal ions, EDTA and citrate were tested at the final concentrations of 0.5 and 5 mM; SDS, phenylmethanesulfonyl fluoride (PMSF) and urea were tested with 30, 5 and 2,000 mM, respectively. Inhibition by incremental of maltose (25–500 mM) and α-cyclodextrin (5–20 mM) (both from Sigma-Aldrich) was assayed under normal conditions. The extent of inhibition or activation of enzyme activity was described as percentage of the ratio of residual activity to complete enzyme activity in the control sample without adding metal ions or chemical agents.
The purified enzyme was examined for its ability to hydrolyze various carbohydrates at 50 °C and at pH 6.0 in 0.05 M sodium phosphate buffer. Among the substrates, glycogen, amylopectin and pullulan were from Sigma; sticky rice starch, maize starch, soluble starch, maltodextrin were purchased from Sinopharm Chemical Reagent Co, Ltd (Shanghai, China).
Internal amino acids sequencing
An in-gel trypsin digestion approach was used prior to sequencing the internal amino acids. The target band stained by CBR was cut from the gel and rinsed three times, in double distilled water. Then, the gel slice was destained twice with 0.2 ml of 100 mM NH4HCO3/50 % acetonitrile (ACN; chromatography grade, Sinopharm Chemical Reagent, Shanghai, China) for 45 min each treatment, at 37 °C to remove CBR. To dehydrate the destained gel slice, 100 % ACN was used at room temperature for 5 min, followed by drying the gel slice in a Speed Vac (Thermo Scientific, Marietta, Ohio, USA) concentrator for 10–15 min at room temperature to remove the ACN. After that, the gel slice was incubated in 40 mM NH4HCO3/10 % ACN containing 20 μg/ml Trypsin Gold (Promega, Wisconsin, USA) at 37 °C overnight to perform digestion. Released peptides were concentrated by freeze drying and analysed by nanoflow liquid chromatography coupled to electrospray ionisation quadrupole time-of-flight tandem mass spectrometry (nanoLC-ESI-Q-TOF/MS/MS). The mass spectrometry was performed on a CapLC with splitting from 6.0 μl/min to 190 nl/min, connected with a nano electrospray interface to a QTOF micro (Waters, Manchester, UK) using MassLynx v4.1 software as operating software. The peptide separation was performed on an Atlantis dC18 NanoEase column (150 × 0.75 mm, 3 μm) (Waters) with an Atlantis dC18 NanoEase precolumn (0.3 × 5 mm, 5 μm particle size) (Waters). The mobile phase A for the nanoLC separation was H2O/ACN (95:5) while the mobile phase B was ACN/H2O (95:5). The chromatographic gradient was set up to give a linear increase after 3 min from 2 % B to 10 % B in 7 min, from 10 % B to 60 % B in 15 min and from 60 % B to 80 % B in 10 min. After 8 min at 80 % B the column is conditioned again at 5 % B for 20 min. The time of a single run was 100 min, and the QTOF was set to scan in profile mode m/z 400–1,800 with 1.9 s per scan and 0.1 s of scan delay. Database search was performed with the MASCOT MS/MS Ion Search against the NCBInr database.
Cloning of the isoamylase gene
Genomic DNA from Bacillus sp. CICIM 304 was prepared by using the DNeasy Kit (Qiagen, Dusseldof, Germany). Based on the two amino acid sequences identified (KIGGEWTE and MGIHDTETM), degenerate primers were designed as follows: forward primer, BIAD1 (5′-AAARTTGGRGRTAAGTGKACYGAAGC-3′); reverse primer, BAID2 (5′-CGYCTCTNTATCATGGATGHCCRT-3′), with Y representing C or T; N representing A, C, G, or T; H representing A, C, or T; R representing A or G and K representing G or T. They were synthesized by Sangon Ltd (Shanghai, China) and then purified with a DNA Refining System (Sangon). PCR was performed in a TP DNA thermal cycler (Biometra, Guttingen, Germany), using each primer (0.2 μg) plus genomic DNA (1.0 μg). The reaction conditions were as follows: 96 °C for 30 s, 52 °C for 30 s, and 72 °C for 2 min for 30 cycles. The reaction mixture contained 200 μM dNTPs, 2.5 mM MgSO4, 25 mM KCL, 5 mM (NH4)2SO4, 1.25 units of ExTaq DNA polymerase (Takara, Dalian, China), and 10 mM Tris–HCl buffer (pH 8.85) in a reaction volume of 50 μl. To determine the complete nucleotide sequence, genomic DNA was digested with VspI and SalI (Takara) respectively, circularized by ligation using T4 polymerase (Takara), and subjected to inverse PCR with the gene-specifice primers Inv-P11 (5′-TTATTGTCGACTCTGTTGCCTATTGG-3′)/Inv-P12 (5′-GTTGAGTCCAGGTCCATTACAACGC-3′) for amplification of 5′-flanking sequence; Inv-P21 (5′-GATCCTGAACAGGCGGTTACGTATG-3′)/Inv-P22 (5′-TGGATACCTTGCAGCACAACCTCTG-3′) for amplification of 3′-flanking sequence. Conditions for inverse PCRs were the same as the above, except for changing the annealing temperature to 53 °C (for primers Inv-P11/P12) or 54 °C (for primers Inv-P21/P22), respectively. The obtained PCR product was purified with a PCR purification kit (Biodev Tech, Beijing, China), cloned and when necessary, DNA fragments that had been amplified were ligated into plasmid pMD18-T simple (Takara). The inserts were sequenced with automated DNA sequencer by Sango Ltd. Analysis of the sequence of gene encoding IAM was performed by a Vienna RNA package software [11].
Preparation of high maltose syrup from starch with IAM
Preparation of high maltose syrup consisted of two stages, namely liquefaction and saccharification. For liquefaction, 1 L of 33 % (w/v) maize starch slurry in 50 mM phosphate buffer (pH 6.5) was mixed with 330 U of bacterial α-amylase (Ruiyang Biotech, Wuxi, China), and incubated at 90 °C for 20 min. Then saccharification was carried out by adding 33 DPo (Diastatic Power) of β-amylase (Ruiyang Biotech), with IAM or pullulanase (Sigma) as auxiliary enzymes, at 60 °C for 12 h.
Chromatography analysis of oligosaccharides
The concentrations of glucose, maltose and maltotriose were determined by an Agilent 1200 HPLC instrument (Waldbronn, Germany) equipped with a quaternary pump with degasser, a series manual injector, a Thermostatted Column Compartment and a refractive index detector (Agilent) under the following conditions: Column, LiChrosorb NH2 (Agilent); solvent, acetonitrile: water (65:35); flow rate, 1 mL/min; column temperature, 40 °C. The reaction mixture (0.5 ml) was added to equal volume of acetonitrile, centrifuged at 10,000g for 10 min at room temperature, filtered through a 0.45 μm filter and injected with a 20 μl loop.
Results
Strain identification and production of extracellular isoamylase
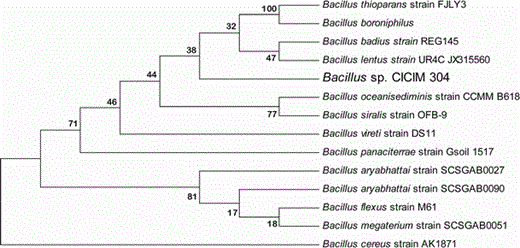
Neighbor-joining phylogenetic tree showing the position of isolated Bacillus sp. CICIM 304 relative to other members of the genus Bacillus. Accession numbers of the sequences used in this study are as follows: Bacillus thioparans strain FJLY3, JN999834.1; Bacillus boroniphilus, AM992181.1; Bacillus badius strain REG145, GQ844977.1; Bacillus lentus strain UR4C, JX315560.1; Bacillus oceanisediminis strain CCMM B618, JN208067.1; Bacillus siralis strain OFB-9, HM439461.1; Bacillus vireti strain DS11, EU834241.1; Bacillus panaciterrae strain Gsoil 1517, NR041379.1; Bacillus aryabhattai strain SCSGAB0027, JX315295.1; Bacillus aryabhattai strain SCSGAB0090, JX315306.1; Bacillus flexus strain M61, JX312636.1; Bacillus megaterium strain SCSGAB0051, JX315300.1; Bacillus cereus strain AK1871, FJ573187.1. Numbers at nodes are percentage bootstrap values based on 1,000 replications
Crude enzyme was prepared in a 30 L fermentor. The whole fermentation process continued for 38 h, i.e. log phase of 10 h and stationary phase of 26 h (S. Fig. 1). After 29 h, isoamylase activity in culture peaked at 13.9 U/ml, then it began to decrease, possibly due to proteases secreted. Finally, 15 L of ferment broth containing 184,500 U of total enzyme activity and 10,423 mg protein was prepared. Supernatant was collected after centrifugation at 8,000×g, 4 °C and stored at 4 °C for IAM purification.
IAM purification
Purification of IAM from Bacillus sp. strain CICIM 304
| Purification step | Total protein (mg) | Total activity (U)a | Specific activity (U/mg) | Purification fold | Yield (%) |
|---|---|---|---|---|---|
| Culture supernatant | 84.7 ± 3.2 | 1,490 ± 40 | 17.7 ± 1.0 | 1 | 100 |
| Ammonium sulfate precipitation | 17.0 ± 0.9 | 1,042 ± 43 | 61.2 ± 2.5 | 3.46 | 69.5 |
| Sephacryl S-100 | 14.1 ± 0.2 | 979 ± 27 | 69.6 ± 2.1 | 3.93 | 65.3 |
| HiPrep 16/10 DEAE FF | 0.4 ± 0.01 | 527 ± 12 | 1,510 ± 33 | 85.5 | 35.1 |
| Mono Q 4.6/100 PE | 0.04 ± 0.01 | 287 ± 10 | 6,535 ± 96 | 369 | 19.2 |
| Purification step | Total protein (mg) | Total activity (U)a | Specific activity (U/mg) | Purification fold | Yield (%) |
|---|---|---|---|---|---|
| Culture supernatant | 84.7 ± 3.2 | 1,490 ± 40 | 17.7 ± 1.0 | 1 | 100 |
| Ammonium sulfate precipitation | 17.0 ± 0.9 | 1,042 ± 43 | 61.2 ± 2.5 | 3.46 | 69.5 |
| Sephacryl S-100 | 14.1 ± 0.2 | 979 ± 27 | 69.6 ± 2.1 | 3.93 | 65.3 |
| HiPrep 16/10 DEAE FF | 0.4 ± 0.01 | 527 ± 12 | 1,510 ± 33 | 85.5 | 35.1 |
| Mono Q 4.6/100 PE | 0.04 ± 0.01 | 287 ± 10 | 6,535 ± 96 | 369 | 19.2 |
a1 U = the amount of enzyme capable of resulting 0.01 increase in absorbance at 600 nm per hour at 50 °C
Purification of IAM from Bacillus sp. strain CICIM 304
| Purification step | Total protein (mg) | Total activity (U)a | Specific activity (U/mg) | Purification fold | Yield (%) |
|---|---|---|---|---|---|
| Culture supernatant | 84.7 ± 3.2 | 1,490 ± 40 | 17.7 ± 1.0 | 1 | 100 |
| Ammonium sulfate precipitation | 17.0 ± 0.9 | 1,042 ± 43 | 61.2 ± 2.5 | 3.46 | 69.5 |
| Sephacryl S-100 | 14.1 ± 0.2 | 979 ± 27 | 69.6 ± 2.1 | 3.93 | 65.3 |
| HiPrep 16/10 DEAE FF | 0.4 ± 0.01 | 527 ± 12 | 1,510 ± 33 | 85.5 | 35.1 |
| Mono Q 4.6/100 PE | 0.04 ± 0.01 | 287 ± 10 | 6,535 ± 96 | 369 | 19.2 |
| Purification step | Total protein (mg) | Total activity (U)a | Specific activity (U/mg) | Purification fold | Yield (%) |
|---|---|---|---|---|---|
| Culture supernatant | 84.7 ± 3.2 | 1,490 ± 40 | 17.7 ± 1.0 | 1 | 100 |
| Ammonium sulfate precipitation | 17.0 ± 0.9 | 1,042 ± 43 | 61.2 ± 2.5 | 3.46 | 69.5 |
| Sephacryl S-100 | 14.1 ± 0.2 | 979 ± 27 | 69.6 ± 2.1 | 3.93 | 65.3 |
| HiPrep 16/10 DEAE FF | 0.4 ± 0.01 | 527 ± 12 | 1,510 ± 33 | 85.5 | 35.1 |
| Mono Q 4.6/100 PE | 0.04 ± 0.01 | 287 ± 10 | 6,535 ± 96 | 369 | 19.2 |
a1 U = the amount of enzyme capable of resulting 0.01 increase in absorbance at 600 nm per hour at 50 °C
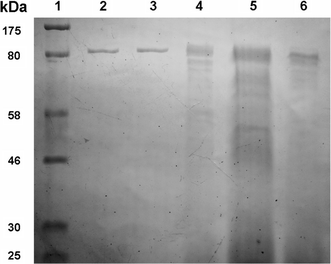
SDS-PAGE profile of IAM purification. Lane 1 molecular mass standard (size as indicated); Lanes 2, 3 active fraction by Mono Q ion exchange chromatography; Lane 4 active fraction by Sephacryl S-100 gel filtration chromatography; Lane 5 active fraction by ammonium sulfate precipitation; Lane 6 active fraction by DEAE ion exchange chromatography
Biochemical characterization of IAM
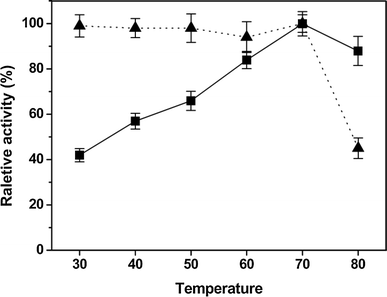
Effects of temperature on activity (filled square) and stability (filled triangle) of purified IAM. Experiments were performed in triplicate and relative activity was calculated by activity at 70 °C. The average of relative value (N = 3) and error bars are shown
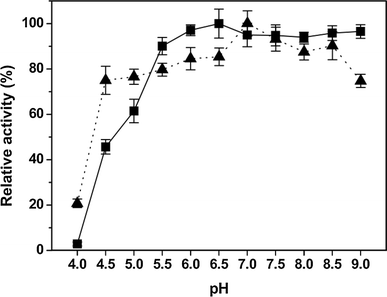
Effects of pH on activity (filled square) and stability (filled triangle) of purified IAM. Experiments were performed in triplicate and relative activity was calculated by activity at pH 6.5. The average of relative value (N = 3) and error bars are shown
IAM followed typical Michaelis–Menten kinetics. Using the Lineweaver–Burk plot, a K m value of 0.403 ± 0.018 mg/mg and a V max value of 0.018 ± 0.001 mg/(min mg) was established for glycogen hydrolysis. Comparison of these two kinetic parameters with those of previously characterized IAM did not provide great interesting or prominent information, whereas it was obvious that IAM could efficiently hydrolyze substrates with high ratio of branched chains such as glycogen, amylopectin, sticky rice, maize starch, soluble starch and maltodextrin (Table 2). So it was noteworthy that the IAM had scarcely any effect on pullulan, which supported it to be an isoamylase-type DBE.
Relative activity of the purified IAM on different carbohydrate compounds
| Substrate | Relative activitya |
|---|---|
| Glycogen | 100 ± 4 |
| Amylopectin | 82 ± 2 |
| Sticky rice | 73 ± 3 |
| Maize starch | 72 ± 2 |
| Soluble starch | 68 ± 3 |
| Maltodextrin | 54 ± 1 |
| Pullulan | 4 ± 0.1 |
| Substrate | Relative activitya |
|---|---|
| Glycogen | 100 ± 4 |
| Amylopectin | 82 ± 2 |
| Sticky rice | 73 ± 3 |
| Maize starch | 72 ± 2 |
| Soluble starch | 68 ± 3 |
| Maltodextrin | 54 ± 1 |
| Pullulan | 4 ± 0.1 |
aExpressed as a percentage of the activity against glycogen, which was given a value of 100 %
Relative activity of the purified IAM on different carbohydrate compounds
| Substrate | Relative activitya |
|---|---|
| Glycogen | 100 ± 4 |
| Amylopectin | 82 ± 2 |
| Sticky rice | 73 ± 3 |
| Maize starch | 72 ± 2 |
| Soluble starch | 68 ± 3 |
| Maltodextrin | 54 ± 1 |
| Pullulan | 4 ± 0.1 |
| Substrate | Relative activitya |
|---|---|
| Glycogen | 100 ± 4 |
| Amylopectin | 82 ± 2 |
| Sticky rice | 73 ± 3 |
| Maize starch | 72 ± 2 |
| Soluble starch | 68 ± 3 |
| Maltodextrin | 54 ± 1 |
| Pullulan | 4 ± 0.1 |
aExpressed as a percentage of the activity against glycogen, which was given a value of 100 %
Effect of metal ions and different reagents on enzyme activity were examined (Table 3). At 50 °C, Mg2+, Ca2+ and Co2+ as chloride salts at a concentration of 0.5 mM and 5 mM, slightly enhanced the enzyme activity. The enzyme was stimulated strongly by Mn2+ (up to 141.31 % at a concentration of 5 mM). However, Zn2+, Fe3+ and Sn2+ reduced the activity to less than 50 % of control levels, while Cu2+ and Hg2+ completely inactivated the enzyme. EDTA, citrate and phenylmethanesulfonyl fluoride (PMSF) were not inhibitory, on the contrary, 2 M urea and 30 mM SDS led to significant loss of activity. Maltose and α-cyclodextrin up to a concentration of 20 % and 20 mM did not influence the activity of the enzyme. A 4 % inhibition of the activity was observed in the presence of 50 mM α-cyclodextrin. In contrast to this, the effect of β-cyclodextrin on isoamylase was more pronounced. In the presence of 20 mM of this compound, only 37 % of the residual activity was measured.
Effect of different reagents on the purified IAM
| Compound | Concentration (mM) | Relative activity (% ± SD)a |
|---|---|---|
| Control | – | 100.8 ± 2.1 |
| Li+ | 0.5 | 97.9 ± 3.0 |
| 5 | 95.3 ± 1.9 | |
| Mg2+ | 0.5 | 102.3 ± 3.2 |
| 5 | 107.7 ± 3.1 | |
| Zn2+ | 0.5 | 36.4 ± 1.7 |
| 5 | 11.9 ± 0.1 | |
| Ca2+ | 0.5 | 110.0 ± 2.4 |
| 5 | 125.4 ± 1.9 | |
| Mn2+ | 0.5 | 128.0 ± 2.7 |
| 5 | 141.4 ± 2.7 | |
| Fe3+ | 0.5 | 41.1 ± 0.5 |
| 5 | 9.9 ± 0.1 | |
| Na+ | 0.5 | 101.8 ± 2.2 |
| 5 | 105.7 ± 2.5 | |
| K+ | 0.5 | 100.6 ± 1.7 |
| 5 | 105.2 ± 3.0 | |
| Co2+ | 0.5 | 101.3 ± 3.2 |
| 5 | 112.3 ± 2.1 | |
| Sn2+ | 0.5 | 72.4 ± 2.4 |
| 5 | 27.3 ± 0.4 | |
| Cu2+ | 0.5 | ND |
| Hg2+ | 0.5 | ND |
| EDTA | 0.5 | 100.9 ± 1.2 |
| 5 | 97.9 ± 1.6 | |
| Citrate | 0.5 | 99.2 ± 0.9 |
| 5 | 93.7 ± 1.5 | |
| SDS | 30 | 22.9 ± 0.2 |
| PMSF | 5 | 91.0 ± 1.8 |
| Urea | 2,000 | 16.8 ± 0.1 |
| Compound | Concentration (mM) | Relative activity (% ± SD)a |
|---|---|---|
| Control | – | 100.8 ± 2.1 |
| Li+ | 0.5 | 97.9 ± 3.0 |
| 5 | 95.3 ± 1.9 | |
| Mg2+ | 0.5 | 102.3 ± 3.2 |
| 5 | 107.7 ± 3.1 | |
| Zn2+ | 0.5 | 36.4 ± 1.7 |
| 5 | 11.9 ± 0.1 | |
| Ca2+ | 0.5 | 110.0 ± 2.4 |
| 5 | 125.4 ± 1.9 | |
| Mn2+ | 0.5 | 128.0 ± 2.7 |
| 5 | 141.4 ± 2.7 | |
| Fe3+ | 0.5 | 41.1 ± 0.5 |
| 5 | 9.9 ± 0.1 | |
| Na+ | 0.5 | 101.8 ± 2.2 |
| 5 | 105.7 ± 2.5 | |
| K+ | 0.5 | 100.6 ± 1.7 |
| 5 | 105.2 ± 3.0 | |
| Co2+ | 0.5 | 101.3 ± 3.2 |
| 5 | 112.3 ± 2.1 | |
| Sn2+ | 0.5 | 72.4 ± 2.4 |
| 5 | 27.3 ± 0.4 | |
| Cu2+ | 0.5 | ND |
| Hg2+ | 0.5 | ND |
| EDTA | 0.5 | 100.9 ± 1.2 |
| 5 | 97.9 ± 1.6 | |
| Citrate | 0.5 | 99.2 ± 0.9 |
| 5 | 93.7 ± 1.5 | |
| SDS | 30 | 22.9 ± 0.2 |
| PMSF | 5 | 91.0 ± 1.8 |
| Urea | 2,000 | 16.8 ± 0.1 |
aExperiments were performed in triplicate and relative activity was calculated by activity untreated control. The average of relative value (N = 3) and standard deviations (SD) are shown. ND not detected
Effect of different reagents on the purified IAM
| Compound | Concentration (mM) | Relative activity (% ± SD)a |
|---|---|---|
| Control | – | 100.8 ± 2.1 |
| Li+ | 0.5 | 97.9 ± 3.0 |
| 5 | 95.3 ± 1.9 | |
| Mg2+ | 0.5 | 102.3 ± 3.2 |
| 5 | 107.7 ± 3.1 | |
| Zn2+ | 0.5 | 36.4 ± 1.7 |
| 5 | 11.9 ± 0.1 | |
| Ca2+ | 0.5 | 110.0 ± 2.4 |
| 5 | 125.4 ± 1.9 | |
| Mn2+ | 0.5 | 128.0 ± 2.7 |
| 5 | 141.4 ± 2.7 | |
| Fe3+ | 0.5 | 41.1 ± 0.5 |
| 5 | 9.9 ± 0.1 | |
| Na+ | 0.5 | 101.8 ± 2.2 |
| 5 | 105.7 ± 2.5 | |
| K+ | 0.5 | 100.6 ± 1.7 |
| 5 | 105.2 ± 3.0 | |
| Co2+ | 0.5 | 101.3 ± 3.2 |
| 5 | 112.3 ± 2.1 | |
| Sn2+ | 0.5 | 72.4 ± 2.4 |
| 5 | 27.3 ± 0.4 | |
| Cu2+ | 0.5 | ND |
| Hg2+ | 0.5 | ND |
| EDTA | 0.5 | 100.9 ± 1.2 |
| 5 | 97.9 ± 1.6 | |
| Citrate | 0.5 | 99.2 ± 0.9 |
| 5 | 93.7 ± 1.5 | |
| SDS | 30 | 22.9 ± 0.2 |
| PMSF | 5 | 91.0 ± 1.8 |
| Urea | 2,000 | 16.8 ± 0.1 |
| Compound | Concentration (mM) | Relative activity (% ± SD)a |
|---|---|---|
| Control | – | 100.8 ± 2.1 |
| Li+ | 0.5 | 97.9 ± 3.0 |
| 5 | 95.3 ± 1.9 | |
| Mg2+ | 0.5 | 102.3 ± 3.2 |
| 5 | 107.7 ± 3.1 | |
| Zn2+ | 0.5 | 36.4 ± 1.7 |
| 5 | 11.9 ± 0.1 | |
| Ca2+ | 0.5 | 110.0 ± 2.4 |
| 5 | 125.4 ± 1.9 | |
| Mn2+ | 0.5 | 128.0 ± 2.7 |
| 5 | 141.4 ± 2.7 | |
| Fe3+ | 0.5 | 41.1 ± 0.5 |
| 5 | 9.9 ± 0.1 | |
| Na+ | 0.5 | 101.8 ± 2.2 |
| 5 | 105.7 ± 2.5 | |
| K+ | 0.5 | 100.6 ± 1.7 |
| 5 | 105.2 ± 3.0 | |
| Co2+ | 0.5 | 101.3 ± 3.2 |
| 5 | 112.3 ± 2.1 | |
| Sn2+ | 0.5 | 72.4 ± 2.4 |
| 5 | 27.3 ± 0.4 | |
| Cu2+ | 0.5 | ND |
| Hg2+ | 0.5 | ND |
| EDTA | 0.5 | 100.9 ± 1.2 |
| 5 | 97.9 ± 1.6 | |
| Citrate | 0.5 | 99.2 ± 0.9 |
| 5 | 93.7 ± 1.5 | |
| SDS | 30 | 22.9 ± 0.2 |
| PMSF | 5 | 91.0 ± 1.8 |
| Urea | 2,000 | 16.8 ± 0.1 |
aExperiments were performed in triplicate and relative activity was calculated by activity untreated control. The average of relative value (N = 3) and standard deviations (SD) are shown. ND not detected
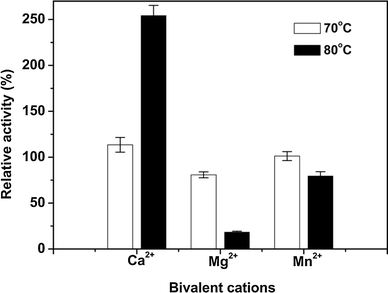
Effects of metal ions on thermostability of purified enzyme. IAM was incubated at 70 °C (empty square) and 80 °C (filled square) for 1 h with 5 mM bivalent cations of Ca2+, Mg2+ and Mn2+, respectively, as protector. The residual activity was expressed as a percentage of that obtained before incubation, which was given a value of 100 %. Identical results were obtained in two independent experiments; data from one experiment are presented
Internal amino acids sequencing
The N-terminal amino acid sequencing of IAM was firstly attempted by the automated Edman degradation method but was not successful, suggesting that the N terminus of IAM might be blocked. Therefore, trypsin digestion of IAM and analysis of the resulting peptides by nanoLC-ESI-Q-TOF/MS/MS was attempted. Search results by NCBInr database showed that sequences of six peptides was highly homologous to that of the proteins relative to debranching enzymes. Two of them were KIGGEWTE and MGIHDTETM based on which degenerate primers were designed.
Cloning and sequence analysis of isoamylase gene
Sequence of a 2,940-bp section in genomic DNA from Bacillus sp. CICIM 304 containing isoamylase encoding gene (BsIam, Genbank accession number JX018171) was identified, with an open reading frame of 2,655 bp, started from an ATG codon and ended with a TAA stop codon. A putative ribosome-binding site (Shine Delgarno sequence, AAGAAGGAGA) was observed at 6–15 bp upstream from the ATG codon. The sequence would have a free energy (Δ G) of −1,013.17 kcal/mol (4,240.98 kJ/mol), as calculated by the method of Gruber et al. [11]. Analysis of the sequence upstream of the initiation codon showed that no presumable promoter region could be identified. According to the system proposed by Henrissat and Bairoch [14], the amino acid sequence of IAM reveals it to be a member of glycoside hydrolase family 13. Further analysis of the conserved domains suggested that IAM had a carbohydrate-binding module family 48 (CBM48) domain, which was often found in a range of enzymes that acted on branched substrates, i.e. isoamylase, pullulanase and branching enzymes. CBM48 specifically binds glycogen [15, 18, 22, 34]. The deduced amino acid sequence showed significant identity with several debranching enzymes: pullulanase from Bacillus megaterium QM B1551 (64 %) and Bacillus cereus Rock1-3(61 %), and alpha-dextran endo-1,6-alpha-glucosidase from Bacillus cereus G9241 (60 %), while less than 40 % identity with microbial isoamylase was ever reported, which further indicated that this enzyme is a novel isoamylase.
Preparation of high maltose syrup from starch using isoamylase
High maltose syrup was prepared from 33 % (w/v) maize starch by means of a liquefaction-saccharification strategy. Liquefaction was carried out with 1 U/gstarch α-amylase at 90 °C for 20 min and immediately followed by a sterilization course to denature liquefying enzymes. At this time, reducing sugar accounted for about 18 % (w/w) in liquefied solution by Benedict’s test. Then, a set of saccharifications was made by using β-amylase combined with pullulanase (Aerobacter aerogenes) or purified isoamylase from Bacillus sp. CICIM 304. From HPLC diagram, maltose was the main product, accompanied by a certain amount of maltotriose. As expected, the yield of both maltose and maltotriose increased when isoamylase was cooperated with β-amylase as an auxiliary enzyme. And 28.2 g/l more maltose and 16.5 g/l more maltotriose were released from starch (Supplemental Table 1; Supplemental Fig. 4). Compared with that done by the same amount of pullulanase (equal debranching activity), 6.6 and 0.6 % promotion could be observed in yield of maltose and maltotriose.
Discussion
Thermoactive and thermostable debranching enzyme is valuable in starch processing industry [10], as high efficient enzymatic liquefaction and saccharification are performed at elevated temperature (up to 117 °C). The current starch conversion techniques need a cooling step to 60 °C or below before saccharification [26] because the commercially available saccharification enzymes are unstable above 60 °C. The application of thermostable debranching enzymes could dramatically improve the energy- and time-consuming process. Here, a novel thermostable isoamylase, IAM from a newly isolated strain Bacillus sp. CICIM 304 was purified and characterized, which worked well at 70 °C and preferred alkaline and neutral conditions. The enzyme can hydrolyze glycogen completely and hydrolyze part of starch. Therefore, the enzyme will be very helpful in starch debranching process.
Purified IAM was a monomer with a molecular mass of about 100 kDa, which was similar to that of P. amyloderamosa [3] or Xanthomonas maltophilia [35] isoamylase. Its molecular mass was quite different from that of isoamylase in bacilli strains, whose molecular mass ranges from 40 to 60 kDa. Meanwhile, IAM exhibited an invariable maximal activity from pH 5.5 to 9.0, whose catalyzing pH range (4.5–9.0) was wider than that of most isoamylases ever reported. The enzymes of bacterial origin usually share a neutral and weak acid catalytic pH ranges. IAM’s most prominent properties were concerning temperature. Its optimal activity occurred at 70 °C and it was highly stable between 30 and 70 °C, clearly superior to most isoamylases ever reported. Ca2+ was shown to be both an activator and a protector of the IAM at high temperature, while heavy metal ions inhibit the enzyme a lot. What is more, EDTA was not inhibitory, indicating that bivalent cations such as Ca2+ and Mn2+ were not necessary although they could stimulate the enzyme.
Amylolytic enzymes are widely distributed among microorganisms and all of their members have different catalytic properties and structures. As they share similar substrates and modes of action, strong similarities exist in their amino acid sequences between substrate binding sites and catalytic sites [23]. Comparing IAM’s amino acid sequence with that of other starch degrading enzymes revealed that the enzyme appeared to be a novel one displaying less than 55 % sequence identity with the debranching enzymes known. The deduced amino acid sequence of IAM suggested that the enzyme had six calcium binding sites (http://www.ncbi.nlm.nih.gov/Structure/), similarly, Pseudomonas isoamylase contains calcium binding sites according to the three-dimensional structure determined by X-ray diffraction analysis [18]. Therefore, calcium is required to maintain the structural integrity, and removal of calcium leads to decreased thermostability and/or decreased enzymatic activity [31, 33]. Suzuki et al. [28] stated that enhanced thermostability of Bacillus oligo-1,6-glucosidases could be gained by increasing the frequency of praline occurrence at β-turns and the total hydrophobic residues, which was referred to as the proline theory. In amino acid composition of IAM, proline, phenylalanine and leucine account for 15 % (w/w), which may help improve the potential of hydrophobic interaction and thus contribute to enhancing thermostability. It was worth noting that addition of calcium strengthens the IAM both in catalysis and thermostability (unpublished results). Three of the six calcium binding sites are found in catalytic domain, resembling the three found in the A domain of pullulanase from Klebsiella pneumoniae [21]. They are thought to be involved in constitution of an octahedral geometry being interacted with water molecules or other amino acid residues nearby, which may contribute to increasing thermostability of the enzyme.
Our screening method by using amylopectin plates was not designed specifically for isoamylase or pullulanase, but the enzyme was preliminarily designated as an isoamylase for its little effect on pullulan after characterizing. Furthermore, IAM could not be inhibited by cyclodextrins and maltose (data not shown), which supports Marshall’s [19] suggestion about sensitivity toward cyclodextrins as an indicator for differentiating isoamylase and pullulanase. In starch processing industries, maltose is usually produced by β-amylase. When β-amylase acts on liquefied starch, amylose is converted almost completely into maltose, whereas amylopectin and other branched polymers are hydrolyzed to various extents depending on the degree of branching. As it is invalid to α-1,6 linkages, conversion yield of starch to maltose by β-amylase is only about 50 % [12]. Debranching enzymes (DBE), including isoamylase and pullulanase, are common auxiliary enzyme of β-amylase in maltose production for its capability of hydrolyzing branches in starch. Efficiency of the two DBEs in maltose production has been studied with different techniques. Shaw [27] tested the two enzymes with rice starch as substrate. He found that pullulanase alone was better than isoamylase alone, 5 U of pullulanase being more effective than 2,000 U of P. amyloderamosa isoamylase. The inefficiency of P. amyloderamosa isoamylase is due to the fact that it is thermolabile and looses its activity quickly at 50 °C, a universal temperature for saccharification by β-amylase. In this study, IAM from Bacillus sp. CICIM 304 was used in combination with β-amylase to produce maltose with liquefied starch at 60 °C for 12 h (Supplemental Fig. 4). Unlike the one from P. amyloderamosa, IAM obtained a higher yield of maltose than that of pullulanase, which displayed IAM’s advantages over pullulanase. That results proved what was suggested above, i.e. isoamylase excels pullulanase in cutting ways and reversibility, so theoretically the conversion yield of the former enzyme is higher in maltose production. IAM may prove superior to be an auxiliary enzyme for β-amylase in saccharification. The novel isoamylase may also be useful for liquefaction for its thermostability and thermoactivity.
Acknowledgments
This study was granted by China-South Africa Joint project (2009DFA31300), the ‘863’ program (2011AA100905), Program for New Century Excellent Talents in University (NCET-11-0665), Innovative Research Team of Jiangsu Province, the Priority Academic Program Development of Jiangsu Higher Education Institutions and the 111 Project (No. 111-2-06).

{kind=link}
{kind=link}
{kind=link}
{kind=link}