





Abstract
Experiments were conducted to determine chemicals at low concentrations, which can be utilized for selective release of periplasmic proteins. It was revealed that 80–100 % of the activity of alpha-amylase, beta-lactamase, and Fab D1.3 was retained in the presence of 0.05 and 0.1 % Triton X-100, 0.1 % Tween 20, 0.1 % DOC, 0.01 % BAC, 0.01 % CTAB, 10 mM EDTA, 1 mM and 10 mM DEA, 10 mM NTA, 0.1 and 1 % SHMP, 200 mM urea, 100–500 mM GndCl, and 1 % solvents (hexane, xylene, toluene, benzene, pyridine and isoamyl alcohol). Performance of these chemicals, recognized as generally safe, for selective release of proteins from the periplasm of Escherichia coli was investigated. DOC was a general and very efficient agent, and at concentrations as low as 0.05, 0.1, and 0.025 %, released beta-lactamase, alpha-amylase, and Fab D1.3 selectively with yield factors of 2.7, 2.3, and 3.6 times greater than osmotic shock procedure, respectively. EDTA (1 and 10 mM) discharged Fab D1.3 with efficiency more than osmotic shock (target protein yield of 110 and 138 %, correspondingly). Isoamyl alcohol (10 % v/v) was effective for periplasmic release of alpha-amylase and particularly Fab D1.3, with target protein yields of 75 and 168 %, respectively.
Electronic supplementary material
The online version of this article (doi:10.1007/s10295-013-1307-1) contains supplementary material, which is available to authorized users.
Introduction
Disulfide bonds are essential for the folding and stability of many proteins and correct formation of disulfide bridges is a main concern, particularly when expressing recombinant proteins having multiple non-consecutive disulfide bonds in their final folded structure, in Escherichia coli (E. coli) [2, 32]. Proper disulfide bond formation is not usually achieved by cytoplasmic expression in this host [7, 11, 39]. The periplasm of E. coli has been the site of choice for the production of soluble recombinant proteins having disulfide bonds in their structures [1, 5, 6, 16–19, 23, 24, 26, 27, 42, 43] because it proffers many advantages. The oxidizing environment of the periplasm, the periplasmic disulfide bond-forming (Dsb) proteins, isomerases, and chaperones assist the formation of properly folded target protein [2, 3, 7, 35]. The oxidizing environment of the periplasm in cooperation with periplasmic proteins, including Dsb proteins and isomerases, assists the formation and isomerization of disulfide bonds, resulting in a firmly folded structure resistant to proteases [2, 7, 32]. Chaperon properties of certain periplasmic proteins can also hamper the aggregation of target products [3, 35]. Also, the outer membrane (OM) of Gram-negative bacteria is penetrable to small (≤600-Da) solutes [31] and as a result the periplasm can be manipulated by being exposed to extracellular chemical agents, which assist the folding and modification of periplasmically expressed proteins [1, 36, 41]. Moreover, the periplasmic space of E. coli contains only 4–8 % of the total cell protein [34] whereas problematic cytoplasmic components (particularly nucleic acids and proteases), which pose additional purification challenges during product recovery, are not present.
Despite these advantages, periplasmic expression systems have not yet fulfilled their true potential at a large scale, due to the lack of reliable general methods for efficient selective release of periplasmically expressed proteins. Therefore, in the beginning of a downstream process for the extraction and purification of periplasmically expressed proteins, the selective recovery of periplasmically located proteins is of great interest, as this will reduce the number, and as a result, the cost, of downstream operation units. Osmotic shock, based on destabilization of the outer envelope with ethylenediaminetetraacetic acid (EDTA) and lysozyme, is the only general technique for selective extraction of periplasmic proteins; however, this technique can be used only on small volumes and is not feasible at a large scale since the large volumes of liquid have to be handled at low temperatures, and lysozyme is expensive [18]. Besides, osmotic shock is time-consuming as an extended time is required to resuspend a large amount of cells pellet.
No other general method for selective release of periplasmically expressed proteins has been reported. There are limited reports in literature in which the recovery of recombinant proteins from the periplasm of E. coli using few chemical agents has been investigated [11, 30]; however, the influence of such chemical agents on the biological activity and structure of the pure target proteins has not been investigated prior to E. coli cell permeabilization, and the selection of the chemicals has been subjective. The chemicals demonstrated effective in those reports have not been successful for the selective and efficient recovery of the target proteins in this study, as reported later. In the present research, subjective selection of various concentrations of the chemicals for E. coli cell permeabilization has been avoided by investigating the influence of varying chemical concentrations on the biological activity and the secondary structure of proteins. The objective of this work was to study the performance of a wide range of chemicals at low concentrations, including ionic and non-ionic detergents, chelators, chaotropic agents, and organic solvents, in the permeabilization of E. coli cells and the selective release of periplasmically produced proteins. As these chemicals may have adverse impacts on biological activity and structure of periplasmically located proteins and consequently they may compromise the production of active compound; therefore, the influence of their various concentrations on the biological activity and structure of three pure target proteins (namely beta-lactamase, alpha-amylase, and Fab D1.3 fragment) was firstly investigated and safe chemicals were recognized. Secondly, the recognized safe chemicals were utilized in the permeabilization of E. coli cells, and their performance in the selective release of the periplasmically produced proteins was evaluated.
Materials and methods
Escherichia coli W3110 (ATCC 27325) transformed with a plasmid containing the gene for the anti-lysozyme antibody fragment (Fab D1.3) under the control of the lac-based expression system was provided by Merck Sharp and Dohme Ltd. (Billingham, UK). Escherichia coli JM107 PQR126 producing alpha-amylase and E. coli JM109 (ATCC 53323) producing beta-lactamase in the periplasm [12] were received from Professor John Ward (Research Department of Structural & Molecular Biology, University College London, UK), and Dr. Tim Overton (School of Chemical Engineering, University of Birmingham, UK), respectively. The bacterial strains were maintained in Luria–Bertani (LB) broth supplemented with 20 % v/v glycerol at −80 °C.
Fed-batch fermentation and chromatographic purification of Fab D1.3 fragment
Starting culture was prepared by inoculating 200 ml of LB broth containing 15 mg/l tetracycline with a fresh single colony of E. coli and shaking at 37 °C and 200 rpm for 13 h. A complex medium was employed for the growth of E. coli in a 10-l Electrolab fermentor (Tewkesbury, UK). The composition of the complex medium is as follows: 14 g (NH4)2SO4/l; 35 g glycerol/l; 20 g yeast extract/l; 2.0 g KH2PO4/l; 16.5 g K2HPO4/l; 7.5 g citric acid/l; and 1.5 ml H3PO4/l. Antifoam AF204 (Sigma-Aldrich, St. Louis, MO, USA) was added at 0.2 ml/l to control foaming. The medium was sterilized in an autoclave at 121 °C for 0.5 h. After cooling to 37 °C, four separate solutions were filter-sterilized and aseptically added to the fermentation medium: 10 ml/l of 1 M MgSO4·7H2O solution; 2 ml/l of 1 M CaCl2·2H2O solution; 1 ml tetracycline/l (of a 15 mg/ml stock); and 34 ml/l trace element solution. The trace element solution contained: 3.36 g FeSO4·7H2O/l; 0.84 g ZnSO4·7H2O/l; 0.51 g MnSO4·H2O/l; 0.25 g Na2MoO4·2H2O/l; 0.12 g CuSO4·5H2O/l; 0.36 g H3BO3/l; and 48 ml concentrated H3PO4/l. The pH of the medium was adjusted to 7.0 using NH4OH. The fermentor was inoculated with 200 ml (~4 % inoculum/medium ratio) from a 13-h shake flask inoculum, and operated in batch mode. The starting conditions were: temperature 37 °C; agitator speed 250 rpm; air-flow rate 1.0 VVM; pH 7.0. Agitation rate was increased during the fermentation (up to maximum value of 1,500 rpm) in order to sustain the dissolved oxygen tension on a set point of 30 %. The pH was maintained at 7.0 throughout the fermentation by the automatic addition of NH4OH. Once glycerol was depleted, being indicated by an increase in the dissolved oxygen measured in the fermentor, fermentation was carried out in the fed-batch mode with the feed (714 g glycerol/l and 30 g MgSO4·7H2O/l) at a constant rate of 15 ml/l h. After 10-h cultivation (OD600nm~50), Fab D1.3 expression was induced by the addition of Isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM.
Purification from cell-free culture broth (after 11-h post-induction with 0.1 mM IPTG) was performed on a 5-ml HiTrap SP Sepharose XL column (GE Healthcare, Uppsala, Sweden). Prior to loading of the cell-free culture fluid onto the column, the feedstock was extensively dialyzed overnight against 15 mM sodium acetate, pH 5.0, at 4 °C with changing the buffer in order to reduce both the conductivity (from ca. 40 to 5.0 mS/cm) and pH (from 7.0 to 5.0) for optimal sorption of the Fab on the cation exchange (CEX) matrix. The dialyzed culture broth sample was re-centrifuged (9,390 × g for 10 min) and filtered through 0.45-μm Millipore filters. The sample (480 ml) was then loaded at a flow rate of 3.4 ml/min on the HiTrap SP XL column pre-equilibrated with 15 mM sodium acetate buffer, pH 5.0. At the same flow velocity, the column was then washed with the equilibration buffer, and elution made with 20 mM MES (2-morpholinoethanesulfonic acid, 4.5 mM sodium MES, 1.5 M NaCl, pH 5.5). Eluted fractions containing Fab D1.3 from chromatography on HiTrap SP XL column were pooled and desalted on PD10 columns (GE Healthcare, Uppsala, Sweden) prior to manual loading onto a 1-ml HiTrap Protein G column (GE Healthcare, Uppsala, Sweden) pre-equilibrated with 20 mM sodium phosphate, pH 7.0. After extensive washing with the equilibration buffer, bound material was eluted with 0.1 M glycine–HCl, pH 2.7. The eluates were neutralized immediately with 1 M Tris–HCl, pH 9.0, and stored at −20 °C.
Biological activity of pure proteins in the presence of various chemicals
The retention activity of the proteins in the presence of various concentrations of chemicals was assessed as mentioned below.
Beta-lactamase assay
Beta-lactamase activity was assayed according to a method adapted from Sargent [37], in which penicilloic acid, the product of reaction between beta-lactamase and penicillin G, makes the iodine colorless, and consequently, the decrease in absorbance due to iodine can be used as an estimation for enzyme activity. The following solutions were made freshly prior to the start of the assay: 4 mg of salt of benzylpenicillin per ml of 50 mM sodium phosphate buffer, pH 7.0; stock iodine solution containing 20.3 g I2 and 100 g KI in 500 ml of distilled water; iodine reagent was prepared by adding 1.25 ml stock iodine solution to 98.25 ml sodium acetate buffer (500 mM, pH 4.0). Routinely, 100 μl of sample was added to 20 μl of sodium salt of benzylpenicillin solution in a 96-well microtiter plate and following incubation at 37 °C, for 180 s, the reaction was stopped by adding 200 μl of iodine reagent. The absorbance of the solution was then read immediately at 450 nm. For each chemical, an individual blank containing 0.1 ml of chemical, 20 μl of 50 mM Na-phosphate buffer pH 7.0, and 200 μl of iodine reagent was prepared.
Alpha-amylase assay
Alpha-amylase activity was measured using a modified assay based on the method of Blanchin-Roland and Masson [4], in which activity is determined by measuring the rate of decrease in absorbance of a colored starch/I2 complex. For this purpose, 0.5 % soluble starch (w/v) was made in 15 mM sodium phosphate buffered pH 5.8, previously heated to boiling point and filtered through No. 1 grade Whatman filter paper (Whatman International Ltd, Maidstone, UK) while hot. The iodine reagent was prepared fresh by diluting 0.2 ml of stock solution [2.2 % (w/v) iodine and 4.4 % (w/v) KI] into 100 ml of 2 % (w/v) potassium iodine solution. Routinely, 150 μl of the preincubated starch solution was added to an equal volume of the preincubated sample in a 96-well microtiter plate and the mixture was incubated at 50 °C for 180 s. Thereafter, 15 μl of the reaction mixture was pipetted into 300 μl of the iodine reagent and the absorbance of the solution was measured at 600 nm. For each chemical, an individual blank containing 150 μl of chemical and 150 μl of starch solution was prepared.
Sandwich enzyme-linked immunosorbent assay (ELISA) for Fab D1.3
For this purpose, 96-well microtiter plate was coated with 100 μl of 0.01 % (w/v) hen egg white lysozyme (HEWL), made in coating buffer (1.59 g/l Na2CO3 and 2.93 g/l NaHCO3, pH 9.6), overnight at 4 °C. Blocking was made with 200 μl of PBS-BSA (1 PBS tablet and 1 g BSA/100 ml distilled water) per well, with shaking (500 rpm) in a microplate incubator shaker for 1 h at 37 °C. The wells were then washed three times with 300 μl wash buffer (1 PBS tablet and 100 μl Tween 20/100 ml distilled water) per well. Samples (100 μl) were transferred to the washed ELISA plate, incubated at 37 °C, 500 rpm for 1 h and washed again with the wash buffer three times. The plate was then loaded with 100 μl anti-IgG (Fab specific)–peroxidase antibody, incubated at 37 °C, 500 rpm for 1 h, and then washed three times with the wash buffer. Peroxidase substrate (100 μl) was added to each well and the reaction was stopped after 600 s by adding 100 μl of 1 M H3PO4. The plate was read at 450 nm.
Circular dichroism (CD) studies
Total protein quantification
The amount of total protein present in the samples was measured using bicinchoninic acid assay (BCA assay kit, Thermo Scientific, Rockford, IL, USA).
Shake-flask cultivation for the production of proteins in the periplasm
Production of beta-lactamase
For this purpose, a single colony of E. coli JM109, grown on nutrient agar containing 100 μg/ml ampicillin, was added to 100 ml of LB broth supplemented with 100 μg/ml ampicillin. The culture broth was incubated overnight (13 h) at 37 °C on an orbital shaker at 200 rpm. Flasks (1 l) containing 200 ml of LB and 100 μg/ml ampicillin were inoculated with 0.2 ml (0.1 % v/v) of the 13-h preculture and placed on an orbital shaker at 200 rpm and 37 °C for 18 h. The cells were collected by centrifugation at 4,120 × g for 600 s, and the pellets were frozen at −20 °C for further cell fractionation experiments.
Production of alpha-amylase
For this purpose, a single colony of E. coli JM107 PQR126, grown on nutrient agar containing 1 % (w/v) potato starch and 20 μg/ml kanamycin, was suspended into 100 ml of nutrient broth containing 10 μg/ml of kanamycin and incubated overnight (13 h) at 200 rpm and 37 °C. A flask (1 l) containing 200 ml of nutrient broth supplemented with 10 μg/ml kanamycin was inoculated with 8 ml (4 % v/v) of the 13-h preculture and placed on an orbital shaker at 200 rpm and 37 °C for 18 h. The cells were then harvested by centrifugation at 4,120 × g for 600 s, and the pellets were frozen at −20 °C for further cell fractionation experiments.
Production of Fab D1.3
For this purpose, a single colony of E. coli W3110, propagated overnight on nutrient agar supplemented with 15 μg/ml tetracycline, was added to 100 ml of LB broth containing 15 μg/ml of tetracycline. Four milliliters of the LB broth preculture was added to an Erlenmeyer flask containing 200 ml of the complex medium described earlier (i.e., as a 4 % inoculum), and incubated at 37 °C and 200 rpm. Fab D1.3 expression was induced by the addition of IPTG to a final concentration of 0.1 mM/l after 6.5 h cultivation (OD600nm of ~10) and the culture was left at 30 °C for another 4.5 h, with shaking at 200 rpm. After 11 h, the bacterial cells were harvested by centrifugation at 4,120 × g for 600 s, and the pellets were frozen at −20 °C for further cell fractionation experiments.
Cell fractionation
Osmotic shock method
A modified osmotic shock treatment [12] was used for releasing periplasmic proteins. Briefly, the bacterial cell pellets (except the one obtained from E. coli W3110), harvested from 1 ml of culture medium in Eppendorf tubes, were resuspended in 0.5 ml of 200 mM Tris buffer pH 7.5 containing 1 mM EDTA, 20 % sucrose, and 500 μg/ml lysozyme. The tubes were then incubated statically 15 min at room temperature, and 0.5 ml cold distilled water was next added. After 15 min of static incubation, the pellet was separated from supernatant by centrifugation at 15,800 × g for 300 s.
For recovery of Fab D1.3 from the periplasm by osmotic shock, it was not feasible to add lysozyme in the osmotic shock solution because lysozyme is specific antigen for Fab D1.3, and its integration to the osmotic shock solution compromises the Fab assay by ELISA, being based on attachment of Fab D1.3 to immobilized lysozyme on microplate as described below. Thus, the osmotic shock procedure for the bacterial cell pellets of E. coli W3110 was performed without using lysozyme.
The supernatants containing the periplasmic proteins were kept frozen at −20 °C for further enzyme activity assay.
High-pressure homogenization
The bacterial cell pellets were resuspended in 50 ml of 200 mM Tris buffer pH 7.5, to a final OD600nm of ~5, and cells were subsequently disrupted using a continuous flow TS-Series benchtop disrupter (Constant Systems Ltd, Low March, UK) operated at 2,400 bar and 5 °C. The supernatant was separated from the cell debris by centrifugation at 15,800 × g for 300 s, and stored at −20 °C for further analyses.
Chemical permeabilization
The bacterial cell pellets, harvested from 1 ml culture medium in Eppendorf tubes, were resuspended in 1 ml of various chemicals. Following incubation for 1 h at room temperature with brief intermittent vortexing every 600 s, the supernatants were separated from the cells by centrifugation at 15,800 × g for 300 s and frozen at −20 °C for further analyses.
Results and discussion
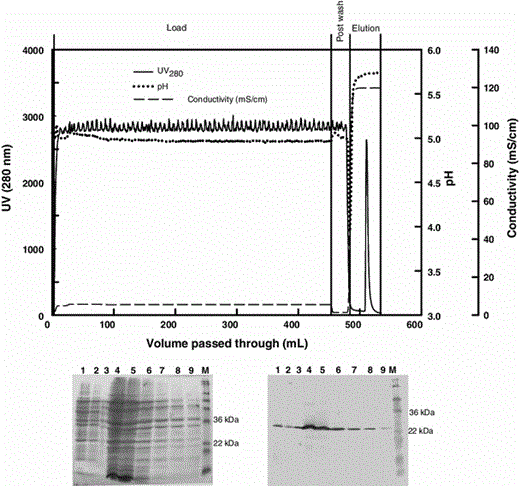
Chromatogram (top), SDS-PAGE (bottom left), and corresponding Western blot (bottom right) analyses for the purification of Fab D1.3 from culture broth (21-h culture) on a HiTrap SP Sepharose XL column. Sodium acetate (15 mM, pH 5.0) was employed for equilibration and the post wash. Elution was achieved by 20 mM MES, 4.5 mM sodium MES, 1.5 M NaCl (pH 5.5). Lane 1 load, 2 post wash, 3–9 elution fractions, M molecular weight markers
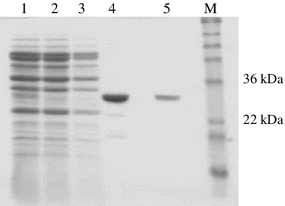
SDS-PAGE analysis following purification of partially purified Fab D1.3 on HiTrap protein G. Fractions (3–9) from chromatography on HiTrap SP Sepharose XL column (Fig. 1) were pooled and desalted on PD10 columns prior to loading into a HiTrap protein G column. Lane 1 PD10 desalted load, 2 flow through, 3 post wash, 4 and 5 eluate 1 and 2, respectively, M molecular weight markers
Highly pure Fab D1.3, obtained from this purification process, and pure beta-lactamase and alpha-amylase obtained from Sigma-Aldrich (Gillingham, UK) were used in further experiments to investigate the effect of chemicals on biological activity and secondary structure of the proteins. The retention activities of the proteins in the presence of various concentrations of chemicals, including ionic and non-ionic detergents, chelators, chaotropic agents and organic solvents, were assessed (Table 1). In most cases, each chemical had a different impact on the biological activity of each protein. For instance, the activity of Fab D1.3 was less dropped by cationic detergents (BAC and CTAB), whereas it was further reduced by chelators and solvents when compared with the activity of alpha-amylase and beta-lactamase. Also in comparison with two other proteins, beta-lactamase was less vulnerable, in terms of biological activity, to the non-ionic, anionic (except DOC), and chelators. The chemicals in the presence of which all three proteins retained ≥80 % of their activities were categorized as generally safe reagents and were used for further permeabilization experiments explained later.
Effect of various chemicals on the activity and mean residue ellipticity of beta-lactamase, alpha-amylase, and Fab D1.3
| Chemical | Beta-lactamase | Alpha-amylase | Fab D1.3 | |||
|---|---|---|---|---|---|---|
| Retention activity (%) | [θ]222 k | Retention activity (%) | [θ]222 k | Retention activity (%) | [θ]215 l | |
| 25 mM phosphate buffer pH 7.0a | 100 | −312.2 | 100 | −279.8 | 100 | −207.8 |
| 0.05 % Triton X−100* | 100 | −222.6 (−29) | 99.2 | −171.2 (−39) | 100 | −103 (−51) |
| 0.1 % Triton X-100* | 100 | −193.7 (−38) | 88.9 | −36.5 (−87) | 95.7 | −94.5 (−55) |
| 0.1 % Tween 20* | 100 | −289 (−7.4) | 83.7 | −195.3 (−30) | 98.9 | −67.1 (−68) |
| 1 % Tween 20 | 100 | 31.2 | 90.6 | |||
| 0.1 % Brij 35 | 100 | 44.5 | 90.5 | |||
| 1 % Brij 35 | 100 | 8.6 | 79 | |||
| 0.01 % SDSb | 83.7 | 52.2 | 57.8 | |||
| 0.1 % SDS | 69 | 46.8 | 43 | |||
| 0.1 % DOCc* | 90 | −310 (−0.7) | 100 | −204 (−27) | 94.3 | −123 (−41) |
| 1 % DOC | 0 | 0 | 89.2 | |||
| 0.01 % BACd* | 100 | −277.1 (−11) | 95.9 | −216.7 (−23) | 100 | −61.4 (−70) |
| 0.05 % BAC | 0 | 70.4 | 100 | |||
| 0.1 % BAC | 0 | 0 | 100 | |||
| 1 % BAC | 0 | 0 | 77.6 | |||
| 0.01 % CTABe* | 100 | −319.2 (+2) | 89.8 | −129.6 (−54) | 100 | 6.3 (−103) |
| 0.05 % CTAB | 0 | 3.25 | 100 | |||
| 0.1 % CTAB | 0 | 0 | 100 | |||
| 1 % CTAB | 0 | 0 | 89.9 | |||
| 10 mM EDTAf* | 100 | −293.9 (−6) | 100 | −124.7 (−55) | 95.5 | −69.5 (−67) |
| 1 mM DEAg* | 100 | −308.6 (−1.1) | 100 | −154.9 (−45) | 89 | |
| 10 mM DEA* | 100 | −248.7 (−20) | 99.6 | −160.5 (−43) | 81.5 | −91.4 (−56) |
| 10 mM NTAh* | 100 | −348.7 (+12) | 100 | −176 (−37) | 98.9 | −29.1 (−86) |
| 0.1 % SHMPi* | 100 | −295.3 (−5.4) | 99.7 | −136.7 (−51) | 95 | |
| 1 % SHMP* | 100 | −386.8 (+24) | 99.2 | −107.9 (−62) | 90.78 | −34.9 (−84) |
| 200 mM Urea* | 96 | −296.3 (−5) | 100 | −261.2 (−7) | 85.8 | −146.7 (−30) |
| 2 M Urea | 42.5 | 100 | 85 | |||
| 100 mM GndClj | 100 | 100 | 98.9 | |||
| 200 mM GndClj* | 100 | −309.9 (−0.7) | 98.6 | −233.8 (−17) | 85.2 | 10.2 (−105) |
| 500 mM GndCl* | 100 | −339.5 (+8.8) | 98.4 | −204 (−27) | 84.4 | 23.5 (−111) |
| 1 % Hexane* | 96.5 | −312.1 (0) | 100 | −230.5 (−18) | 85.8 | −77.5 (−63) |
| 1 % Xylene* | 98.7 | −289.9 (−7) | 100 | −173.4 (−38) | 80 | −104.7 (−50) |
| 1 % Toluene* | 100 | −311 (−0.4) | 100 | −175.2 (−39) | 80.1 | −69.3 (−67) |
| 1 % Benzene* | 100 | −307 (−1.6) | 100 | −176.5 (−37) | 100 | −89.1 (−58) |
| 1 % Diethyl ether | 98 | 100 | 69.6 | |||
| 1 % Acetone | 99.8 | 100 | 78.5 | |||
| 1 % Chloroform | 98.3 | 100 | 68 | |||
| 1 % Pyridine* | 100 | 100 | 83.2 | |||
| 1 % Ethanol | 90.3 | 100 | 70.7 | |||
| 1 % Isoamyl alcohol* | 100 | −357.1 (+14.4) | 100 | −317.6 (+13) | 84.1 | −173.3 (−17) |
| 10 % Isoamyl alcohol* | 100 | −400.5 (+28.3) | 99.1 | −284.9 (+1.8) | 80 | −272.3 (+31) |
| Chemical | Beta-lactamase | Alpha-amylase | Fab D1.3 | |||
|---|---|---|---|---|---|---|
| Retention activity (%) | [θ]222 k | Retention activity (%) | [θ]222 k | Retention activity (%) | [θ]215 l | |
| 25 mM phosphate buffer pH 7.0a | 100 | −312.2 | 100 | −279.8 | 100 | −207.8 |
| 0.05 % Triton X−100* | 100 | −222.6 (−29) | 99.2 | −171.2 (−39) | 100 | −103 (−51) |
| 0.1 % Triton X-100* | 100 | −193.7 (−38) | 88.9 | −36.5 (−87) | 95.7 | −94.5 (−55) |
| 0.1 % Tween 20* | 100 | −289 (−7.4) | 83.7 | −195.3 (−30) | 98.9 | −67.1 (−68) |
| 1 % Tween 20 | 100 | 31.2 | 90.6 | |||
| 0.1 % Brij 35 | 100 | 44.5 | 90.5 | |||
| 1 % Brij 35 | 100 | 8.6 | 79 | |||
| 0.01 % SDSb | 83.7 | 52.2 | 57.8 | |||
| 0.1 % SDS | 69 | 46.8 | 43 | |||
| 0.1 % DOCc* | 90 | −310 (−0.7) | 100 | −204 (−27) | 94.3 | −123 (−41) |
| 1 % DOC | 0 | 0 | 89.2 | |||
| 0.01 % BACd* | 100 | −277.1 (−11) | 95.9 | −216.7 (−23) | 100 | −61.4 (−70) |
| 0.05 % BAC | 0 | 70.4 | 100 | |||
| 0.1 % BAC | 0 | 0 | 100 | |||
| 1 % BAC | 0 | 0 | 77.6 | |||
| 0.01 % CTABe* | 100 | −319.2 (+2) | 89.8 | −129.6 (−54) | 100 | 6.3 (−103) |
| 0.05 % CTAB | 0 | 3.25 | 100 | |||
| 0.1 % CTAB | 0 | 0 | 100 | |||
| 1 % CTAB | 0 | 0 | 89.9 | |||
| 10 mM EDTAf* | 100 | −293.9 (−6) | 100 | −124.7 (−55) | 95.5 | −69.5 (−67) |
| 1 mM DEAg* | 100 | −308.6 (−1.1) | 100 | −154.9 (−45) | 89 | |
| 10 mM DEA* | 100 | −248.7 (−20) | 99.6 | −160.5 (−43) | 81.5 | −91.4 (−56) |
| 10 mM NTAh* | 100 | −348.7 (+12) | 100 | −176 (−37) | 98.9 | −29.1 (−86) |
| 0.1 % SHMPi* | 100 | −295.3 (−5.4) | 99.7 | −136.7 (−51) | 95 | |
| 1 % SHMP* | 100 | −386.8 (+24) | 99.2 | −107.9 (−62) | 90.78 | −34.9 (−84) |
| 200 mM Urea* | 96 | −296.3 (−5) | 100 | −261.2 (−7) | 85.8 | −146.7 (−30) |
| 2 M Urea | 42.5 | 100 | 85 | |||
| 100 mM GndClj | 100 | 100 | 98.9 | |||
| 200 mM GndClj* | 100 | −309.9 (−0.7) | 98.6 | −233.8 (−17) | 85.2 | 10.2 (−105) |
| 500 mM GndCl* | 100 | −339.5 (+8.8) | 98.4 | −204 (−27) | 84.4 | 23.5 (−111) |
| 1 % Hexane* | 96.5 | −312.1 (0) | 100 | −230.5 (−18) | 85.8 | −77.5 (−63) |
| 1 % Xylene* | 98.7 | −289.9 (−7) | 100 | −173.4 (−38) | 80 | −104.7 (−50) |
| 1 % Toluene* | 100 | −311 (−0.4) | 100 | −175.2 (−39) | 80.1 | −69.3 (−67) |
| 1 % Benzene* | 100 | −307 (−1.6) | 100 | −176.5 (−37) | 100 | −89.1 (−58) |
| 1 % Diethyl ether | 98 | 100 | 69.6 | |||
| 1 % Acetone | 99.8 | 100 | 78.5 | |||
| 1 % Chloroform | 98.3 | 100 | 68 | |||
| 1 % Pyridine* | 100 | 100 | 83.2 | |||
| 1 % Ethanol | 90.3 | 100 | 70.7 | |||
| 1 % Isoamyl alcohol* | 100 | −357.1 (+14.4) | 100 | −317.6 (+13) | 84.1 | −173.3 (−17) |
| 10 % Isoamyl alcohol* | 100 | −400.5 (+28.3) | 99.1 | −284.9 (+1.8) | 80 | −272.3 (+31) |
Negative and positive values in parentheses indicate decreased and increased percentages of the mean residue ellipticity, respectively
* Generally safe chemicals the impact of which on the secondary structure of the proteins was examined using CD. Chemicals in the presence of which three proteins retained ≥80 % of their activities
aEstimation for activity of each protein in 25 mM phosphate buffer, pH 7.0, was set as 100 % value
bSodium dodecyl sulphate
cSodium deoxycholate
dBenzalkonium chloride
eCetyltrimethylammonium bromide
fEthylenediaminetetraacetic acid
gDiethanolamine
hNitrilotriacetic acid
iSodium hexametaphosphate
jGuanidinium chloride
k, lMean residue ellipticities (deg cm2/dmol) at 222 and 215 nm, respectively
Effect of various chemicals on the activity and mean residue ellipticity of beta-lactamase, alpha-amylase, and Fab D1.3
| Chemical | Beta-lactamase | Alpha-amylase | Fab D1.3 | |||
|---|---|---|---|---|---|---|
| Retention activity (%) | [θ]222 k | Retention activity (%) | [θ]222 k | Retention activity (%) | [θ]215 l | |
| 25 mM phosphate buffer pH 7.0a | 100 | −312.2 | 100 | −279.8 | 100 | −207.8 |
| 0.05 % Triton X−100* | 100 | −222.6 (−29) | 99.2 | −171.2 (−39) | 100 | −103 (−51) |
| 0.1 % Triton X-100* | 100 | −193.7 (−38) | 88.9 | −36.5 (−87) | 95.7 | −94.5 (−55) |
| 0.1 % Tween 20* | 100 | −289 (−7.4) | 83.7 | −195.3 (−30) | 98.9 | −67.1 (−68) |
| 1 % Tween 20 | 100 | 31.2 | 90.6 | |||
| 0.1 % Brij 35 | 100 | 44.5 | 90.5 | |||
| 1 % Brij 35 | 100 | 8.6 | 79 | |||
| 0.01 % SDSb | 83.7 | 52.2 | 57.8 | |||
| 0.1 % SDS | 69 | 46.8 | 43 | |||
| 0.1 % DOCc* | 90 | −310 (−0.7) | 100 | −204 (−27) | 94.3 | −123 (−41) |
| 1 % DOC | 0 | 0 | 89.2 | |||
| 0.01 % BACd* | 100 | −277.1 (−11) | 95.9 | −216.7 (−23) | 100 | −61.4 (−70) |
| 0.05 % BAC | 0 | 70.4 | 100 | |||
| 0.1 % BAC | 0 | 0 | 100 | |||
| 1 % BAC | 0 | 0 | 77.6 | |||
| 0.01 % CTABe* | 100 | −319.2 (+2) | 89.8 | −129.6 (−54) | 100 | 6.3 (−103) |
| 0.05 % CTAB | 0 | 3.25 | 100 | |||
| 0.1 % CTAB | 0 | 0 | 100 | |||
| 1 % CTAB | 0 | 0 | 89.9 | |||
| 10 mM EDTAf* | 100 | −293.9 (−6) | 100 | −124.7 (−55) | 95.5 | −69.5 (−67) |
| 1 mM DEAg* | 100 | −308.6 (−1.1) | 100 | −154.9 (−45) | 89 | |
| 10 mM DEA* | 100 | −248.7 (−20) | 99.6 | −160.5 (−43) | 81.5 | −91.4 (−56) |
| 10 mM NTAh* | 100 | −348.7 (+12) | 100 | −176 (−37) | 98.9 | −29.1 (−86) |
| 0.1 % SHMPi* | 100 | −295.3 (−5.4) | 99.7 | −136.7 (−51) | 95 | |
| 1 % SHMP* | 100 | −386.8 (+24) | 99.2 | −107.9 (−62) | 90.78 | −34.9 (−84) |
| 200 mM Urea* | 96 | −296.3 (−5) | 100 | −261.2 (−7) | 85.8 | −146.7 (−30) |
| 2 M Urea | 42.5 | 100 | 85 | |||
| 100 mM GndClj | 100 | 100 | 98.9 | |||
| 200 mM GndClj* | 100 | −309.9 (−0.7) | 98.6 | −233.8 (−17) | 85.2 | 10.2 (−105) |
| 500 mM GndCl* | 100 | −339.5 (+8.8) | 98.4 | −204 (−27) | 84.4 | 23.5 (−111) |
| 1 % Hexane* | 96.5 | −312.1 (0) | 100 | −230.5 (−18) | 85.8 | −77.5 (−63) |
| 1 % Xylene* | 98.7 | −289.9 (−7) | 100 | −173.4 (−38) | 80 | −104.7 (−50) |
| 1 % Toluene* | 100 | −311 (−0.4) | 100 | −175.2 (−39) | 80.1 | −69.3 (−67) |
| 1 % Benzene* | 100 | −307 (−1.6) | 100 | −176.5 (−37) | 100 | −89.1 (−58) |
| 1 % Diethyl ether | 98 | 100 | 69.6 | |||
| 1 % Acetone | 99.8 | 100 | 78.5 | |||
| 1 % Chloroform | 98.3 | 100 | 68 | |||
| 1 % Pyridine* | 100 | 100 | 83.2 | |||
| 1 % Ethanol | 90.3 | 100 | 70.7 | |||
| 1 % Isoamyl alcohol* | 100 | −357.1 (+14.4) | 100 | −317.6 (+13) | 84.1 | −173.3 (−17) |
| 10 % Isoamyl alcohol* | 100 | −400.5 (+28.3) | 99.1 | −284.9 (+1.8) | 80 | −272.3 (+31) |
| Chemical | Beta-lactamase | Alpha-amylase | Fab D1.3 | |||
|---|---|---|---|---|---|---|
| Retention activity (%) | [θ]222 k | Retention activity (%) | [θ]222 k | Retention activity (%) | [θ]215 l | |
| 25 mM phosphate buffer pH 7.0a | 100 | −312.2 | 100 | −279.8 | 100 | −207.8 |
| 0.05 % Triton X−100* | 100 | −222.6 (−29) | 99.2 | −171.2 (−39) | 100 | −103 (−51) |
| 0.1 % Triton X-100* | 100 | −193.7 (−38) | 88.9 | −36.5 (−87) | 95.7 | −94.5 (−55) |
| 0.1 % Tween 20* | 100 | −289 (−7.4) | 83.7 | −195.3 (−30) | 98.9 | −67.1 (−68) |
| 1 % Tween 20 | 100 | 31.2 | 90.6 | |||
| 0.1 % Brij 35 | 100 | 44.5 | 90.5 | |||
| 1 % Brij 35 | 100 | 8.6 | 79 | |||
| 0.01 % SDSb | 83.7 | 52.2 | 57.8 | |||
| 0.1 % SDS | 69 | 46.8 | 43 | |||
| 0.1 % DOCc* | 90 | −310 (−0.7) | 100 | −204 (−27) | 94.3 | −123 (−41) |
| 1 % DOC | 0 | 0 | 89.2 | |||
| 0.01 % BACd* | 100 | −277.1 (−11) | 95.9 | −216.7 (−23) | 100 | −61.4 (−70) |
| 0.05 % BAC | 0 | 70.4 | 100 | |||
| 0.1 % BAC | 0 | 0 | 100 | |||
| 1 % BAC | 0 | 0 | 77.6 | |||
| 0.01 % CTABe* | 100 | −319.2 (+2) | 89.8 | −129.6 (−54) | 100 | 6.3 (−103) |
| 0.05 % CTAB | 0 | 3.25 | 100 | |||
| 0.1 % CTAB | 0 | 0 | 100 | |||
| 1 % CTAB | 0 | 0 | 89.9 | |||
| 10 mM EDTAf* | 100 | −293.9 (−6) | 100 | −124.7 (−55) | 95.5 | −69.5 (−67) |
| 1 mM DEAg* | 100 | −308.6 (−1.1) | 100 | −154.9 (−45) | 89 | |
| 10 mM DEA* | 100 | −248.7 (−20) | 99.6 | −160.5 (−43) | 81.5 | −91.4 (−56) |
| 10 mM NTAh* | 100 | −348.7 (+12) | 100 | −176 (−37) | 98.9 | −29.1 (−86) |
| 0.1 % SHMPi* | 100 | −295.3 (−5.4) | 99.7 | −136.7 (−51) | 95 | |
| 1 % SHMP* | 100 | −386.8 (+24) | 99.2 | −107.9 (−62) | 90.78 | −34.9 (−84) |
| 200 mM Urea* | 96 | −296.3 (−5) | 100 | −261.2 (−7) | 85.8 | −146.7 (−30) |
| 2 M Urea | 42.5 | 100 | 85 | |||
| 100 mM GndClj | 100 | 100 | 98.9 | |||
| 200 mM GndClj* | 100 | −309.9 (−0.7) | 98.6 | −233.8 (−17) | 85.2 | 10.2 (−105) |
| 500 mM GndCl* | 100 | −339.5 (+8.8) | 98.4 | −204 (−27) | 84.4 | 23.5 (−111) |
| 1 % Hexane* | 96.5 | −312.1 (0) | 100 | −230.5 (−18) | 85.8 | −77.5 (−63) |
| 1 % Xylene* | 98.7 | −289.9 (−7) | 100 | −173.4 (−38) | 80 | −104.7 (−50) |
| 1 % Toluene* | 100 | −311 (−0.4) | 100 | −175.2 (−39) | 80.1 | −69.3 (−67) |
| 1 % Benzene* | 100 | −307 (−1.6) | 100 | −176.5 (−37) | 100 | −89.1 (−58) |
| 1 % Diethyl ether | 98 | 100 | 69.6 | |||
| 1 % Acetone | 99.8 | 100 | 78.5 | |||
| 1 % Chloroform | 98.3 | 100 | 68 | |||
| 1 % Pyridine* | 100 | 100 | 83.2 | |||
| 1 % Ethanol | 90.3 | 100 | 70.7 | |||
| 1 % Isoamyl alcohol* | 100 | −357.1 (+14.4) | 100 | −317.6 (+13) | 84.1 | −173.3 (−17) |
| 10 % Isoamyl alcohol* | 100 | −400.5 (+28.3) | 99.1 | −284.9 (+1.8) | 80 | −272.3 (+31) |
Negative and positive values in parentheses indicate decreased and increased percentages of the mean residue ellipticity, respectively
* Generally safe chemicals the impact of which on the secondary structure of the proteins was examined using CD. Chemicals in the presence of which three proteins retained ≥80 % of their activities
aEstimation for activity of each protein in 25 mM phosphate buffer, pH 7.0, was set as 100 % value
bSodium dodecyl sulphate
cSodium deoxycholate
dBenzalkonium chloride
eCetyltrimethylammonium bromide
fEthylenediaminetetraacetic acid
gDiethanolamine
hNitrilotriacetic acid
iSodium hexametaphosphate
jGuanidinium chloride
k, lMean residue ellipticities (deg cm2/dmol) at 222 and 215 nm, respectively
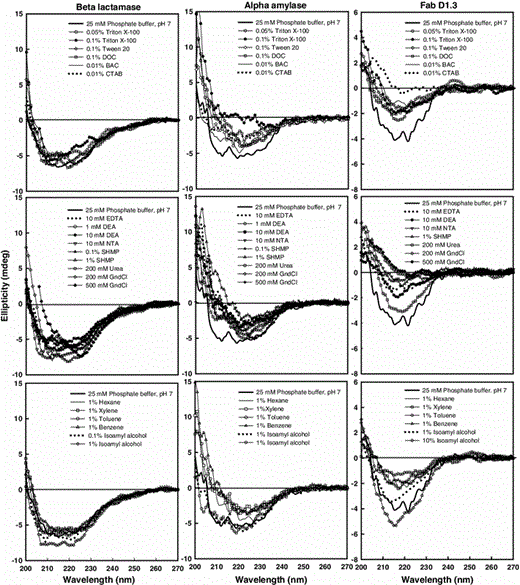
Intrinsic CD spectra of pure proteins (1 mg/ml in 25 mM sodium phosphate buffer, pH 7.0) in the presence of various chemicals. The spectra were recorded using a 0.2-cm path length cell and averaged over three scans
For the far-UV CD measurements, the mean residue ellipticity is often used as a unit for the presentation of CD data [8, 20, 21]; therefore, this parameter was used to make estimates for the changes in the signal of the alpha helix- and beta sheet-rich proteins at 222 and 215 nm, respectively (Table 1). Based on the values of the mean residue ellipticities at the key wavelengths, it is evident that changes in the CD signals of the proteins by same chemicals were not identical. The secondary structure of beta-lactamase was less vulnerable than that of alpha-amylase to majority of the chemicals. Also, beta sheet-rich Fab D1.3 gave rise to less intense signals than alpha helix rich proteins, and showed more variations in the spectral signals in the presence of chemicals when compared to the alpha helix-rich proteins. This was in agreement with what has been reported by Miles and Wallace [29]. Despite more sensitivity of its secondary structure to chemicals such as 0.1 % Tween 20, 0.1 % DOC, 0.01 % BAC, 0.01 % CTAB, 10 mM NTA, and 1 % Benzene, Fab D1.3 activity was equal or more than two other proteins in the presence of such chemicals.
Based on the protein activity and the mean residue ellipticity values shown in this table, there are several chemicals, such as 0.05 % Triton X-100, 0.01 % BAC, 10 mM NTA, 0.1 % SHMP, and 1 % Benzene, in presence of which the activity of the proteins remained ~100 %, but the secondary structure of the proteins changed significantly. This shows that the altered structure detected by CD is outside the proteins’ active sites; hence, proteins with significantly modified secondary structure may or may not maintain their native activity depending on the site in which alteration is occurred.
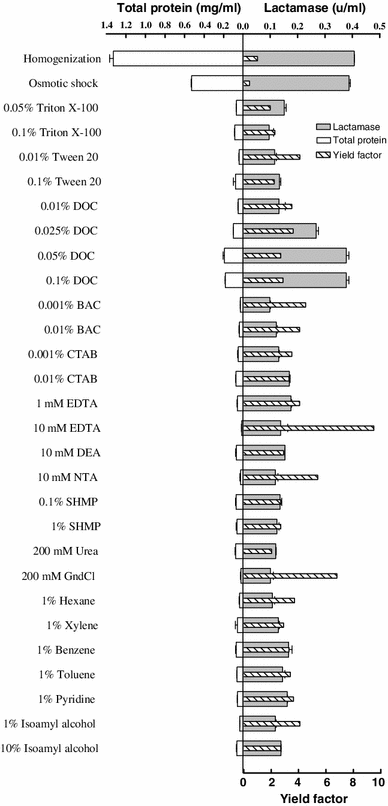
Release of beta-lactamase from transformed E. coli cells by homogenization, osmotic shock, and various chemicals made in 200 mM Tris buffer pH 7.5. Results are averages of two measurements. The yield factor was calculated by dividing target protein yield by total protein yield. Target protein yield and total protein yield are relative to the extraction by osmotic shock; the amounts of target and total proteins recovered from the bacterial cells by osmotic shock were considered as 100 % target and total protein yields, respectively
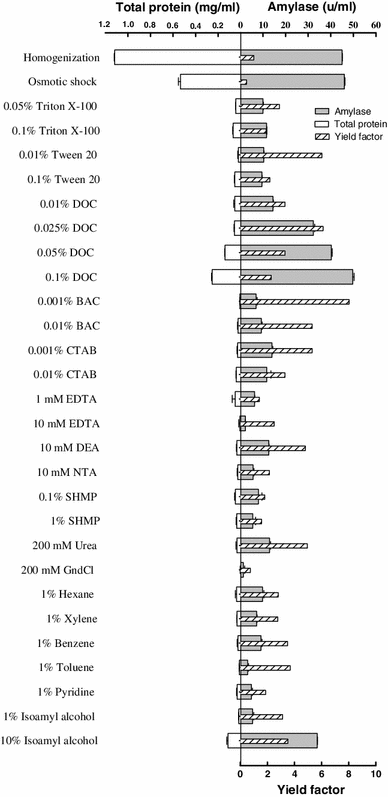
Release of alpha-amylase from E. coli cells by homogenization, osmotic shock, and various chemicals made in 200 mM Tris buffer pH 7.5. Results are averages of two measurements. The yield factor was calculated by dividing target protein yield by total protein yield. Target protein yield and total protein yield are relative to the extraction by osmotic shock; the amounts of target and total proteins recovered from the bacterial cells by osmotic shock were considered as 100 % target and total protein yields, respectively
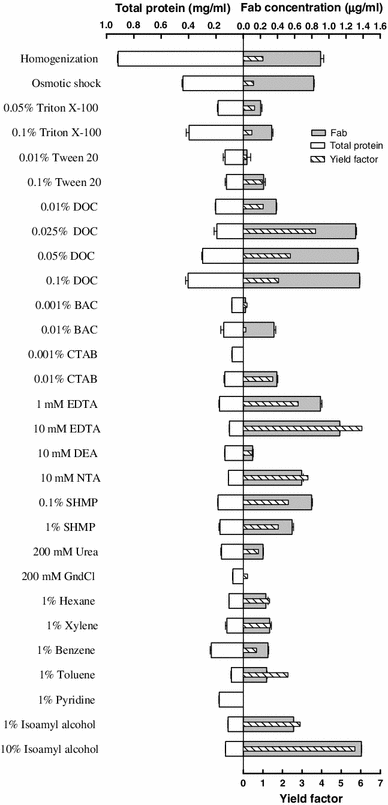
Release of Fab D1.3 from E. coli cells by homogenization, osmotic shock, and various chemicals made in 200 mM Tris buffer pH 7.5. Results are averages of two measurements. The yield factor was calculated by dividing target protein yield by total protein yield. Target protein yield and total protein yield are relative to the extraction by osmotic shock; the amounts of target and total proteins recovered from the bacterial cells by osmotic shock were considered as 100 % target and total protein yields, respectively
After ensuring that the osmotic shock could effectively release the target proteins from the periplasm of E. coli cells, the release of these target proteins by various chemicals, which were previously acknowledged safe, was investigated. Tris (hydroxymethyl) aminomethane (Tris) at a concentration of 200 mM and pH 7.5 was chosen as the buffer in which the chemicals were made, because it has been reported that Tris at moderately high concentrations can interact with lipopolysaccharide (LPS), replace stabilizing cations and consequently sensitize Gram-negative bacterial cells to other agents [15, 33, 40]. The selectivity and efficiency of different categories of chemicals (detergents, chelators, chaotropic agents; and organic solvents) were assessed by comparing the quantities of target proteins and total protein released by them with those figures obtained from the osmotic shock fractionation.
Comparisons of the target protein and total protein release by the osmotic shock and various chemicals are shown in Figs. 4, 5, and 6. Detergents (nonionic, anionic, and cationic) resulted in different extent of target proteins release from the periplasm of the bacterial cells. LPS and phospholipids molecules of E. coli cell OM form a bilayer, in which the hydrophobic tails reside in the bilayer and the hydrophobic heads are pointing to the solution environment, and the peripheral and integral membrane proteins are attached to the surface and the inner part of the bilayer, respectively [28]. Having an ionic hydrophilic region and a hydrophobic hydrocarbon portion and hence being amphipathic molecules, detergents (surfactants) can interact both with water and lipid to form micelles containing lipid. Detergent binds to the biomembrane lipid until saturation is attained, and more addition of detergent results in the formation of mixed micelles containing lipid from the biomembrane. It is evident from these results that, among detergents, sodium deoxycholate (DOC) at concentrations ranged from 0.025 to 0.1 % is a general and very efficient agent for releasing three periplasmic proteins. Although increasing the concentration of this anionic detergent resulted in a gradual rise in total protein release from the bacterial cells and consequently lowered the yields factor, the total protein released even by the highest concentration of DOC (i.e., 0.1 %) was much lower than that released by the osmotic shock procedure, and as a result yield factor values obtained from cell permeabilization by ≤0.1 % DOC were reasonably higher than that acquired by osmotic shock. The most effective concentrations of DOC for releasing beta-lactamase, alpha-amylase, and Fab D1.3 were 0.05, 0.1, and 0.025 %, with yield factor values of 2.7, 2.3, and 3.6, respectively. Hence, DOC at these concentrations is a very effective chemical, enabling to release the periplasmic proteins more efficient than osmotic shock procedure. As it has been earlier reported, even at concentration of 0.1 % DOC, 90–100 % of the proteins’ biological activity is retained. In terms of physical properties, the formation of micelles is dependent on the critical micelle concentration (CMC) of a detergent. DOC has lower aggregation number (the average number of monomers in a micelle) and lower CMC (which is the monomer concentration at which micelles begin to form) compared to other probed detergents [41], and mechanism by which DOC permeabilizes the OM and releases the periplasmic proteins in a higher rate than the other experimented detergents is probably attributed to these low values, which result in smaller and more stable DOC micelles. Also, If the target protein molecules exist in DOC micelles, they would be faced with low concentration of the detergent monomers, compared to the micelles originated from other detergents; consequently, the deleterious effect on protein biological activity and structure would be minimize in DOC micelles.
Chelating agents can permeabilize the OM by chelating Mg2+ and Ca2+ crossbridging adjacent LPS molecules and as a result releasing a proportion of LPS from the bacterial cells [33]. In this work, 1 mM EDTA and 10 mM DEA released 44 and 27 % beta-lactamase and alpha-amylase from the periplasm, correspondingly. EDTA at concentrations of 1 mM and 10 mM discharged the Fab from the periplasm more efficient than osmotic shock (108 and 138 %, respectively). NTA (10 mM) and SHMP (0.1 %), releasing 81 and 95 % of the periplasmic Fab D1.3, were less effectual than EDTA. These chelators caused much less total protein release from the cells expressing Fab D1.3 when compared to the osmotic shock treatment, and consequently led to much higher yield factor (Figs. 4, 5, 6).
Chaotropes, such as urea and guanidinium chloride, disorganize the structure of water by interfering with hydrogen bonds, rendering it less hydrophilic, and weaken solute–solute interactions. Therefore, hydrophobic compounds such as membrane proteins can be solubilized in reasonably concentrated chaotropic solutions. Chaotropes should usually be employed at high concentration while they are exploited singly for extraction of cytoplasmic proteins [9, 10]. Nonetheless, it has been evinced that the OM in the presence of low concentrations of chaotropes (e.g., 0.2 mM GndCl) can be disrupted leading to selective protein recovery with high purity from the periplasm [30]. In recent work, urea at concentrations up to 200 mM released the periplasmic recombinant proteins with a product yield less than that of osmotic shock, (31.5 % beta-lactamase, 28 % alpha-amylase, and Fab D1.3) although less total protein released by urea caused higher specific activity and yield factor when compared to the osmotic shock treatment. By and large, the efficiency of 200 mM GndCl for permeabilizing the cells producing beta-lactamase was the same as urea; however, this concentration of GndCl had no or negligible permeabilization effect on the strains producing alpha-amylase and Fab D1.3.
The last class of chemicals used for cell permeabilization in this work included solvents. Solvents act by dissolving hydrophobic components, such as the inner and OM of Gram-negative bacteria [28]. Geckil and colleagues [13] investigated membrane permeabilization for the release of periplasmic l-asparaginase from Enterobacter aerogenes and Pseudomonas aeruginosa using various solvents (hexane, toluene, xylene, benzene, and diethyl ether) made in potassium phosphate buffer, and showed that 1 % hexane was the most efficient solvent for permeabilization in both strains, releasing the periplasmic enzyme with much higher specific activity than that achieved by mechanical disruption (sonication). In the present work, solvents (hexane, xylene, toluene, benzene, pyridine, ethanol, and isoamyl alcohol) were utilized for cell permeabilization. In terms of periplasmic protein release, in general solvents at 1 % concentration were not as effective as osmotic shock treatment, as shown in Figs. 4, 5, and 6. Ten percent (v/v) isoamyl alcohol was exceptionally effective for periplasmic release of alpha-amylase (75 %) and Fab D1.3 (168 %), and yielded much yield factor due to nominal amount of total protein release, when compared to the osmotic shock treatment.
Conclusions
The chemicals, which affected the proteins in order that ≥80 % of their biological activity sustained were categorized as safe, were prepared in 200 mM Tris buffer pH 7.5 and utilized in cell permeabilization experiments; their efficiency for periplasmic release of the target proteins was compared to that of osmotic shock procedure. It was demonstrated that selective extraction of periplasmic proteins, with releasing only small amount of total cell protein, from E. coli could be achieved. DOC at the concentrations ranged from 0.025 to 0.1 % was a very efficient permeabilizer, and released three periplasmic proteins as efficient as and more selective than osmotic shock. Isoamyl alcohol (10 %) was able to effectively release alpha-amylase (75 %) and Fab D1.3 (168 %). Chelating agents, such as 1 mM and 10 mM EDTA, 10 mM NTA, and 0.1 % and 1 % SHMP discharged Fab D1.3 from the periplasm very efficiently. All of these chemicals resulted in higher yield factor due to less total protein release from the cells, compared to the osmotic shock procedure. Obviously, this is very important as the higher yield factor, at the stage of protein extraction from the periplasm, improves the yield of target protein purification in downstream processing.
The potential of the periplasmic expression system would be completed as a result of an efficient selective release of the expressed proteins, at large scale. The present study demonstrates that the above-mentioned chemicals, particularly DOC at concentrations as low as 0.025 to 0.1 %, can be considered as suitable alternatives to the osmotic shock treatment for the release of target proteins from the periplasm. These chemicals can be superior selective agents, particularly at a large scale, where the osmotic shock can not be used because of being expensive and unfeasible.
Acknowledgments
The author would like to thank Owen Thomas and Eirini Theodosiou (The University of Birmingham, UK) for their valuable assistance. The contribution of Timothy Dafforn (The University of Birmingham, UK) in CD spectropolarimetry is gratefully acknowledged. Financial support received from the Ministry of Health and Medical Education of Iran for this study is highly appreciated.

{kind=link}
{kind=link}
{kind=link}