






Abstract
Toxoplasma gondii infection of Alzheimer's disease model mice decreases amyloid β plaques. We aimed to determine if there is a brain regional difference in amyloid β reduction in the brains of T. gondii-infected compared to control mice.
Three-month-old 5xFAD (AD model) mice were injected with T. gondii or with phosphate-buffered saline as a control. Intact brains were harvested at 6 weeks postinfection, optically cleared using iDISCO+, and brain-wide amyloid burden was visualized using volumetric light-sheet imaging. Amyloid signal was quantified across each brain and computationally mapped to the Allen Institute Brain Reference Atlas to determine amyloid density in each region.
A brain-wide analysis of amyloid in control and T. gondii-infected 5xFAD mice revealed that T. gondii infection decreased amyloid burden in the brain globally as well as in the cortex and hippocampus, and many daughter regions. Daughter regions that showed reduced amyloid burden included the prelimbic cortex, visual cortex, and retrosplenial cortex. The olfactory tubercle, a region known to have increased monocytes following T. gondii infection, also showed reduced amyloid after infection.
T. gondii infection of AD mice reduces amyloid burden in a brain region-specific manner that overlaps with known regions of T. gondii infection and peripheral immune cell infiltration.
Alzheimer's disease (AD) is a devastating neurodegenerative disease characterized by the presence of amyloid β plaques, tau tangles, and glial cell activation in the brain [1]. For many years, the prevailing hypothesis describing the cause of AD has been the amyloid hypothesis—the notion that amyloid plaque accumulation causes neuronal damage, resulting in neuroinflammation and cognitive deficits [2]. However, mounting evidence pointing toward a more complex narrative, including anti-amyloid drug trial failures [3] and the existence of cognitively resilient patients with significant amyloid levels [4], have prompted a revaluation of this hypothesis. To more fully understand the complexities of AD pathology, a new idea has emerged: infectious agents may influence neuroinflammation and AD pathology. This idea has arisen from the research of Ruth Itzhaki and others, and has been called the infectious etiology of AD [5–11]. However, in considering this hypothesis, a variety of other factors should be taken into account, including the infectious agent, the timing and localization of infection, and the confluence of factors that may influence the interactions between microbial infection and AD progression.
Recent studies have shown a link between infection with the obligate intracellular protozoan parasite Toxoplasma gondii and protection from AD-associated pathology in mouse models. T. gondii is estimated to infect 13.3% of the US human population and up to one-third of the world's human population [12, 13]. It is most commonly transmitted to humans by ingestion of contaminated food, after which the parasites disseminate throughout the body to establish life-long infection in the brain [14]. Interestingly, several studies have shown that infection of AD mouse models with T. gondii results in reduced amyloid plaque number, plaque area, or plaque volume, and soluble and insoluble amyloid aggregates in the cortex, hippocampus, or whole brain [15–18]. This effect is durable, lasting up to 10 months after infection [15, 17]. Furthermore, T. gondii infection can alleviate learning and memory deficits in AD mice [18]. In contrast, it was reported that T. gondii infection in wild-type C57BL/6 mice may induce low-level amyloid production, as measured by an antibody labeling amyloid β amino acids 1–16 (6E10), and Balb/c mice that are infected with T. gondii and injected with amyloid exhibit increased learning and memory deficits [19, 20]. As a result, the effects of infection on amyloid in AD mice compared to wild-type C57BL/6 or Balb/c strains of mice may differ.
In investigating the mechanisms underlying plaque reduction in AD mice infected with T. gondii, it has been proposed that monocytes and microglia may phagocytose and degrade amyloid: Ly6C+ monocytes that are recruited to the brains of T. gondii-infected AD mice exhibit high amyloid phagocytosis ex vivo, and the depletion of these cells in infected mice results in increased amyloid β in the brain [16, 17]. In a separate study, it was found that T. gondii-infected AD mice have an increase in homeostatic microglia and plaque-associated microglia, which may also contribute to amyloid clearance [15]. Notably, the reduction in amyloid β due to infection is specific to the type II strain of T. gondii [17], a strain that accounts for significant infection in humans [21, 22]. An increase in anti-inflammatory cytokines interleukin 10 (IL-10) and transforming growth factor-β (TGF-β), and reduced neuronal degeneration was observed in the brains of T. gondii-infected compared to control AD mice, suggesting that infection may also promote an anti-inflammatory neuroimmune environment [18].
These prior studies have demonstrated a significant reduction in amyloid β in the brains of T. gondii-infected AD mice compared to uninfected control AD mice, but how T. gondii infection influences amyloid β plaque burden in different regions of the brain remains poorly understood. Herein, we use the 5xFAD AD amyloidosis mouse model, coupled with intact brain imaging and amyloid quantification to comprehensively investigate how T. gondii infection prior to significant amyloidosis impacts amyloid deposition throughout the brain. The 5xFAD mice have 5 mutations found in familial AD (PSEN1 and APP mutations) and develop plaques at approximately 3 months of age in the subiculum and midbrain before spreading through upper layers of the cortex [23]. By employing whole-hemispheric optical clearing, volumetric light-sheet imaging, and a bioinformatics imaging analysis platform, we quantified amyloid plaques in intact brain hemispheres of infected and uninfected mice and mapped plaque densities to the Allen Institute Brain Reference Atlas to generate a brain-wide analysis of amyloid burden in control and infected mice. Our findings reveal that during T. gondii infection, amyloid burden is reduced across the entire brain, and most notably in specific regions of the cortex, including the prelimbic cortex, retrosplenial cortex, and visual cortex. A greater understanding of how T. gondii influences amyloid deposition throughout the brain may enhance our understanding of mechanisms of amyloid clearance, and how plaque degradation may be elicited through targeted therapeutics.
METHODS
Mice and Infection Experiments
All mouse husbandry and experimentation was approved by University of California, Irvine Institutional Animal Care and Use Committee. 5xFAD hemizygous (B6.Cg-Tg(APPSwFlLon, PSEN1*M146L*L286V)6799Vas/Mmjax) mice were purchased from the National Institutes of Health Mutant Mouse Resource and Research Centers, or Jackson Laboratories, or bred in house. All mice were infected at 3 months of age (12 weeks) with 200 green fluorescent protein-expressing Prugniaud (PA7) tachyzoites in 200 µL of phosphate-buffered saline (PBS) injected intraperitoneally. Control mice were similarly injected with 200 µL of PBS. Female mice were used in all experiments, as females have been reported to have a more aggressive time line for the development of AD pathology [24]. Parasites were serially passaged in human foreskin fibroblasts, as previously described [25].
Optical Clearing, Whole Hemispheric Mounting, and Light-Sheet Imaging
5xFAD mice were injected with T. gondii or PBS at 3 months of age and euthanized 6 weeks later at 4.5 months of age. At harvest, mice were transcardially perfused with 50 mL of ice cold PBS at 6 mL/min to remove cells in the vasculature, followed by 4% paraformaldehyde (PFA) in the same volume and at the same rate. Brains were removed, and 1 hemisphere was placed in 4% PFA overnight. Brains were then washed 3 times in 1 × PBS for 30 minutes each. Optical clearing was completed according to the iDISCO+ protocol [26]. Amyloid was labeled using anti-Aβ1-16 antibody 6E10 (BioLegend). The hemisphere was mounted in dibenzyl ether (Sigma) and imaged with a Z.1 light-sheet fluorescence microscope (LSFM; Zeiss) using the EC Plan-Neofluar 5×/0.164908000162 detection objective (Zeiss) and LSFM5×/.1 illumination objectives (Zeiss). Excitation was done with a 561-nm laser and captured with a 575–615-nm bandpass emission filter. Tiling of z-stacks was completed in Stitchy (Translucence Biosystems).
Light-Sheet Image Amyloid Segmentation and Analysis
Amyloid plaques were segmented using BrainQuant3D. Machine learning and deterministic filters were tuned to maximize plaque segmentation in 5xFAD mice while minimizing false-positive labels in wild-type mice. The segmentation workflows do not segment all amyloid plaques, which is necessary to produce the best assay window (signal to background). Amyloid was assigned to each region of the Allen Institute Brain Reference Atlas by Translucence Biosystems. Amyloid density was generated by dividing the amyloid counts for each region by the total region volume. Forty-seven regions were chosen for further analysis based on image resolution and regional hierarchical organization. Multiple 2-tailed Student t tests were conducted to compare density between conditions in each region, followed by Benjamini-Hochburg false discovery rate correction, α = .1 to generate Q values.
RESULTS
Chronic T. gondii Infection Reduces Amyloid in Cortical and Hippocampal Regions of 5xFAD Mice
To examine the effects of T. gondii infection on amyloid pathology in a brain-wide manner, we intraperitoneally infected 12-week-old 5xFAD mice with 200 type II (Prugniaud strain) T. gondii, a parasite strain that has previously been shown to reduce amyloid burden in AD mice [17], or injected an equivalent volume of PBS as a control. We combined this infection model with whole-hemispheric optical clearing, immunofluorescent labeling, and volumetric light-sheet imaging to quantify amyloid in the brains of infected and control animals. At the chronic infection timepoint of 6 weeks postinfection, the mice were euthanized, and the brains were harvested, optically cleared using the iDISCO+ technique, and imaged using an LSFM [26]. Individual plaques were segmented across the hemispheres using open-source BrainQuant3D software, and automated quantification of plaques in each region of the brain was determined (Figure 1A). For each brain, plaque density was calculated based on the numbers of plaques per brain region and then computationally mapped to the Allen Institute Mouse Brain Reference Atlas. Average amyloid density for each region and each condition was done on a voxel basis and weighted on the sum (intensity value) of the objects within a given voxel. Average amyloid density heat maps were then generated for the samples in each condition (Figure 1B). Amyloid was observed in both infected and uninfected animals throughout the brain, but the highest density was observed in regions where significant amyloid accumulation has been described in the 5xFAD model, such as the subiculum of the hippocampus (Figure 1B and Supplementary Figure 1). T. gondii-infected mice had significantly reduced amyloid density across the entire hemisphere of each brain (Figure 1C).
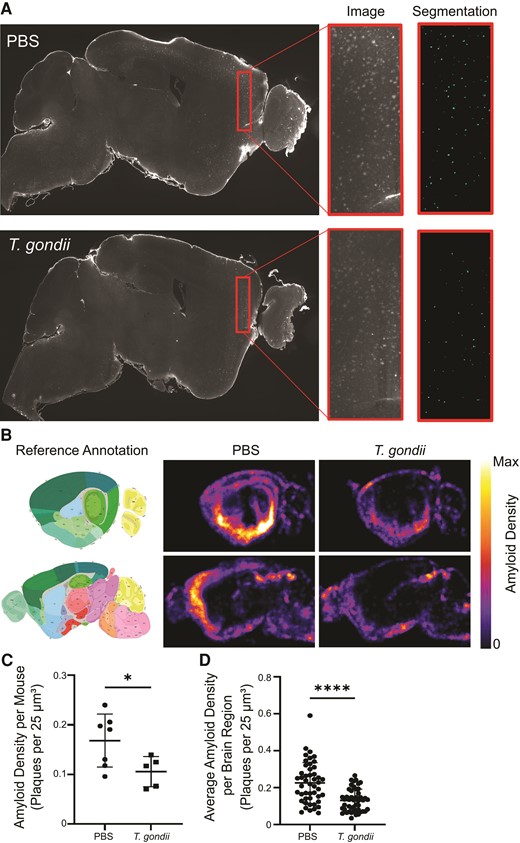
Amyloid burden is reduced throughout the brain in 5xFAD mice infected with Toxoplasma gondii. Three-month-old 5xFAD mice were injected with PBS or infected with type II T. gondii, and at 6 weeks postinfection brains were imaged using light-sheet fluorescence microscopy. A, Representative z-planes showing amyloid signal from PBS-injected or T. gondii-infected 5xFAD mice. Inset shows segmentation of amyloid signal using BrainQuant3D. B, Amyloid density was quantified in each mouse and density maps were computationally aligned to the Allen Institute Brain Reference Atlas. Mean density of amyloid at a lateral and medial sagittal view are shown. C, Amyloid density as the number of plaques per 25 μm3 voxel in the entire root brain is displayed. Each dot represents 1 animal. D, Mean amyloid density displayed for 47 brain regions. Each point represents 1 brain region. B–D, nPBS = 7, nT. gondii = 5, reflective of 2 independent experiments. C and D, Error bars represent SD and groups were compared using Student t test. * P < .05, **** P < .0001.
To analyze amyloid distribution in specific brain regions, we next examined a subset of 47 regions of the brain based on representative hierarchical order and regions of interest (cortex and hippocampus). This analysis also revealed significantly reduced amyloid density in infected animals (Figure 1D). To identify the regions most profoundly affected by infection, amyloid density for each of the 47 regions was compared between PBS- and T. gondii-injected mice. This analysis yielded 33 regions in which amyloid density was significantly different (Q < .1), with all 33 regions showing a reduction in amyloid in the infected mice (Figure 2A). Hierarchical clustering, significance (as represented by Q value), and average density values for each condition were graphed to highlight the 33 regions showing a significant reduction in amyloid density (Figure 2B). Amyloid density was significantly decreased in the isocortex overall, with 11 of 16 direct daughter regions also showing significant reductions in amyloid during infection, including in the prelimbic cortex, visual cortex, and retrosplenial cortex (Figure 2B and 2C, and Supplementary Figure 2A). There was also a decline in amyloid density in other regions, including the hippocampal formation and many daughter regions (Figure 2B and 2C, and Supplementary Figure 2B). Notably, we also detected a reduction in amyloid density in the olfactory tubercle (Figure 2B and 2C, and Supplementary Figure 2C), a brain region previously found to have a significant increase in inflammatory monocyte recruitment during T. gondii infection [27].
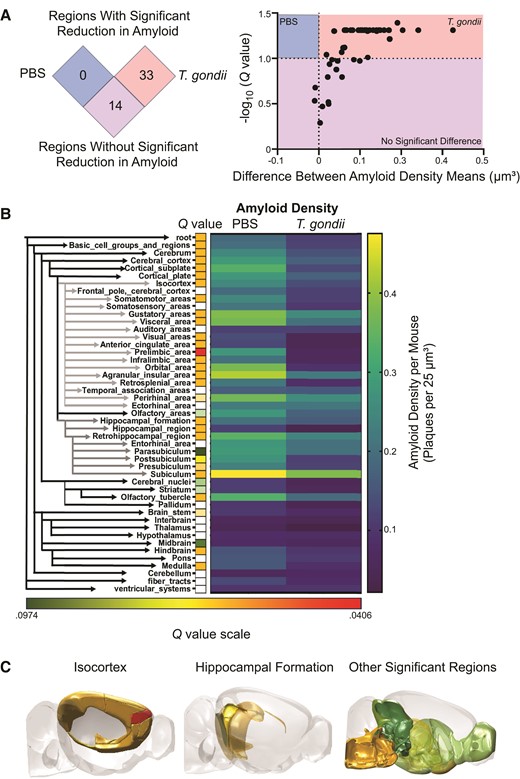
Amyloid density is reduced in the cortex after Toxoplasma gondii infection. A, The differences between the average amyloid densities of PBS-treated or T. gondii-infected mice in each of the 47 brain regions were plotted against the adjusted P value (Q value) for that region. B, Forty-seven regions were hierarchically clustered, with isocortex and hippocampal formation indicated by gray arrows. Directly to the right are squares reflecting Q values of < .1. White squares do not meet this Q value cut off threshold. Heatmaps (right) display the average amyloid density for each of the 47 regions across each condition. C, Regions significantly reduced in amyloid density (Q < .1) at 6 weeks postinfection with T. gondii. nPBS = 7, nT. gondii = 5, reflective of 2 independent experiments. Student t tests followed by Benjamini-Hochberg false discovery rate correction.
DISCUSSION
Toxoplasma gondii has long been known to induce a neuroimmune response in the brain, but it is only in the past 8 years that researchers have appreciated its potential to influence levels of amyloid β and the pathology associated with AD, by examining infection in transgenic AD mouse models [15–18]. Prior studies have largely focused on the effect of T. gondii infection on amyloid plaque burden in the hippocampus and cortex [15, 17]. Although confocal fluorescence microscopy imaging of thin brain sections from control or infected mice enables high-resolution analysis of cellular and molecular processes, the use of volumetric light-sheet imaging of intact brain tissues allowed us to comprehensively identify brain regions with significantly reduced amyloid. This approach complements and builds on the prior studies and begins to enable an analysis of how parasite localization may impact amyloid reduction across the entire brain.
T. gondii disseminates throughout the body via the circulatory system and infects the brain during acute infection. The parasites then establish a chronic infection by undergoing a stage conversion into slow-growing bradyzoites that encyst within neurons in the brain. As the immune system controls the proliferation and spread of the parasites, and long-term chronic infection develops, T. gondii cysts are most commonly found in the cortex, the hippocampus, and diencephalic regions, including the thalamus and hypothalamus [20, 28, 29], but some studies have shown enrichment of parasites in the amygdala [30]. Within the cortex, one study showed that type II cysts were most enriched in the visual areas, orbital areas, anterior cingulate areas, retrosplenial areas, as well as the somatosensory area and frontal pole [31]. Additionally, by 15 days after infection, inflammatory monocytes infiltrate the brain from the bloodstream and are highly enriched in the olfactory tubercle [27]; although as chronic infection continues, inflammation does not necessarily correlate with cyst location [31]. However, much less is known about how the localization of parasites in the brain is related to the development of Alzheimer's pathology and, in particular, the development of amyloid β plaques. As noted above, previously published studies have focused on the cortex and the hippocampus, showing reductions in amyloid in both of these brain regions after infection [15, 17]. In accordance with these data, we found that amyloid is reduced throughout the 5xFAD mouse cortex and subiculum after infection. The volumetric light-sheet imaging, coupled with automated quantification of amyloid signal and bioinformatic mapping to the Allen Institute Brain Reference Atlas, has allowed us to interrogate this question on a deeper level, and to identify daughter regions within the regional hierarchical organization. For example, the prelimbic cortical region was identified as a brain region with one of the largest and most significant reductions in amyloid after T. gondii infection. The prelimbic cortex is part of the medial prefrontal cortex and is involved in memory consolidation [32].
Although the 5xFAD model is a valuable tool to study amyloidosis in AD, it is important to note that plaque development in this model occurs in a region-specific manner due to the Thy1 promoter controlling the familial AD transgenes. These animals develop plaques in the subiculum and the deeper levels of the cortex in early life (as early as 2 months), then plaques spread to the rest of the hippocampus, cortex, thalamus, brainstem, and olfactory bulb [23]. This pattern of plaque spreading from the subiculum and midbrain to the rest of the brain has also been detected via light-sheet imaging modalities, such as vDISCO, in the 5xFAD mouse model [33]. This pattern differs from humans, in which plaques form in the neocortex, then spread to the allocortex (including the hippocampus), brainstem, and finally the cerebellum [34, 35]. However, by knowing the established localization and severity of pathology in the 5xFAD mouse, we can draw comparisons with sites of T. gondii infection, the localization of immune cell recruitment and infiltration, and amyloid burden. Importantly, our data indicate that in regions where amyloid is known to be enriched in the 5xFAD model, there is a reduction in amyloid during infection, and this reduction is greatest in regions where T. gondii has been observed in prior studies. These findings suggest that the parasite may influence changes in amyloid burden depending on the site of infection in the brain. Moreover, a recent review of case reports on T. gondii infection in immunocompetent humans revealed that T. gondii-induced lesions often occur in supratentorial regions of the brain, including the cortex [36]. This raises intriguing parallels, suggesting that T. gondii infection, encountered later in life (>60 years) in patients in the United States [37], may coincide with regions where plaques may have already begun to develop or will soon develop, and could mirror the development (or degradation) of amyloid observed in infected mouse models. Although some epidemiological studies and meta-analyses examining correlations between T. gondii infection and cognitive function in humans have claimed a positive association of infection with the development of AD [38–42], others have shown no association at all [43–47]. In all studies, AD or dementia was diagnosed according to clinical symptoms [48, 49] and was not based on postmortem analysis of AD pathology in brain tissue. Notably, there are no studies showing a protective effect of T. gondii infection against dementia in humans. It should also be noted that, thus far, no study has investigated AD pathology (plaques or tangles) during life or at end of life in humans seropositive for T. gondii infection. Another component of the host-microbe interaction that has not been examined is the potential effect of parasite genetics on the development of AD or AD pathology in humans. Different strains of T. gondii are associated with varying clinical outcomes in humans. The North American and European type II strain is most highly associated with infection in these regions. Type I and more genetically diverse strains found in Canada, the United States, and Brazil, are associated with ocular toxoplasmosis [22, 50, 51]. The extent to which parasite strain may influence the development or delay of neurodegenerative disease in humans remains unknown.
In conclusion, we have shown that chronic T. gondii infection of 5xFAD mice elicits a decrease in amyloid throughout the brain, and that this reduction is most significant in the cortex. Our work adds to the growing body of research demonstrating that chronic T. gondii infection of amyloidosis AD mouse models leads to a reduction in amyloid β in the cortex; however, it is the first to use volumetric brain imaging and a computational platform to quantify amyloid in all regions of the brains of mice with and without T. gondii infection. Future studies may help to determine the brain region-specific mechanisms leading to this marked reduction in amyloid during T. gondii infection.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the members of the Lodoen, Andrade, and Morrissette laboratories for their helpful suggestions on the project, as well as conceptual framing and figure design. We also thank Dr Andrea Tenner and her laboratory for their ongoing input on the project and Dr Adeela Syed for her guidance in light-sheet imaging. This study was made possible in part through access to the Optical Biology Core Facility of the Developmental Biology Center, a shared resource supported by the Cancer Center Support Grant (CA-62203) and Center for Complex Biological Systems Support Grant (GM-076516) at the University of California, Irvine.
Author contributions. K. J. O. Y. and M. B. L. designed the experiments. K. J. O. Y. conducted the infection experiments. K. J. O. Y. and N. A. G. performed the imaging techniques. K. J. O. Y., N. A. G., R. A., D. G. W., S. P. G., and M. B. L. generated and analyzed data. K. J. O. Y., N. A. G., D. G. W., and M. B. L. wrote the manuscript.
Financial support. This work was supported by the National Institutes of Health (grant numbers 3R01AI120846-05S1 to M. B. L. and 5T32AG000096-38 to K. J. O. Y); Infectious Diseases Society of America (grant number 05677169 to M. B. L.); and the University of California, Irvine, Institute for Memory Impairments and Neurological Disorders, Research and Mentoring Program (to K. J. O. Y.).
Supplement sponsorship. This article appears as part of the supplement “Advances in Identifying Microbial Pathogenesis in Alzheimer's Disease,” sponsored by the Infectious Diseases Society of America.
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
Author notes
Presented in part: Alzheimer's Association International Conference, July 16–20, 2023, Amsterdam, the Netherlands, https://doi.org/10.1002/alz.080427.


{kind=link}
{kind=link}