





Abstract
α-Ketoglutarate is accumulated as the main byproduct during the aerobic succinate production from glycerol by Escherichia coli BL21(DE3) in minimal medium. To address this issue, here a strategy of directed pathway evolution was developed to enhance the alternative succinate production route—the glyoxylate shunt. Via the directed pathway evolution, the glyoxylate shunt was recruited as the primary anaplerotic pathway in a ppc mutant, which restored its viability in glycerol minimal medium. Subsequently, the operon sdhCDAB was deleted and the gene ppc was reverted in the evolved strain for succinate production. The resulting strain E2-Δsdh-ppc produced 30 % more succinate and 46 % less α-ketoglutarate than the control strain. A G583T mutation in gene icdA, which significantly decreased the activity of isocitrate dehydrogenase, was identified in the evolved strain as the main mutation responsible for the observed phenotype. Overexpression of α-ketoglutarate dehydrogenase complex in E2-Δsdh-ppc further reduced the amount of byproduct and improved succinate production. The final strain E2-Δsdh-ppc-sucAB produced 366 mM succinate from 1.3 M glycerol in minimal medium in fed-batch fermentation. The maximum and average succinate volumetric productivities were 19.2 and 6.55 mM h−1, respectively, exhibiting potential industrial production capacity from the low-priced substrate.
Introduction
As the biodiesel industry increases significantly worldwide, glycerol has become an abundant and inexpensive feedstock for industrial microbiology as the major by-product of biodiesel [10]. The production of fuels and valuable chemicals through microbial fermentation of glycerol by many microorganisms such as Citrobacter freundii, Klebsiella pneumonia, Clostridium pasteurianum, Clostridium butyricum, Enterobacter aerogenes, and Lactobacillus reuteri [8] is becoming an attractive field, including organic acids, 1,3-propanediol, ethanol, butanol, and so on [11].
Succinate as a valued platform compound, which has significant applications in agricultural, food, and pharmaceutical industries, has been studied for many years in the aspect of microbial production using several different microorganisms [2]. Production of succinate from glycerol fermentation has been primarily focusing on A. succiniciproducens [18, 19], B. succiniciproducens DD1 [37, 38], A. succinogenes [41], Y. lipolytica [45], and E. coli [3, 47]. In particular, the succinate production performance of E. coli from glycerol under anaerobic conditions was examined recently. Metabolically engineered E. coli strain (XZ721) showed the capability to produce 102 mM succinate with a succinate yield of 0.8 mol/mol glycerol under anaerobic conditions [47]. On the other hand, E. coli was engineered to produce succinate from glycerol under microaerobic conditions with a yield of 0.54 mol/mol glycerol [3].
The metabolism of glycerol in E. coli under anaerobic conditions in the absence of external electron acceptors was previously reported [10, 13, 28]. However, the growth of E. coli is extremely slow with an average specific growth rate of 0.040 ± 0.003 h−1 in glycerol low-supplement minimal medium [28]. In accordance with this, metabolically engineered E. coli strain (XZ721) had a long fermentation period of 6 days [47]. Even though the fermentation period was shortened under microaerobic conditions, the productivity was still not sufficient for the industrial application [3]. On the other hand, designing aerobic succinate production system can overcome the drawbacks of anaerobic fermentation, with the advantages of fast growth, no need of rich supplements, and high productivity.
Strategies employed for the construction of aerobic succinate producers primarily focus on the interruption of TCA cycle for accumulation succinate as the end product, adoption of the glyoxylate cycle as alternative succinate route, overexpression of carboxylation enzymes, and blocking competing by-product pathways [21–24, 35, 45]. Although the maximum theoretical yield of 1.0 mol/mol glucose has been achieved by metabolic engineered E. coli in rich nutrient medium [23], the succinate yield in defined minimal medium using glycerol as the carbon source has not been reported. Being one of the two potential routes for succinate production under aerobic conditions, the glyoxylate shunt is significantly important for improving succinate production. Several efforts has been made to functionalize the glyoxylate cycle, which is generally active with acetate as the sole carbon source [42], including deletion of the repressor gene iclR in E. coli [22] and overexpression of the glyoxylate shunt genes in Corynebacterium glutamicum [24]. However, due to the existence of some other regulation of intracellular metabolic flux, these efforts had limited effect.
Evolutionary engineering as a widely used tool for strain development in industrial microbiology has many applications including latent pathway activation, phenotype optimization, and environmental adaptation [34]. Instead of rational engineering, evolutionary engineering has the capacity to address issues in a nonintuitive way. Nowadays, the aim of evolutionary engineering generally focused on the improvement of nonnative substrates utility or tolerance to fermentative products. Here we adopted evolutionary engineering to improve the flux of the glyoxylate cycle for improved aerobic succinate production from glycerol in engineered E. coli. This was accomplished by deletion of phosphoenolpyruvate carboxylase (PPC) and couple the glyoxylate shunt to cell viability in glycerol minimal medium.
Materials and methods
Strains, plasmids, and primers
The strains and plasmids used in this study are listed in Table 1. The primers used for gene knockout and promoter replacement are listed in Table 2. E. coli strain BL21(DE3) was used as a host strain for succinate production and E. coli DH5α was used for gene cloning. For plasmids construction and genetic manipulations, Luria–Bertani (LB) broth was used for culture. As appropriate, ampicillin (100 μg/ml), kanamycin (24 μg/ml), and chloramphenicol (4 μg/ml) were supplemented.
Escherichia coli strains and plasmids used in this study
| Strains/plasmids | Relevant characteristics | Reference/source |
|---|---|---|
| Strains | ||
| DH5α | Cloning host | Lab collection |
| BL21(DE3) | F’ ompT gal dcm lon hsdSB(rB- mB-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | Novagen |
| CP | BL21(DE3), ΔsdhCDAB, ppc:trc, aceBAK:trc | This study |
| C1 | BL21(DE3), Δppc | This study |
| C2 | C1, aceBAK:trc | This study |
| E1 | Evolved C2 | This study |
| E2 | Evolved E1 | This study |
| E2-ΔaceBA | E2, ΔaceBA | This study |
| E2-Δpck | E2, Δpck | This study |
| E2-ΔmaeA | E2, ΔmaeA | This study |
| E2-ΔmaeB | E2, ΔmaeB | This study |
| E2-Δsdh | E2, ΔsdhCDAB | This study |
| E2-Δsdh-ppc | E2, ΔsdhCDAB, ppc:trc | This study |
| E2-Δsdh-ppc-sucAB | E2, ΔsdhCDAB, ppc:trc,sucAB:trc | This study |
| C2-583 | C2, G583T icdA | This study |
| CP-583 | CP, G583T icdA | This study |
| E2-Δsdh-ppc-583 | E2-Δsdh-ppc, wild-type icdA | This study |
| C2-583-gltA | C2-583, gltA:trc | This study |
| CP-583-gltA | CP-583, gltA:trc | This study |
| Plasmids | ||
| pUC18 | bla, pBR322 ori | Lab collection |
| pEL04 | cat-sacB cassette | [17] |
| pTrc99a | bla, pBR322 ori, trc promoter, lacIq | Invitrogen |
| pKD3 | bla, FRT-cat-FRT | [9] |
| pKD4 | bla, FRT-kan-FRT | [9] |
| pCP20 | bla, cat, yeast Flp recombinase | [6] |
| pKD46 | bla, λ-Red recombinase under araBAD promoter, temperature-conditional replicon | [9] |
| pET-28a(+) | Kan, pBR322 ori, T7 promoter, lacI | Novagen |
| pCM01 | Bla, cat, cat from pKD3 cloned into pUC18 | This study |
| pCMppcHR | bla, cat, homologous regions of 600 bp positioned upstream and downstream of the gene ppc cloned into pCM01 | This study |
| pUCtrc | Bla, trc promoter from pTrc99a cloned into pUC18 | This study |
| pKMtrc | Bla, kan, kan from pKD4 cloned into pUCtrc | This study |
| pCMtrc | Bla, cat, cat from pKD3 cloned into pUCtrc | This study |
| pCMtrcHR | Bla, cat, homologous regions of 500 bp and 900 bp positioned upstream and downstream of the gene sucAB cloned into pCMtrc | This study |
| pCSHR | Bla, cat-sacB, homologous regions of 500 bp positioned upstream and downstream of the gene icdA and cat-sacB cassette cloned into pUC18 | This study |
| Strains/plasmids | Relevant characteristics | Reference/source |
|---|---|---|
| Strains | ||
| DH5α | Cloning host | Lab collection |
| BL21(DE3) | F’ ompT gal dcm lon hsdSB(rB- mB-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | Novagen |
| CP | BL21(DE3), ΔsdhCDAB, ppc:trc, aceBAK:trc | This study |
| C1 | BL21(DE3), Δppc | This study |
| C2 | C1, aceBAK:trc | This study |
| E1 | Evolved C2 | This study |
| E2 | Evolved E1 | This study |
| E2-ΔaceBA | E2, ΔaceBA | This study |
| E2-Δpck | E2, Δpck | This study |
| E2-ΔmaeA | E2, ΔmaeA | This study |
| E2-ΔmaeB | E2, ΔmaeB | This study |
| E2-Δsdh | E2, ΔsdhCDAB | This study |
| E2-Δsdh-ppc | E2, ΔsdhCDAB, ppc:trc | This study |
| E2-Δsdh-ppc-sucAB | E2, ΔsdhCDAB, ppc:trc,sucAB:trc | This study |
| C2-583 | C2, G583T icdA | This study |
| CP-583 | CP, G583T icdA | This study |
| E2-Δsdh-ppc-583 | E2-Δsdh-ppc, wild-type icdA | This study |
| C2-583-gltA | C2-583, gltA:trc | This study |
| CP-583-gltA | CP-583, gltA:trc | This study |
| Plasmids | ||
| pUC18 | bla, pBR322 ori | Lab collection |
| pEL04 | cat-sacB cassette | [17] |
| pTrc99a | bla, pBR322 ori, trc promoter, lacIq | Invitrogen |
| pKD3 | bla, FRT-cat-FRT | [9] |
| pKD4 | bla, FRT-kan-FRT | [9] |
| pCP20 | bla, cat, yeast Flp recombinase | [6] |
| pKD46 | bla, λ-Red recombinase under araBAD promoter, temperature-conditional replicon | [9] |
| pET-28a(+) | Kan, pBR322 ori, T7 promoter, lacI | Novagen |
| pCM01 | Bla, cat, cat from pKD3 cloned into pUC18 | This study |
| pCMppcHR | bla, cat, homologous regions of 600 bp positioned upstream and downstream of the gene ppc cloned into pCM01 | This study |
| pUCtrc | Bla, trc promoter from pTrc99a cloned into pUC18 | This study |
| pKMtrc | Bla, kan, kan from pKD4 cloned into pUCtrc | This study |
| pCMtrc | Bla, cat, cat from pKD3 cloned into pUCtrc | This study |
| pCMtrcHR | Bla, cat, homologous regions of 500 bp and 900 bp positioned upstream and downstream of the gene sucAB cloned into pCMtrc | This study |
| pCSHR | Bla, cat-sacB, homologous regions of 500 bp positioned upstream and downstream of the gene icdA and cat-sacB cassette cloned into pUC18 | This study |
Escherichia coli strains and plasmids used in this study
| Strains/plasmids | Relevant characteristics | Reference/source |
|---|---|---|
| Strains | ||
| DH5α | Cloning host | Lab collection |
| BL21(DE3) | F’ ompT gal dcm lon hsdSB(rB- mB-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | Novagen |
| CP | BL21(DE3), ΔsdhCDAB, ppc:trc, aceBAK:trc | This study |
| C1 | BL21(DE3), Δppc | This study |
| C2 | C1, aceBAK:trc | This study |
| E1 | Evolved C2 | This study |
| E2 | Evolved E1 | This study |
| E2-ΔaceBA | E2, ΔaceBA | This study |
| E2-Δpck | E2, Δpck | This study |
| E2-ΔmaeA | E2, ΔmaeA | This study |
| E2-ΔmaeB | E2, ΔmaeB | This study |
| E2-Δsdh | E2, ΔsdhCDAB | This study |
| E2-Δsdh-ppc | E2, ΔsdhCDAB, ppc:trc | This study |
| E2-Δsdh-ppc-sucAB | E2, ΔsdhCDAB, ppc:trc,sucAB:trc | This study |
| C2-583 | C2, G583T icdA | This study |
| CP-583 | CP, G583T icdA | This study |
| E2-Δsdh-ppc-583 | E2-Δsdh-ppc, wild-type icdA | This study |
| C2-583-gltA | C2-583, gltA:trc | This study |
| CP-583-gltA | CP-583, gltA:trc | This study |
| Plasmids | ||
| pUC18 | bla, pBR322 ori | Lab collection |
| pEL04 | cat-sacB cassette | [17] |
| pTrc99a | bla, pBR322 ori, trc promoter, lacIq | Invitrogen |
| pKD3 | bla, FRT-cat-FRT | [9] |
| pKD4 | bla, FRT-kan-FRT | [9] |
| pCP20 | bla, cat, yeast Flp recombinase | [6] |
| pKD46 | bla, λ-Red recombinase under araBAD promoter, temperature-conditional replicon | [9] |
| pET-28a(+) | Kan, pBR322 ori, T7 promoter, lacI | Novagen |
| pCM01 | Bla, cat, cat from pKD3 cloned into pUC18 | This study |
| pCMppcHR | bla, cat, homologous regions of 600 bp positioned upstream and downstream of the gene ppc cloned into pCM01 | This study |
| pUCtrc | Bla, trc promoter from pTrc99a cloned into pUC18 | This study |
| pKMtrc | Bla, kan, kan from pKD4 cloned into pUCtrc | This study |
| pCMtrc | Bla, cat, cat from pKD3 cloned into pUCtrc | This study |
| pCMtrcHR | Bla, cat, homologous regions of 500 bp and 900 bp positioned upstream and downstream of the gene sucAB cloned into pCMtrc | This study |
| pCSHR | Bla, cat-sacB, homologous regions of 500 bp positioned upstream and downstream of the gene icdA and cat-sacB cassette cloned into pUC18 | This study |
| Strains/plasmids | Relevant characteristics | Reference/source |
|---|---|---|
| Strains | ||
| DH5α | Cloning host | Lab collection |
| BL21(DE3) | F’ ompT gal dcm lon hsdSB(rB- mB-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | Novagen |
| CP | BL21(DE3), ΔsdhCDAB, ppc:trc, aceBAK:trc | This study |
| C1 | BL21(DE3), Δppc | This study |
| C2 | C1, aceBAK:trc | This study |
| E1 | Evolved C2 | This study |
| E2 | Evolved E1 | This study |
| E2-ΔaceBA | E2, ΔaceBA | This study |
| E2-Δpck | E2, Δpck | This study |
| E2-ΔmaeA | E2, ΔmaeA | This study |
| E2-ΔmaeB | E2, ΔmaeB | This study |
| E2-Δsdh | E2, ΔsdhCDAB | This study |
| E2-Δsdh-ppc | E2, ΔsdhCDAB, ppc:trc | This study |
| E2-Δsdh-ppc-sucAB | E2, ΔsdhCDAB, ppc:trc,sucAB:trc | This study |
| C2-583 | C2, G583T icdA | This study |
| CP-583 | CP, G583T icdA | This study |
| E2-Δsdh-ppc-583 | E2-Δsdh-ppc, wild-type icdA | This study |
| C2-583-gltA | C2-583, gltA:trc | This study |
| CP-583-gltA | CP-583, gltA:trc | This study |
| Plasmids | ||
| pUC18 | bla, pBR322 ori | Lab collection |
| pEL04 | cat-sacB cassette | [17] |
| pTrc99a | bla, pBR322 ori, trc promoter, lacIq | Invitrogen |
| pKD3 | bla, FRT-cat-FRT | [9] |
| pKD4 | bla, FRT-kan-FRT | [9] |
| pCP20 | bla, cat, yeast Flp recombinase | [6] |
| pKD46 | bla, λ-Red recombinase under araBAD promoter, temperature-conditional replicon | [9] |
| pET-28a(+) | Kan, pBR322 ori, T7 promoter, lacI | Novagen |
| pCM01 | Bla, cat, cat from pKD3 cloned into pUC18 | This study |
| pCMppcHR | bla, cat, homologous regions of 600 bp positioned upstream and downstream of the gene ppc cloned into pCM01 | This study |
| pUCtrc | Bla, trc promoter from pTrc99a cloned into pUC18 | This study |
| pKMtrc | Bla, kan, kan from pKD4 cloned into pUCtrc | This study |
| pCMtrc | Bla, cat, cat from pKD3 cloned into pUCtrc | This study |
| pCMtrcHR | Bla, cat, homologous regions of 500 bp and 900 bp positioned upstream and downstream of the gene sucAB cloned into pCMtrc | This study |
| pCSHR | Bla, cat-sacB, homologous regions of 500 bp positioned upstream and downstream of the gene icdA and cat-sacB cassette cloned into pUC18 | This study |
Primers used in this study
| Primer name | Sequence |
|---|---|
| sdhCDAB knockout | 5′ ATGATAAGAAATGTGAAAAAACAAAGACCTGTTAATCTGGACCTAGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACGCATTACGCTGCAACAACATCGACTTGATATGGCCGATGGCCATATGAATATCCTCCTTAG 3′ | |
| aceBA knockout | 5′ ATGACTGAACAGGCAACAACAACCGATGAACTGGCTTTCACAAGGCCGTACCTGTGACGGAAGATCACTTCG 3′ |
| 5′ TTAGAACTGCGATTCTTCGGTGGAGCCGGTCAGCGCTGTAACCGACGACGTTACGCCCCGCCCTGCCACT 3′ | |
| pck knockout | 5′ ATGCGCGTTAACAATGGTTTGACCCCGCAAGAACTCGAGGCTTATGGTATGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACAGCTTCGGACCAGCCGCTACCAGCGCGGCACCCGCAGGGGTGTCGGCATATGAATATCCTCCTTAG 3′ | |
| maeA knockout | 5′ ACGATAAAAGCCCCCCAGGGATGGATATTCAAAAAAGAGTGAGTTACATGGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ CCCGGTAGCCTTCACTACCGGGCGCAGGCTTAGATGGAGGTACGGCGGTACATATGAATATCCTCCTTAG 3′ | |
| maeB knockout | 5′ ATGGATGACCAGTTAAAACAAAGTGCACTTGATTTCCATGAATTTCCAGTGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACAGCGGTTGGGTTTGCGCTTCTACCACGGCCAGCGCCACCATGTTGACATATGAATATCCTCCTTAG 3′ | |
| aceBA promoter replacement | 5′ CACCTTACCTCAGGCACCTTCGGGTGCCTTTTTTATTTCCGAAACGTACCGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TACGGCCTTGTGAAAGCCAGTTCATCGGTTGTTGTTGCCTGTTCAGTCATGGTCTGTTTCCTGTGTGAAA 3′ | |
| ppc promoter replacement | 5′ GGCGATTTTTTAACATTTCCATAAGTTACGCTTATTTAAAGCGTCGTGAGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTGCCGAGCATACTGACATTACTACGCAATGCGGAATATTGTTCGTTCATGGTCTGTTTCCTGTGTGAAA 3′ | |
| ppc knockout | 5′ GTACAAGCTTATGAATGCCCACCGAATC 3′ |
| 5′ GTACAAGCTTTTGCCGAGCATACTGACA 3′ | |
| 5′ GGACGGATCCCCTCTTCTGCAAACCCTC 3′ | |
| 5′ GTAGGAGCTCGCGTGTCCACAGCAGAAC 3′ | |
| 5′ ATGAATGCCCACCGAATC 3′ | |
| 5′ GCGTGTCCACAGCAGAAC 3′ | |
| Trc promoter | 5′ GTAGGGATCCCAGCTTATCATCGACTGCAC 3′ |
| 5′ GCGCGAATTCCCATGGTCTGTTTCCTGTGT 3′ | |
| Kan or Cm amplification | 5′ GTCGAAAGCTTGATTGCAGCATTACACGTCT 3′ |
| 5′ ACGTGGTCGACCATATGAATATCCTCCTTAG 3′ | |
| sucAB promoter replacement | 5′ AGAGAAGCTTCGGGCAAGAAGATTGTGAT 3′ |
| 5′ AGAGAAGCTTTTACGCATTACGCTGCAAC 3′ | |
| 5′ AGAGGAATTCCGATGCAGAACAGCGCTTTG 3′ | |
| 5′ AGAGGAATTCAGAAGCCCATGTGGTATTTCA 3′ | |
| 5′ CGGGCAAGAAGATTGTGAT 3′ | |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| icdA allele replacement | 5′ GTGTAAGCTTCCTTCGCTATCGCGGTCAA 3′ |
| 5′ GACTAAGCTTAGCGCTACTGGTTTGCTCG 3′ | |
| 5′ CTACGTCGACATGTAATCACTACATGTG 3′ | |
| 5′ GCAGGGTACCTTGGCACAAAGTAGCATA 3′ | |
| 5′ CCTTCGCTATCGCGGTCAA 3′ | |
| 5′ TTGGCACAAAGTAGCATA 3′ | |
| 5′ CTAGCTGCAGCCTGTGACGGAAGATCACTTCG 3′ | |
| 5′ CTAGCTGCAGCTGAGGTTCTTATGGCTCTTG 3′ | |
| gltA promoter replacement | 5′ GATGTGCGAAGGCAAATTTAAGTTCCGGCAGTCTTACGCAATAAGGCGCAAGATCCCCTCACGCTGC 3′ |
| 5′ CCGTTGAGGGTGAGTTTTGCTTTTGTATCAGCCATTTAAGGTCTCCTTAGTGTGAAATTGTTATCCGC 3′ | |
| CD28A | 5′ CGATGAGCTCATGGAAAGTAAAGTAGTTGTTCC 3′ |
| CD28B | 5′ GACGCTCGAGTTACATGTTCTTGATGATCGC 3′ |
| Verification | |
| sdhCDAB knockout | 5′ ACCTCTGTGCCCGTAGTCC 3′ |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| aceBA knockout | 5′ GTAGGGATCCCAGCTTATCATCGACTGCAC 3′ |
| 5′ GCATTTAGCGCCCTCATC 3′ | |
| pck knockout | 5′ AAAAGTTAGCGTGGTGAATC 3′ |
| 5′ TTAGCGGCACGGGATT 3′ | |
| maeA knockout | 5′ TCAGTGATATCTACCAGCAAACGA 3′ |
| 5′ GTATCCGGCATTTTTAACTGAACG 3′ | |
| maeB knockout | 5′ AAAAGTAAAGTGTTAGATGAGTGC 3′ |
| 5′ GATTTTCTTCGCCAGTTCCTCGC 3′ | |
| ppc knockout | 5′ ATGAATGCCCACCGAATC 3′ |
| 5′ GCGTGTCCACAGCAGAAC 3′ | |
| aceBA promoter replacement | 5′ TACGGCACATGAATCCAACG 3′ |
| 5′ TTTGCCTGCTTCATTAGTGT 3′ | |
| sucAB promoter replacement | 5′ CGGGCAAGAAGATTGTGAT 3′ |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| ppc promoter replacement | 5′ ACACCTTTGGTGTTATGTATG 3′ |
| 5′ TGATGGTGTCTTCGCTCAGTT 3′ | |
| icdA allele replacement | 5′ CCTTCGCTATCGCGGTCAA 3′ |
| 5′ TTGGCACAAAGTAGCATA 3′ | |
| gltA promoter replacement | 5′ AGTTTGTCGGGTTTTATCCT 3′ |
| 5′ CAGGATGCGGTTGAAGTGAA 3′ | |
| Primer name | Sequence |
|---|---|
| sdhCDAB knockout | 5′ ATGATAAGAAATGTGAAAAAACAAAGACCTGTTAATCTGGACCTAGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACGCATTACGCTGCAACAACATCGACTTGATATGGCCGATGGCCATATGAATATCCTCCTTAG 3′ | |
| aceBA knockout | 5′ ATGACTGAACAGGCAACAACAACCGATGAACTGGCTTTCACAAGGCCGTACCTGTGACGGAAGATCACTTCG 3′ |
| 5′ TTAGAACTGCGATTCTTCGGTGGAGCCGGTCAGCGCTGTAACCGACGACGTTACGCCCCGCCCTGCCACT 3′ | |
| pck knockout | 5′ ATGCGCGTTAACAATGGTTTGACCCCGCAAGAACTCGAGGCTTATGGTATGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACAGCTTCGGACCAGCCGCTACCAGCGCGGCACCCGCAGGGGTGTCGGCATATGAATATCCTCCTTAG 3′ | |
| maeA knockout | 5′ ACGATAAAAGCCCCCCAGGGATGGATATTCAAAAAAGAGTGAGTTACATGGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ CCCGGTAGCCTTCACTACCGGGCGCAGGCTTAGATGGAGGTACGGCGGTACATATGAATATCCTCCTTAG 3′ | |
| maeB knockout | 5′ ATGGATGACCAGTTAAAACAAAGTGCACTTGATTTCCATGAATTTCCAGTGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACAGCGGTTGGGTTTGCGCTTCTACCACGGCCAGCGCCACCATGTTGACATATGAATATCCTCCTTAG 3′ | |
| aceBA promoter replacement | 5′ CACCTTACCTCAGGCACCTTCGGGTGCCTTTTTTATTTCCGAAACGTACCGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TACGGCCTTGTGAAAGCCAGTTCATCGGTTGTTGTTGCCTGTTCAGTCATGGTCTGTTTCCTGTGTGAAA 3′ | |
| ppc promoter replacement | 5′ GGCGATTTTTTAACATTTCCATAAGTTACGCTTATTTAAAGCGTCGTGAGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTGCCGAGCATACTGACATTACTACGCAATGCGGAATATTGTTCGTTCATGGTCTGTTTCCTGTGTGAAA 3′ | |
| ppc knockout | 5′ GTACAAGCTTATGAATGCCCACCGAATC 3′ |
| 5′ GTACAAGCTTTTGCCGAGCATACTGACA 3′ | |
| 5′ GGACGGATCCCCTCTTCTGCAAACCCTC 3′ | |
| 5′ GTAGGAGCTCGCGTGTCCACAGCAGAAC 3′ | |
| 5′ ATGAATGCCCACCGAATC 3′ | |
| 5′ GCGTGTCCACAGCAGAAC 3′ | |
| Trc promoter | 5′ GTAGGGATCCCAGCTTATCATCGACTGCAC 3′ |
| 5′ GCGCGAATTCCCATGGTCTGTTTCCTGTGT 3′ | |
| Kan or Cm amplification | 5′ GTCGAAAGCTTGATTGCAGCATTACACGTCT 3′ |
| 5′ ACGTGGTCGACCATATGAATATCCTCCTTAG 3′ | |
| sucAB promoter replacement | 5′ AGAGAAGCTTCGGGCAAGAAGATTGTGAT 3′ |
| 5′ AGAGAAGCTTTTACGCATTACGCTGCAAC 3′ | |
| 5′ AGAGGAATTCCGATGCAGAACAGCGCTTTG 3′ | |
| 5′ AGAGGAATTCAGAAGCCCATGTGGTATTTCA 3′ | |
| 5′ CGGGCAAGAAGATTGTGAT 3′ | |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| icdA allele replacement | 5′ GTGTAAGCTTCCTTCGCTATCGCGGTCAA 3′ |
| 5′ GACTAAGCTTAGCGCTACTGGTTTGCTCG 3′ | |
| 5′ CTACGTCGACATGTAATCACTACATGTG 3′ | |
| 5′ GCAGGGTACCTTGGCACAAAGTAGCATA 3′ | |
| 5′ CCTTCGCTATCGCGGTCAA 3′ | |
| 5′ TTGGCACAAAGTAGCATA 3′ | |
| 5′ CTAGCTGCAGCCTGTGACGGAAGATCACTTCG 3′ | |
| 5′ CTAGCTGCAGCTGAGGTTCTTATGGCTCTTG 3′ | |
| gltA promoter replacement | 5′ GATGTGCGAAGGCAAATTTAAGTTCCGGCAGTCTTACGCAATAAGGCGCAAGATCCCCTCACGCTGC 3′ |
| 5′ CCGTTGAGGGTGAGTTTTGCTTTTGTATCAGCCATTTAAGGTCTCCTTAGTGTGAAATTGTTATCCGC 3′ | |
| CD28A | 5′ CGATGAGCTCATGGAAAGTAAAGTAGTTGTTCC 3′ |
| CD28B | 5′ GACGCTCGAGTTACATGTTCTTGATGATCGC 3′ |
| Verification | |
| sdhCDAB knockout | 5′ ACCTCTGTGCCCGTAGTCC 3′ |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| aceBA knockout | 5′ GTAGGGATCCCAGCTTATCATCGACTGCAC 3′ |
| 5′ GCATTTAGCGCCCTCATC 3′ | |
| pck knockout | 5′ AAAAGTTAGCGTGGTGAATC 3′ |
| 5′ TTAGCGGCACGGGATT 3′ | |
| maeA knockout | 5′ TCAGTGATATCTACCAGCAAACGA 3′ |
| 5′ GTATCCGGCATTTTTAACTGAACG 3′ | |
| maeB knockout | 5′ AAAAGTAAAGTGTTAGATGAGTGC 3′ |
| 5′ GATTTTCTTCGCCAGTTCCTCGC 3′ | |
| ppc knockout | 5′ ATGAATGCCCACCGAATC 3′ |
| 5′ GCGTGTCCACAGCAGAAC 3′ | |
| aceBA promoter replacement | 5′ TACGGCACATGAATCCAACG 3′ |
| 5′ TTTGCCTGCTTCATTAGTGT 3′ | |
| sucAB promoter replacement | 5′ CGGGCAAGAAGATTGTGAT 3′ |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| ppc promoter replacement | 5′ ACACCTTTGGTGTTATGTATG 3′ |
| 5′ TGATGGTGTCTTCGCTCAGTT 3′ | |
| icdA allele replacement | 5′ CCTTCGCTATCGCGGTCAA 3′ |
| 5′ TTGGCACAAAGTAGCATA 3′ | |
| gltA promoter replacement | 5′ AGTTTGTCGGGTTTTATCCT 3′ |
| 5′ CAGGATGCGGTTGAAGTGAA 3′ | |
Primers used in this study
| Primer name | Sequence |
|---|---|
| sdhCDAB knockout | 5′ ATGATAAGAAATGTGAAAAAACAAAGACCTGTTAATCTGGACCTAGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACGCATTACGCTGCAACAACATCGACTTGATATGGCCGATGGCCATATGAATATCCTCCTTAG 3′ | |
| aceBA knockout | 5′ ATGACTGAACAGGCAACAACAACCGATGAACTGGCTTTCACAAGGCCGTACCTGTGACGGAAGATCACTTCG 3′ |
| 5′ TTAGAACTGCGATTCTTCGGTGGAGCCGGTCAGCGCTGTAACCGACGACGTTACGCCCCGCCCTGCCACT 3′ | |
| pck knockout | 5′ ATGCGCGTTAACAATGGTTTGACCCCGCAAGAACTCGAGGCTTATGGTATGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACAGCTTCGGACCAGCCGCTACCAGCGCGGCACCCGCAGGGGTGTCGGCATATGAATATCCTCCTTAG 3′ | |
| maeA knockout | 5′ ACGATAAAAGCCCCCCAGGGATGGATATTCAAAAAAGAGTGAGTTACATGGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ CCCGGTAGCCTTCACTACCGGGCGCAGGCTTAGATGGAGGTACGGCGGTACATATGAATATCCTCCTTAG 3′ | |
| maeB knockout | 5′ ATGGATGACCAGTTAAAACAAAGTGCACTTGATTTCCATGAATTTCCAGTGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACAGCGGTTGGGTTTGCGCTTCTACCACGGCCAGCGCCACCATGTTGACATATGAATATCCTCCTTAG 3′ | |
| aceBA promoter replacement | 5′ CACCTTACCTCAGGCACCTTCGGGTGCCTTTTTTATTTCCGAAACGTACCGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TACGGCCTTGTGAAAGCCAGTTCATCGGTTGTTGTTGCCTGTTCAGTCATGGTCTGTTTCCTGTGTGAAA 3′ | |
| ppc promoter replacement | 5′ GGCGATTTTTTAACATTTCCATAAGTTACGCTTATTTAAAGCGTCGTGAGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTGCCGAGCATACTGACATTACTACGCAATGCGGAATATTGTTCGTTCATGGTCTGTTTCCTGTGTGAAA 3′ | |
| ppc knockout | 5′ GTACAAGCTTATGAATGCCCACCGAATC 3′ |
| 5′ GTACAAGCTTTTGCCGAGCATACTGACA 3′ | |
| 5′ GGACGGATCCCCTCTTCTGCAAACCCTC 3′ | |
| 5′ GTAGGAGCTCGCGTGTCCACAGCAGAAC 3′ | |
| 5′ ATGAATGCCCACCGAATC 3′ | |
| 5′ GCGTGTCCACAGCAGAAC 3′ | |
| Trc promoter | 5′ GTAGGGATCCCAGCTTATCATCGACTGCAC 3′ |
| 5′ GCGCGAATTCCCATGGTCTGTTTCCTGTGT 3′ | |
| Kan or Cm amplification | 5′ GTCGAAAGCTTGATTGCAGCATTACACGTCT 3′ |
| 5′ ACGTGGTCGACCATATGAATATCCTCCTTAG 3′ | |
| sucAB promoter replacement | 5′ AGAGAAGCTTCGGGCAAGAAGATTGTGAT 3′ |
| 5′ AGAGAAGCTTTTACGCATTACGCTGCAAC 3′ | |
| 5′ AGAGGAATTCCGATGCAGAACAGCGCTTTG 3′ | |
| 5′ AGAGGAATTCAGAAGCCCATGTGGTATTTCA 3′ | |
| 5′ CGGGCAAGAAGATTGTGAT 3′ | |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| icdA allele replacement | 5′ GTGTAAGCTTCCTTCGCTATCGCGGTCAA 3′ |
| 5′ GACTAAGCTTAGCGCTACTGGTTTGCTCG 3′ | |
| 5′ CTACGTCGACATGTAATCACTACATGTG 3′ | |
| 5′ GCAGGGTACCTTGGCACAAAGTAGCATA 3′ | |
| 5′ CCTTCGCTATCGCGGTCAA 3′ | |
| 5′ TTGGCACAAAGTAGCATA 3′ | |
| 5′ CTAGCTGCAGCCTGTGACGGAAGATCACTTCG 3′ | |
| 5′ CTAGCTGCAGCTGAGGTTCTTATGGCTCTTG 3′ | |
| gltA promoter replacement | 5′ GATGTGCGAAGGCAAATTTAAGTTCCGGCAGTCTTACGCAATAAGGCGCAAGATCCCCTCACGCTGC 3′ |
| 5′ CCGTTGAGGGTGAGTTTTGCTTTTGTATCAGCCATTTAAGGTCTCCTTAGTGTGAAATTGTTATCCGC 3′ | |
| CD28A | 5′ CGATGAGCTCATGGAAAGTAAAGTAGTTGTTCC 3′ |
| CD28B | 5′ GACGCTCGAGTTACATGTTCTTGATGATCGC 3′ |
| Verification | |
| sdhCDAB knockout | 5′ ACCTCTGTGCCCGTAGTCC 3′ |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| aceBA knockout | 5′ GTAGGGATCCCAGCTTATCATCGACTGCAC 3′ |
| 5′ GCATTTAGCGCCCTCATC 3′ | |
| pck knockout | 5′ AAAAGTTAGCGTGGTGAATC 3′ |
| 5′ TTAGCGGCACGGGATT 3′ | |
| maeA knockout | 5′ TCAGTGATATCTACCAGCAAACGA 3′ |
| 5′ GTATCCGGCATTTTTAACTGAACG 3′ | |
| maeB knockout | 5′ AAAAGTAAAGTGTTAGATGAGTGC 3′ |
| 5′ GATTTTCTTCGCCAGTTCCTCGC 3′ | |
| ppc knockout | 5′ ATGAATGCCCACCGAATC 3′ |
| 5′ GCGTGTCCACAGCAGAAC 3′ | |
| aceBA promoter replacement | 5′ TACGGCACATGAATCCAACG 3′ |
| 5′ TTTGCCTGCTTCATTAGTGT 3′ | |
| sucAB promoter replacement | 5′ CGGGCAAGAAGATTGTGAT 3′ |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| ppc promoter replacement | 5′ ACACCTTTGGTGTTATGTATG 3′ |
| 5′ TGATGGTGTCTTCGCTCAGTT 3′ | |
| icdA allele replacement | 5′ CCTTCGCTATCGCGGTCAA 3′ |
| 5′ TTGGCACAAAGTAGCATA 3′ | |
| gltA promoter replacement | 5′ AGTTTGTCGGGTTTTATCCT 3′ |
| 5′ CAGGATGCGGTTGAAGTGAA 3′ | |
| Primer name | Sequence |
|---|---|
| sdhCDAB knockout | 5′ ATGATAAGAAATGTGAAAAAACAAAGACCTGTTAATCTGGACCTAGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACGCATTACGCTGCAACAACATCGACTTGATATGGCCGATGGCCATATGAATATCCTCCTTAG 3′ | |
| aceBA knockout | 5′ ATGACTGAACAGGCAACAACAACCGATGAACTGGCTTTCACAAGGCCGTACCTGTGACGGAAGATCACTTCG 3′ |
| 5′ TTAGAACTGCGATTCTTCGGTGGAGCCGGTCAGCGCTGTAACCGACGACGTTACGCCCCGCCCTGCCACT 3′ | |
| pck knockout | 5′ ATGCGCGTTAACAATGGTTTGACCCCGCAAGAACTCGAGGCTTATGGTATGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACAGCTTCGGACCAGCCGCTACCAGCGCGGCACCCGCAGGGGTGTCGGCATATGAATATCCTCCTTAG 3′ | |
| maeA knockout | 5′ ACGATAAAAGCCCCCCAGGGATGGATATTCAAAAAAGAGTGAGTTACATGGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ CCCGGTAGCCTTCACTACCGGGCGCAGGCTTAGATGGAGGTACGGCGGTACATATGAATATCCTCCTTAG 3′ | |
| maeB knockout | 5′ ATGGATGACCAGTTAAAACAAAGTGCACTTGATTTCCATGAATTTCCAGTGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTACAGCGGTTGGGTTTGCGCTTCTACCACGGCCAGCGCCACCATGTTGACATATGAATATCCTCCTTAG 3′ | |
| aceBA promoter replacement | 5′ CACCTTACCTCAGGCACCTTCGGGTGCCTTTTTTATTTCCGAAACGTACCGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TACGGCCTTGTGAAAGCCAGTTCATCGGTTGTTGTTGCCTGTTCAGTCATGGTCTGTTTCCTGTGTGAAA 3′ | |
| ppc promoter replacement | 5′ GGCGATTTTTTAACATTTCCATAAGTTACGCTTATTTAAAGCGTCGTGAGTGTAGGCTGGAGCTGCTTC 3′ |
| 5′ TTGCCGAGCATACTGACATTACTACGCAATGCGGAATATTGTTCGTTCATGGTCTGTTTCCTGTGTGAAA 3′ | |
| ppc knockout | 5′ GTACAAGCTTATGAATGCCCACCGAATC 3′ |
| 5′ GTACAAGCTTTTGCCGAGCATACTGACA 3′ | |
| 5′ GGACGGATCCCCTCTTCTGCAAACCCTC 3′ | |
| 5′ GTAGGAGCTCGCGTGTCCACAGCAGAAC 3′ | |
| 5′ ATGAATGCCCACCGAATC 3′ | |
| 5′ GCGTGTCCACAGCAGAAC 3′ | |
| Trc promoter | 5′ GTAGGGATCCCAGCTTATCATCGACTGCAC 3′ |
| 5′ GCGCGAATTCCCATGGTCTGTTTCCTGTGT 3′ | |
| Kan or Cm amplification | 5′ GTCGAAAGCTTGATTGCAGCATTACACGTCT 3′ |
| 5′ ACGTGGTCGACCATATGAATATCCTCCTTAG 3′ | |
| sucAB promoter replacement | 5′ AGAGAAGCTTCGGGCAAGAAGATTGTGAT 3′ |
| 5′ AGAGAAGCTTTTACGCATTACGCTGCAAC 3′ | |
| 5′ AGAGGAATTCCGATGCAGAACAGCGCTTTG 3′ | |
| 5′ AGAGGAATTCAGAAGCCCATGTGGTATTTCA 3′ | |
| 5′ CGGGCAAGAAGATTGTGAT 3′ | |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| icdA allele replacement | 5′ GTGTAAGCTTCCTTCGCTATCGCGGTCAA 3′ |
| 5′ GACTAAGCTTAGCGCTACTGGTTTGCTCG 3′ | |
| 5′ CTACGTCGACATGTAATCACTACATGTG 3′ | |
| 5′ GCAGGGTACCTTGGCACAAAGTAGCATA 3′ | |
| 5′ CCTTCGCTATCGCGGTCAA 3′ | |
| 5′ TTGGCACAAAGTAGCATA 3′ | |
| 5′ CTAGCTGCAGCCTGTGACGGAAGATCACTTCG 3′ | |
| 5′ CTAGCTGCAGCTGAGGTTCTTATGGCTCTTG 3′ | |
| gltA promoter replacement | 5′ GATGTGCGAAGGCAAATTTAAGTTCCGGCAGTCTTACGCAATAAGGCGCAAGATCCCCTCACGCTGC 3′ |
| 5′ CCGTTGAGGGTGAGTTTTGCTTTTGTATCAGCCATTTAAGGTCTCCTTAGTGTGAAATTGTTATCCGC 3′ | |
| CD28A | 5′ CGATGAGCTCATGGAAAGTAAAGTAGTTGTTCC 3′ |
| CD28B | 5′ GACGCTCGAGTTACATGTTCTTGATGATCGC 3′ |
| Verification | |
| sdhCDAB knockout | 5′ ACCTCTGTGCCCGTAGTCC 3′ |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| aceBA knockout | 5′ GTAGGGATCCCAGCTTATCATCGACTGCAC 3′ |
| 5′ GCATTTAGCGCCCTCATC 3′ | |
| pck knockout | 5′ AAAAGTTAGCGTGGTGAATC 3′ |
| 5′ TTAGCGGCACGGGATT 3′ | |
| maeA knockout | 5′ TCAGTGATATCTACCAGCAAACGA 3′ |
| 5′ GTATCCGGCATTTTTAACTGAACG 3′ | |
| maeB knockout | 5′ AAAAGTAAAGTGTTAGATGAGTGC 3′ |
| 5′ GATTTTCTTCGCCAGTTCCTCGC 3′ | |
| ppc knockout | 5′ ATGAATGCCCACCGAATC 3′ |
| 5′ GCGTGTCCACAGCAGAAC 3′ | |
| aceBA promoter replacement | 5′ TACGGCACATGAATCCAACG 3′ |
| 5′ TTTGCCTGCTTCATTAGTGT 3′ | |
| sucAB promoter replacement | 5′ CGGGCAAGAAGATTGTGAT 3′ |
| 5′ AGAAGCCCATGTGGTATTTCA 3′ | |
| ppc promoter replacement | 5′ ACACCTTTGGTGTTATGTATG 3′ |
| 5′ TGATGGTGTCTTCGCTCAGTT 3′ | |
| icdA allele replacement | 5′ CCTTCGCTATCGCGGTCAA 3′ |
| 5′ TTGGCACAAAGTAGCATA 3′ | |
| gltA promoter replacement | 5′ AGTTTGTCGGGTTTTATCCT 3′ |
| 5′ CAGGATGCGGTTGAAGTGAA 3′ | |
Gene deletion, promoter replacement, and allele replacement
Deletion strains were constructed following a one-step inactivation method developed earlier [9]. Knockout of sdhCDAB, aceBA, pck, maeA, and maeB were performed with homologous regions as long as 50 bp contained in the primers (Table 2). For ppc deletion, chloramphenicol resistance gene amplified from pKD3 was firstly cloned into pUC18 to produce plasmid pCM01. Homologous regions of 600 bp positioned upstream and downstream of the ppc gene were then cloned into plasmid pCM01, respectively. The resulting plasmid pCMppcHR was used as the template of PCR amplification. Amplified PCR fragments comprised of homogenous regions and antibiotic resistance gene were electroporated into E. coli strains harboring plasmid pKD46. The resistance gene flanked by FRT sites was subsequently removed from the chromosome with FLP recombinase using plasmid pCP20 [6].
Replacement of the native promoters of aceBA, sucAB, and ppc with the trc promoter was performed also by λ-Red recombination. Firstly, the trc promoter was amplified using plasmid pTrc99a as template and cloned into pUC18 to produce plasmid pUCtrc. Secondly, the kanamycin resistance gene or chloramphenicol resistance gene amplified from plasmid pKD4 or pKD3 was cloned into pUCtrc to produce plasmid pKMtrc or pCMtrc. For promoter replacement of aceBA and gltA, primers containing 50 bp homologous regions (Table 2) were used to amplify PCR fragments comprised of antibiotic marker and trc promoter using pKMtrc as template. For promoter replacement of sucAB, homologous regions of 500 and 900 bp positioned upstream and downstream of the promoter region of sucAB were used through cloned into plasmid pCMtrc, respectively. The promoter replacement of ppc was performed similar to that of aceBA. Once it was done in wild-type E. coli BL21(DE3), the antibiotic region, the trc promoter, and the coding region of ppc were amplified together using primers Pout5/Pout6 and electroporated into E. coli ppc mutants for recovering the ppc gene.
The allele replacement of icdA was accomplished through a two-step homogenous recombination method [39] with the counter-selectable sacB gene. Homologous regions of 500 bp positioned upstream and downstream of the gene icdA and the cat-sacB cassette from plasmid pEL04 [17] were cloned into pUC18, respectively. The resulting plasmid pCSHR was used as the template for amplification of PCR fragments which were used for accomplishing the first homogenous recombination. The second homogenous recombination was achieved using PCR fragments harboring point mutation amplified from the genome of wild type or the mutant. Cells finishing the second homogenous recombination were selected in the presence of 10 % sucrose, and the allele replacement was confirmed by sequencing.
Medium and cultivation conditions
Flask cultures were performed in M9 minimal medium [40] supplemented with 110 mM glycerol at 37 °C and 220 rpm in a rotary shaker. A single colony was inoculated into a 15-ml test tube containing 4 ml LB medium and cultured at 37 °C and 220 rpm for 12 h. One percent of seed culture was transferred into 25 ml of M9 minimal medium with 110 mM glycerol in a 250-ml flask. When the culture grew to mid-exponential phase, 1 % of it was inoculated into 50 ml M9 medium with 110 mM glycerol in a 500-ml flask. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was supplemented to 0.2 mM in the shake flask culture at the beginning of fermentation to induce gene expression. All the fermentation experiments were performed in triplicate.
Fed-batch fermentation was carried out in a 1.3-l reactor (BioFlo 110, New Brunswick Scientific, Enfield, CT, USA) containing 600 ml M9 minimal medium supplemented with 470 mM glycerol initially. The frozen glycerol stock at −80 °C was firstly streaked on a LB plate, incubated at 37 °C for 12 h, and then a colony was inoculated into a 15-ml test tube containing 4 ml LB medium. After incubating in the test tube at 37 °C and 220 rpm for 12 h, the cells were transferred into a 500-ml shake flask containing 50 ml M9 medium supplemented with 110 mM glycerol. When the culture grew to mid-exponential phase in the shake flask, the cells were inoculated into the reactor with an initial OD600 of 0.18. Fed-batch fermentation was performed at 37 °C and the pH was controlled at 7.0 with 30 % ammonium hydroxide and 1 M sulfuric acid. The dissolved oxygen was maintained above 30 % of air saturation by aerating air at 1 l/min and adjusting the agitation speed from 500 to 850 rpm. IPTG was added to a final concentration of 1 mM during inoculation. When the glycerol concentration was below 110 mM, 25 ml 80 % (v/v) glycerol was supplemented into the reactor. Samples were taken periodically to determine cell growth, glycerol and extracellular metabolites concentration.
Adaptive evolution
Adaptive evolution was performed through sequential transfers in M9 minimal medium under aerobic conditions. The starting strain was initially subjected to incubation in a 500-ml flask containing 50 ml M9 minimal medium with 110 mM glycerol at 37 °C and 220 rpm. Several days were needed for the growth to occur and sequential transfers were subsequently carried out during the mid-exponential phase. When the cell density (OD600) grew to approximately 1.0, the cells were transferred into fresh M9 medium with 2 % inoculum. Each transfer was stored at −80 °C. 0.2 mM IPTG was added to induce the expression of aceBA at the time of inoculation.
Analytical methods
Cell growth was monitored by measuring the optical density at 600 nm (OD600) with a UV–Vis spectrophotometer. Glycerol and organic acids were quantified by HPLC equipped with a cation exchange column (Aminex HPX 87-H, Bio-Rad, Hercules, CA, USA), an UV absorbance detector (Agilent, G1315D), and a refractive index (RI) detector (Agilent, HP1047A); 5 mM H2SO4 was used as the mobile phase at a flow rate of 0.4 ml/min. The column was operated at 65 °C.
Enzyme assays
For preparation of cell extract, all the strains were cultured in 500-ml flasks containing 50 ml M9 minimal medium with 110 mM glycerol at 220 rpm and 37 °C. Cell was harvested by centrifugation at 8,000 × g for 10 min at the mid-exponential phase, washed twice with 100 mM Tris–HCl (pH 7.0) containing 20 mM KCl, 5 mM H2SO4, 2 mM DTT, and 0.1 mM EDTA and then resuspended in the same buffer [33]. All the operations were conducted at 4 °C. Crude extract was prepared through freezing cells with liquid nitrogen and immediately grinding with silica sand at 4 °C for 5 min. Cell debris was removed by centrifugation at 20,000 × g for 15 min. The supernatant was immediately used for enzyme assays. The total protein concentration was measured as previously described [4] using bovine serum albumin as standard. Citrate synthase (CS) activity was determined based on the reaction of 5,5-dithiobis (2-nitrobenzoic acid) and CoA as reported [27]. Isocitrate lyase (ICL) activity was measured according to the formation of glyoxylic acid phenylhydrazone from glyoxylate and phenylhydrazine [5]. Isocitrate dehydrogenase (ICDH) activity was detected through monitoring the formation of NADPH at 340 nm [1]. Enzyme activities were measured spectrophotometrically at 30 °C for ICL and 37 °C for CS and ICDH.
For purification of the wild-type and mutated ICDH, the wild-type and mutated icdA genes were amplified with primers CD28A/CD28B from the genome of wild-type BL21(DE3) and evolved strain E2, and cloned into plasmid pET-28a(+). The resulting plasmids were then transformed into E. coli BL21(DE3). When culture was grown to an OD600 of 0.6 in 50 ml of LB medium, 1 mM IPTG was supplemented and after 3 h of induction the cells was harvested and washed. The pellet was resuspended in 5 ml 50 mM NaH2PO4 (pH 8.0) containing 0.3 M NaCl. Cells were lysed and the cell debris was removed as mentioned above. Protein was purified using Ni–NTA-Sefinose Column (Sangon, Shanghai, China) according to the manufacturer’s protocol. Kinetics parameters of ICDH were determined by altering the concentration of isocitrate while the other substrates were maintained at saturating concentrations. The K M values were determined from Lineweaver–Burk plots.
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted from mid-exponential phase cultures grown in M9 minimal medium with RNAprep Pure Cell/Bacteria Kit (Tiangen, Beijing, China) according to the manufacturer’s protocol. cDNA synthesis was prepared using FastQuant RT Kit (with gDNase) (Tiangen, Beijing, China) with 500 ng total RNA and random primers. Prior to cDNA synthesis, the total RNA was treated with the gDNase in the kit at 42 °C for 3 min to avoid the interference of genome DNA. RT-qPCR was performed in triplicate for each sample in a Light CyclerH 480 II (Roche, Basel, Switzerland) using Real Master Mix (SYBR Green). The 16s rRNA gene was used as the internal control. The relative quantitative method (
Results
Engineering E. coli BL21(DE3) for aerobic succinate production from glycerol
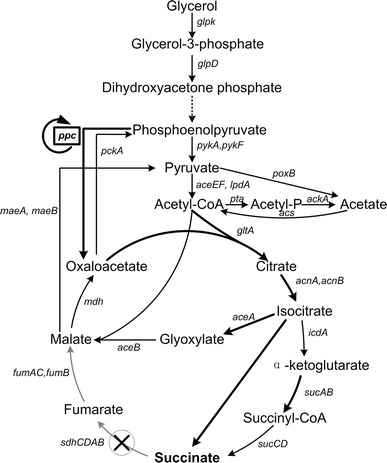
The central carbon metabolism of Escherichia coli BL21(DE3) with glycerol as carbon source and the manipulation strategies for succinate production. The circular arrow around the gene ppc represents that it was firstly deleted for adaptive evolution and then recovered for succinate production. The cross in the circle indicates deleted pathways, which are displayed in grey. The bold arrow represents the upregulated pathways. glpK, glycerol kinase; glpD, glycerol-3-phophate dehydrogenase; pykA/pykF, pyruvate kinase; aceEF/lpdA, pyruvate dehydrogenase complex; poxB, pyruvate oxidase; pta, phosphotransacetylase; ackA, acetate kinase; acs, acetyl-CoA synthase; gltA, citrate synthase; acnA/acnB, aconitate hydratase; icdA, isocitrate dehydrogenase; sucAB, α-ketoglutarate dehydrogenase; sucCD, succinyl-CoA synthase; sdhCDAB, succinate dehydrogenase; fumAC/fumB, aerobic and anaerobic fumarases; mdh, malate dehydrogenase; aceA, isocitrate lyase; aceB, malate synthase; maeA/maeB, NADH/NADPH malic enzymes; ppc, phosphoenolpyruvate carboxylase; pckA, phosphoenolpyruvate carboxykinase
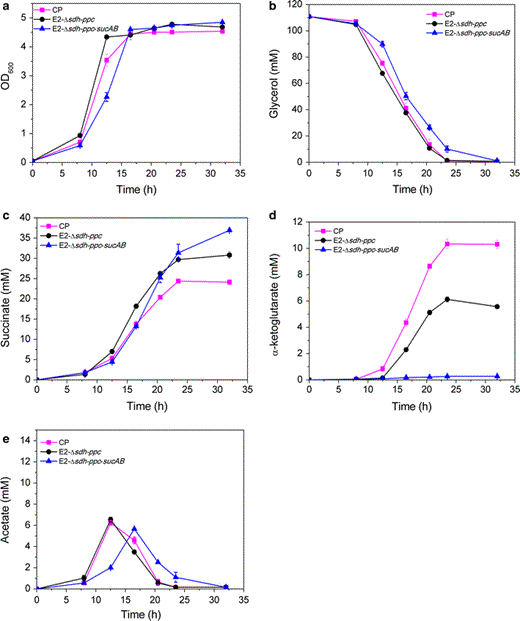
Time profiles of biomass synthesis (a), glycerol consumption (b), succinate production (c), and byproducts accumulation (d, e) for strain CP (square), E2-Δsdh-ppc (circle) and E2-Δsdh-ppc-sucAB (triangle) in minimal medium measured from flask cultures
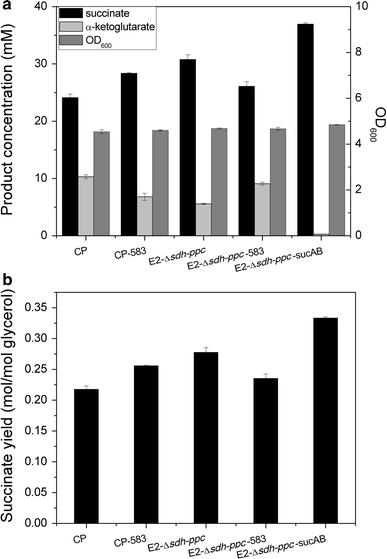
Comparison of succinate production and byproduct accumulation between different strains developed in this study. a Biomass (OD600), succinate and α-ketoglutarate concentrations; b succinate yield (mol/mol glycerol). All the strains were cultured in minimal medium with 110 mM glycerol in 500-ml flasks at 37 °C and 220 rpm
The accumulation of α-ketoglutarate wasted carbon source and resulted in low succinate yield. Redirecting more carbon flux into the glyoxylate bypass and consequently decreasing the flux of the oxidative arm of the TCA cycle were expected to reduce the accumulation of α-ketoglutarate and improve succinate production. Since the transcriptional regulation of key enzymes of the glyoxylate shunt had been removed in strain CP through replacing the native promoter of aceBAK with trc strong promoter, some other efforts needed to be made to further increase the flux of the glyoxylate shunt.
Directed pathway evolution of the glyoxylate shunt
The reaction catalyzed by PPC and the glyoxylate shunt can both function as anaplerotic pathways for replenishing oxaloacetate (OAA) under different conditions. PPC is the primary anaplerotic enzyme and is essential for growth in minimal medium with glycerol as the sole carbon source in E. coli K-12 [15]. However, when acetate is used as the sole carbon source, the glyoxylate shunt becomes essential and functions as the primary anaplerotic pathway for avoiding the loss of carbon as CO2 [42]. As shown in Fig. 1, one OAA and two acetyl-CoA are converted to one malate and one succinate by one round of the glyoxylate shunt, and then malate and succinate can be converted to two OAA by malate dehydrogenase and SDH, respectively. Therefore, the net effect of the glyoxylate shunt is to replenish one OAA. It has been reported that the glyoxylate shunt is inactive in E. coli K-12 but active in E. coli BL21 with 22 % flux of the TCA cycle in the presence of glucose [29]. Therefore, ppc was deleted in E. coli BL21(DE3) to examine if the active glyoxylate shunt could support cell growth in glycerol minimal medium in the absence of PPC. However, the growth of the resulting strain C1 in glycerol minimal medium was completely abolished (Table 3), similar to E. coli K-12 as previously reported [15]. This phenomenon suggested that the flux of the active glyoxylate shunt in E. coli BL21(DE3) was not sufficient for cell growth on glycerol minimum medium. We then replaced the native promoter of the operon aceBAK with trc strong promoter in strain C1 to eliminate the transcriptional regulation by regulators such as iclR, crp [7, 44, 48]. The resulting strain C2 was still unable to grow in glycerol minimal medium, indicating that the flux of the glyoxylate shunt was still insufficient for cell growth. The regulation of the key enzymes of the glyoxylate shunt by inhibitors on enzyme kinetics [14, 30] might be responsible for it.
The growth phenotype and specific growth rate of strains developed in this study in glycerol minimal medium
| Strains | Growth (Y, growth; N, non-growth) | Specific growth rate (h−1) |
|---|---|---|
| C1 | N | – |
| C2 | N | – |
| E1 | Y | 0.18 ± 0.0039 |
| E2 | Y | 0.40 ± 0.00505 |
| E2-ΔaceBA | N | – |
| E2-Δpck | Y | 0.32 ± 0.0049 |
| E2-ΔmaeA | Y | 0.34 ± 0.0012 |
| E2-ΔmaeB | Y | 0.39 ± 0.00035 |
| C2-583 | Y | 0.19 ± 0.00045 |
| C2-583-gltA | Y | 0.26 ± 0.004 |
| Strains | Growth (Y, growth; N, non-growth) | Specific growth rate (h−1) |
|---|---|---|
| C1 | N | – |
| C2 | N | – |
| E1 | Y | 0.18 ± 0.0039 |
| E2 | Y | 0.40 ± 0.00505 |
| E2-ΔaceBA | N | – |
| E2-Δpck | Y | 0.32 ± 0.0049 |
| E2-ΔmaeA | Y | 0.34 ± 0.0012 |
| E2-ΔmaeB | Y | 0.39 ± 0.00035 |
| C2-583 | Y | 0.19 ± 0.00045 |
| C2-583-gltA | Y | 0.26 ± 0.004 |
The specific growth rate was measured at 37 °C and 220 rpm in 500-ml flask. ‘Y’ represents the ability to growth under this condition, and ‘N’ represents that the strain could not grow in glycerol minimal medium
The growth phenotype and specific growth rate of strains developed in this study in glycerol minimal medium
| Strains | Growth (Y, growth; N, non-growth) | Specific growth rate (h−1) |
|---|---|---|
| C1 | N | – |
| C2 | N | – |
| E1 | Y | 0.18 ± 0.0039 |
| E2 | Y | 0.40 ± 0.00505 |
| E2-ΔaceBA | N | – |
| E2-Δpck | Y | 0.32 ± 0.0049 |
| E2-ΔmaeA | Y | 0.34 ± 0.0012 |
| E2-ΔmaeB | Y | 0.39 ± 0.00035 |
| C2-583 | Y | 0.19 ± 0.00045 |
| C2-583-gltA | Y | 0.26 ± 0.004 |
| Strains | Growth (Y, growth; N, non-growth) | Specific growth rate (h−1) |
|---|---|---|
| C1 | N | – |
| C2 | N | – |
| E1 | Y | 0.18 ± 0.0039 |
| E2 | Y | 0.40 ± 0.00505 |
| E2-ΔaceBA | N | – |
| E2-Δpck | Y | 0.32 ± 0.0049 |
| E2-ΔmaeA | Y | 0.34 ± 0.0012 |
| E2-ΔmaeB | Y | 0.39 ± 0.00035 |
| C2-583 | Y | 0.19 ± 0.00045 |
| C2-583-gltA | Y | 0.26 ± 0.004 |
The specific growth rate was measured at 37 °C and 220 rpm in 500-ml flask. ‘Y’ represents the ability to growth under this condition, and ‘N’ represents that the strain could not grow in glycerol minimal medium
The inviability of C1 and C2 in glycerol minimal medium indicated the low flux of the glyoxylate shunt and the need to increase it for succinate production. It has been reported that the glyoxylate shunt was activated in E. coli K-12 ppc mutant as alternative anaplerotic pathway in the presence of glucose [32]. In the ppc mutant, ICL activity increased by about threefold and 18.9 % of the carbon flux flowed into the glyoxylate shunt [32]. In another study, the glyoxylate shunt was also recruited as the anaplerotic pathway in place of PPC in unevolved and evolved E. coli K-12 ppc mutants [12]. Based on the facts mentioned above, we attempted to perform directed pathway evolution using strain C2 as the starting strain and hoped to isolate mutant strains with enhanced glyoxylate shunt.
At the beginning of the directed pathway evolution, strain C2 was incubated in glycerol minimum medium for several days, after which culture of C2 began to grow and a clone was isolated through streaking on agar plate and designated as E1. When cultured in glycerol minimal medium, E1 was able to grow at a specific growth rate of 0.18 h−1. Because of the slow growth of E1, serial transfers were carried out in minimal medium with glycerol as the sole carbon source to further improve the growth. After adaptive evolution of about 100 generations, a mutant (E2) was obtained that was able to grow in glycerol minimal medium at a specific growth rate of 0.40 h−1 (Table 3). When the operon aceBA encoding the key enzymes of the glyoxylate shunt was deleted in E2, the mutant E2-ΔaceBA lost the ability to grow in glycerol minimal medium. This result indicated that the glyoxylate shunt had become essential for cell growth as the primary anaplerotic pathway through adaptive evolution, which was in accordance with the results of previous studies [12, 32]. In order to examine whether mutations had occurred in ICL or malate synthase, the promoter and coding region of the operon aceBAK were sequenced. Unexpectedly, no mutation was observed.
The previously reported results [12, 32] and the above result in this study demonstrated that in the absence of PPC the favored alternative anaplerotic pathway would be the glyoxylate shunt. However, there are three other carboxylation enzymes besides PPC, phosphoenolpyruvate carboxykinase (pck) and two malic enzymes (maeA and maeB), which are involved in gluconeogenesis and physiologically function in reverse direction [36]. In order to confirm that they were not evolved to serve as primary anaplerotic enzymes in this study, they were also deleted individually in strain E2. As depicted in Table 3, all the mutants (E2-Δpck, E2-ΔmaeA and E2-ΔmaeB) retained the ability to grow in glycerol minimal medium with slightly decreased growth rates, suggesting that the three pathways were not evolved to function as the primary anaplerotic pathway in the evolved mutant.
Effects of the enhanced glyoxylate shunt on succinate production
In order to explore the effects of the enhanced glyoxylate shunt on succinate production, SDH was deleted in strain E2 (producing strain E2-Δsdh) to allow succinate to accumulate as an end product. Knockout of SDH resulted in the inviability of strain E2-Δsdh in glycerol minimal medium due to the elimination of the ability of the glyoxylate shunt to replenish OAA. So ppc was recovered in stain E2-Δsdh and at the same time the native promoter of ppc was replaced with trc strong promoter. As expected, the resulting strain E2-Δsdh-ppc was able to grow in glycerol minimal medium. Subsequently, the production performance of E2-Δsdh-ppc was examined, using strain CP as the control strain.
As shown in Fig. 2a, the growth rate of strain E2-Δsdh-ppc was a little higher than that of strain CP. After being cultured in glycerol minimal medium under aerobic conditions for 32 h, strain E2-Δsdh-ppc produced 30.7 mM (3.62 g/l) succinate from 110 mM (10 g/l) glycerol. Succinate yield increased by 30 % for strain E2-Δsdh-ppc in comparison with the control strain CP, which represented 55.5 % of the maximum theoretical yield (Figs. 2c, 3). Meanwhile, α-ketoglutarate level reduced to 5.5 mM (0.8 g/l), which was 46 % lower than strain CP (Figs. 2d, 3). These results suggested that the enhanced glyoxylate shunt acquired by directed pathway evolution successfully improved succinate production and reduced the byproduct level as expected.
Relative gene transcription levels and enzyme activities of key steps of central carbon metabolism
In order to figure out the genetic basis for the phenotypes of the evolved strain, several key reactions were investigated in the aspects of gene transcription levels and enzyme activities. Citrate synthase (CS) is the first enzyme of both the TCA cycle and the glyoxylate shunt and plays an important role in controlling the rate of these pathways. ICL and isocitrate dehydrogenase (ICDH) are key enzymes at isocitrate branch point and control the flux distribution between the glyoxylate shunt and the oxidative arm of the TCA cycle. Due to the importance of these enzymes, the relative gene transcription levels and the activities were evaluated for strain CP and E2-Δsdh-ppc.
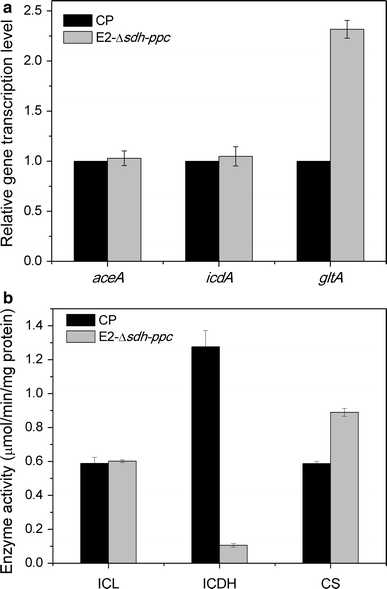
a Changes of gene transcription levels of aceA, icdA, and gltA between strain CP and E2-Δsdh-ppc; b Changes of enzyme activities of ICL, ICDH, and CS between strain CP and E2-Δsdh-ppc. The unit of enzyme activity is μmol/min/mg protein. All the measurements were performed in triplicate
Effects of the mutated ICDH on succinate production
When acetate is the sole carbon source, the flux of the glyoxylate shunt is increased through dramatically decreasing ICDH activity [16]. Hence, the enhanced glyoxylate shunt in the evolved mutant in this study might also be attributed to the extremely low activity of the mutated ICDH. In order to validate this hypothesis, the G195C mutation was introduced to the parental strains C2 and CP to explore the role of the mutated ICDH. The growth performance and fermentation products pattern were evaluated for the resulting strains C2-583 and CP-583, respectively.
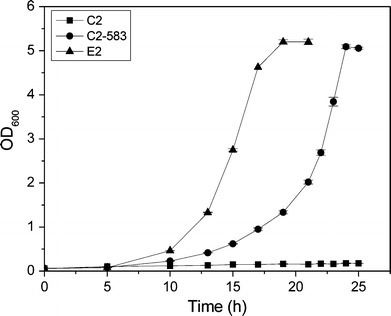
The growth curves of strain C2, C2-583, and E2 in glycerol minimal medium in 500-ml flasks at 37 °C and 220 rpm
As expected, significant differences in the product’s pattern were observed after introducing the G195C mutation into ICDH of the control strain CP (Fig. 3). The resulting strain CP-583 produced 20 % more succinate than the control CP. Meanwhile, α-ketoglutarate level of CP-583 showed a 34 % reduction compared to CP. Cell growth was not affected by the mutated ICDH. These results indicated that the enhanced glyoxylate shunt resulting from the mutated ICDH could reduce α-ketoglutarate level and improve succinate production.
In order to better explore the role of the mutated ICDH, the G195C mutation in strain E2-Δsdh-ppc was reverted to the wild type to produce strain E2-Δsdh-ppc-583. As depicted in Fig. 3, succinate production decreased by 15 % and α-ketoglutarate level increased by 63 % for strain E2-Δsdh-ppc-583 compared to E2-Δsdh-ppc. This suggested that the G195C mutation in ICDH was the main reason for the observed phenotype of E2-Δsdh-ppc and further confirmed the role of the mutated ICDH.
The mutated ICDH and wild-type ICDH were both purified and the kinetics of them were measured and compared. The K M value of wild-type ICDH for isocitrate was 0.035 mM as previously reported [30], whereas that of the mutated ICDH was 0.225 mM. In addition, the k cat value of the mutated ICDH was 9.9 s−1, much lower than that of wild-type ICDH of 57.8 s−1.
Effects of overexpression of CS on cell growth and succinate production
According to the above-mentioned results, CS was upregulated in the evolved strain. In order to explore the effects of the upregulated CS on cell growth and succinate production, the native promoter of gene gltA was replaced with trc strong promoter in strain C2-583 and CP-583, producing strain C2-583-gltA and CP-583-gltA. Strain C2-583-gltA grew faster with a specific growth rate of 0.26 h−1 in glycerol minimal medium, compared to that of strain C2-583 of 0.19 h−1, but still lower than that of the evolved strain E2. Similar phenomenon was also observed for strain CP-583-gltA in comparison with CP-583. Although cell growth was improved for strain CP-583-gltA, succinate production was not affected unexpectedly.
Further improving succinate production by adopting dual enhanced routes
Although the amount of the main byproduct α-ketoglutarate was significantly reduced by the enhanced glyoxylate shunt, it was still accumulated to a concentration of 5.5 mM. With the aim to further improve succinate production, continuing to reduce the level of α-ketoglutarate would probably be effective. α-ketoglutarate dehydrogenase complex is a rate-limiting enzyme of the TCA cycle which catalyzes the dehydrogenation of α-ketoglutarate to generate succinyl-CoA. Overexpression of this enzyme might be able to reduce the accumulation of α-ketoglutarate. Therefore, the native promoter of sucAB was replaced by the strong promoter trc in strain E2-Δsdh-ppc to produce strain E2-Δsdh-ppc-sucAB. As shown in Figs. 2 and 3, E2-Δsdh-ppc-sucAB showed a 20 % increase in succinate yield as compared with E2-Δsdh-ppc, and a 55.8 % increase as compared to the original strain CP, which represented 67 % of the maximum theoretical yield. As expected, the amount of α-ketoglutarate decreased significantly to 0.27 mM (0.04 g/l) for strain E2-Δsdh-ppc-sucAB. Meanwhile, cell growth of E2-Δsdh-ppc-sucAB was retarded by overexpression of α-ketoglutarate dehydrogenase complex (Fig. 2a). Eventually, E2-Δsdh-ppc-sucAB gained two enhanced routes to produce succinate.
Fed-batch fermentation for aerobic succinate production
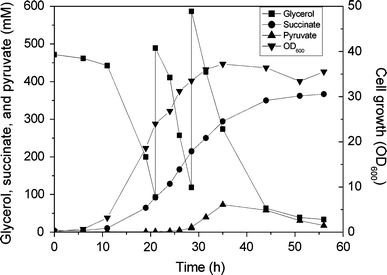
Fed-batch fermentation profile of strain E2-Δsdh-ppc-sucAB for succinate production under aerobic conditions in defined minimal medium
Discussion
For some desired phenotypes that are difficult to acquire through rational design, evolutionary engineering provides us an effective alternative. The flux ratio between the glyoxylate shunt and the oxidative arm of the TCA cycle, which we attempted to alter in this study, is controlled by several factors and is difficult to change. Therefore, a strategy of directed pathway evolution of the glyoxylate shunt was developed in E. coli for improved production of succinate from glycerol under aerobic conditions. The selection pressure of the evolution was provided by the knockout of PPC, which functioned as the primary anaplerotic pathway. The glyoxylate shunt was successfully recruited as the essential anaplerotic pathway in place of PPC for growth in glycerol minimum medium through adaptive evolution, as evidenced by the inviability of the evolved mutant after deletion of the operon aceBA. This result further confirmed the results reported previously that the glyoxylate shunt would be activated as the anaplerotic pathway in response to knockout of PPC [12, 32]. However, in another study, the energy-conserving phosphoenolpyruvate carboxykinase was evolved to function as the primary carboxylation pathway for succinate production in the presence of PPC [46]. Different from our work, the selection pressure of evolution in that study was not the absence of the anaplerotic pathway, but the insufficient supply of ATP for cell growth, which resulted in the recruitment of phosphoenolpyruvate carboxykinase.
When acetate is used as the sole carbon source, the inactivation of ICDH by ICDH kinase allows isocitrate lyse (ICL), whose affinity for isocitrate is relatively lower than ICDH [29], to acquire more substrate isocitrate, directing more carbon flux into the glyoxylate bypass [16]. In this study, the G195C mutation of ICDH discovered in strain E2 could also significantly decrease the activity of ICDH and consequently increase the flux of the glyoxylate shunt. This was evidenced by the viability of C2-583 in glycerol minimal medium rendered by the mutated ICDH. In addition, the G195C mutation lowered the affinity of ICDH for isocitrate, which was also beneficial for ICL to acquire more substrate. However, the growth of C2-583 was much slower than that of strain E2, and succinate production of CP-583 was lower than that of E2-Δsdh-ppc. These results indicated that not all the phenotypes of E2 and E2-Δsdh-ppc were attributed to the mutated ICDH and some other genetic changes might be present.
It was observed that the transcription level and activity of citrate synthase (CS) both increased for strain E2-Δsdh-ppc compared to strain CP. The increased activity of CS was also reported previously in E. coli ppc mutant [32]. In that study, the flux of the TCA cycle was increased by the upregulation of CS for channeling more carbon to replenish OAA. In another study, the flux of the TCA cycle was also increased in unevolved and evolved ppc mutants [12]. As the first enzyme of the TCA cycle and the glyoxylate bypass, CS is subjected to tight regulation [31] and plays a key role in controlling cell growth and carbon flow through the TCA cycle under certain conditions [43]. This probably could explain the faster growth of strain C2-583-gltA and CP-583-gltA observed in this study. However, succinate production was not improved by overexpression of CS. On the contrary, the high CS activity has been previously reported in the efficient aerobic succinate producer HL27659K [20], and succinate production was improved by overexpression of CS in Corynebacterium glutamicum under aerobic conditions [49]. The reason for the different results observed between this work and others’ was not clear. Probably the degree of overexpression was still not sufficient for improving succinate production in this study and this would be examined in future study.
Under oxygen-limited conditions, poor growth and low cell density were commonly observed when glycerol was used to produce succinate by metabolic engineered E. coli, which resulted in long fermentation periods and low volumetric productivities [3, 47]. As an alternative strategy, engineering E. coli for aerobic production of succinate from glycerol would solve the issue mentioned above. The biocatalyst developed in this study achieved relative high cell density and exhibit a maximum glycerol consumption rate of 61.5 mM h−1 during fed-batch fermentation under aerobic conditions. These advantages led to the high succinate volumetric productivity of 6.55 mM h−1, which exhibited potential industrial production capacity from the low-priced substrate.
Rational engineering based on the information obtained from evolutionary engineering would be greatly meaningful. Regulating the expression of key genes which exhibited different patterns between the parental and the evolved strain with promoter libraries or RBS libraries would probably have striking effects on succinate production. In addition, more genetic changes need to be figured out through genome sequencing to discover the underlying reasons of the phenotype of the evolved strain. Moreover, metabolic flux analysis needs to be performed to explore the different flux distribution pattern of the developed strains in this study, to figure out the bottleneck of carbon flow and guide us to further improve succinate production.
Acknowledgments
This work was supported by National 973 Project (2011CBA00804, 2012CB725203), National Natural Science Foundation of China (NSFC-21176182, NSFC-21206112) and National High-tech R&D Program of China (2012AA02A702, 2012AA022103).

{kind=link}
{kind=link}
{kind=link}
{kind=link}