






Abstract
Multisystem inflammatory syndrome in children (MIS-C) is a rare, potentially fatal complication of SARS-CoV-2 infection. Genetic defects in inflammation-related pathways have been linked to MIS-C, but additional research is needed, especially in diverse ethnic groups. The present study aimed to identify genetic variants underlying MIS-C in Brazilian patients. Whole exome sequencing was performed, focusing on genes involved in the host immune response to SARS-CoV-2. Functional assays assessed the impact of selected variants on nuclear factor–κB signaling. Nine rare, potentially deleterious variants were found in 8 of 21 patients, located in the IL17RC, IFNA10, or NLRP12 gene. Unlike the wild type NLRP12 protein, which inhibits nuclear factor–κB activation in HEK 293T cells, the mutant NLRP12 proteins have significantly reduced inhibitory properties. In conclusion, our results indicate that rare autosomal variants in immune-related genes may underlie MIS-C, highlighting the potential role of NLRP12 in its predisposition. These findings provide new insights for the appropriate management of MIS-C.
Multisystem inflammatory syndrome in children (MIS-C) is a life-threatening condition that occurs after primary SARS-CoV-2 infection [1, 2]. It was first identified in April 2020, during the COVID-19 pandemic, when the health systems of several countries observed an increase in the number of infants and adolescents presenting with persistent fever and evidence of systemic hyperinflammation [3]. Initial descriptions revealed that MIS-C affects approximately 3 persons per 10 000 SARS-CoV-2 infections, with a significantly higher proportion observed among non-White individuals [4].
MIS-C exhibits significant clinical heterogeneity, with features overlapping Kawasaki disease and toxic shock syndrome [5, 6]. Furthermore, MIS-C encompasses features of other immune hyperactivation syndromes, such as macrophage activation syndrome or secondary hemophagocytic lymphohistiocytosis [7, 8]. Patients with MIS-C may develop gastrointestinal symptoms, rash, mucocutaneous lesions, cardiac complications, acute kidney injury, and, in severe cases, vascular problems and shock [9, 10]. MIS-C immunopathogenesis involves intense production of inflammatory factors, lymphocytic and myeloid chemotaxis and activation, and autoantibodies production [11–13]. This condition is also characterized by superantigen-like polyclonal expansion of CD4+ and CD8+ T cells expressing the T-cell receptor Vβ21.3+ [13].
Genetic studies are essential to expand knowledge on the molecular and cellular mechanisms underlying severe conditions such as MIS-C. In this regard, variants potentially involved in MIS-C were identified in the SOCS1 gene [14], a negative regulator of type I and II interferons, and in XIAP and CYBB, which may result in dysregulation of the inflammatory response [15]. In addition, an autosomal recessive IFNAR1 deficiency was identified in a child presenting critical COVID-19 pneumonia and MIS-C [16]. Studies with Middle Eastern [17] and Latin American [18, 19] individuals identified variants in several immune-related genes potentially implicated in MIS-C, although these studies have not validated their findings through functional assays. Finally, it was observed that rare recessive OAS1, OAS2, or RNASEL deficiencies promote excessive amounts of inflammatory cytokines in response to SARS-CoV-2, possibly resulting in MIS-C [20].
These genetic studies support the hypothesis that variants in different loci may underlie MIS-C. Nevertheless, the genetic factors identified so far explain only isolated cases of MIS-C or very small proportions of the analyzed cases. In this context, it is highly likely that certain MIS-C–predisposing loci remain unknown. To expand our understanding of the pathophysiologic mechanisms of this life-threatening condition, we recruited patients with MIS-C from Brazil and searched for rare coding variants in pathways related to SARS-CoV-2 infection.
METHODS
Study Design, Sample Selection, and Ethical Considerations
This is a retrospective cross-sectional study of 21 unrelated hospitalized children diagnosed with multisystem inflammatory syndrome (MIS-C). This cohort is referred here as BR-MIS-C. We adopted the MIS-C case definition outlined by the US Centers for Disease Control and Prevention [10]. None of the patients had previously been vaccinated against SARS-CoV-2. All were healthy before the MIS-C episode, without any medical conditions.
This study was approved by the research ethics committee of the Instituto Aggeu Magalhães, Fundação Oswaldo Cruz, Brazil (resolution 4263871). The parents or legal guardians of the children/adolescents agreed with the study and signed an informed consent form.
Demographic, Clinical, and Laboratory Data
Data were extracted from electronic medical records of public hospitals in the northeast region of Brazil from May 2020 to March 2021: Hospital Martagão Gesteira (Salvador), Instituto Couto Maia (Salvador), and Hospital Universitário Oswaldo Cruz (Recife). At hospital admission, patients underwent serum testing (enzyme-linked immunosorbent assay) and molecular testing (reverse transcriptase–polymerase chain reaction) for SARS-CoV-2, and the main clinical symptoms associated with SARS-CoV-2 infection were collected. During hospitalization, data on complications and laboratory findings were also collected. Parents/guardians declared the ethnoracial group of the patients according to the categories established by the Brazilian Institute of Geography and Statistics [21].
Genomic DNA
DNA was extracted from oral mucosal cells with the ReliaPrep Blood gDNA Miniprep System kit (Promega Corp). Samples were processed on the same day of collection and stored at −20 °C until use. DNA concentrations were evaluated with the Qubit DNA Assay kit and a Qubit 2.0 fluorometer (Life Technologies).
Whole Exome Sequencing and Quality Control
One microgram of genomic DNA was used for whole exome sequencing library preparation via the Illumina DNA Prep with Exome version 2 Enrichment kit (Illumina). The libraries were subjected to next-generation sequencing (2 × 150 base pairs) on the NovaSeq 6000 platform with the NovaSeq SP version 1 Reagent kit (Illumina). Data for each sample were analyzed by the Genome Analysis ToolKit (GATK Best Practices). Off-target regions were excluded. The remaining sequences were aligned to the reference human genome version GRCh38, and the mapping files (BAM) were obtained through SAMTools version 1.15. Only target regions with ≥10 reads were retained in the study. Raw data from sequencing experiments are available in the Database of Genotypes and Phenotypes (National Center for Biotechnology Information) under controlled access mode (access phs003512.v1.p1).
Variant Detection and Filtering Strategy
Single-nucleotide variants (SNVs) were detected through GATK Best Practices (germline short variants) with the GATK HaplotypeCaller program. Functional annotations were carried out with ANNOVAR (version 2022Dec) [22]. The present study was focused on human protein-coding genes related to SARS-CoV-2 infection (Reactome R-HSA-9694516). Data from the 1000 Genomes Project (phase 3) and gnomAD (version 2.1.1) were used to determine allele frequencies in human reference populations. The combined annotation–dependent depletion score was used to prioritize potentially deleterious variants in coding regions. A standard cutoff score of 15 for all human genes was implemented here, as proposed by Kircher et al [23]. The gene damage index was also considered to prioritize variants in genes sensitive to purifying selection. The standard cutoff of 13.84 was assumed [24], based on the distribution of all disease-causing human genes.
Ancestry Analyses
To conduct principal component analysis, the full data set of unrelated individuals of the 1000 Genomes Project (phase 3) was used as a reference. This 1000 Genomes panel was merged with the genetic data of BR-MIS-C by using autosomal variants with a minor allele frequency (MAF) >0.1 that were common to both data sets. Then, the merged data were pruned with PLINK 1.9 software [25] with a window size of 50 markers, a step size of 5, and a variance inflation factor threshold of 1.5, leaving 16 005 markers to calculate principal components with PLINK.
The ancestry of each MIS-C case was estimated with ADMIXTURE software [26]. This analysis was conducted under an unsupervised mode with the EUR, AFR, and AMR samples of the 1000 Genomes Project and the same prune approach described previously. K = 3 was chosen, assuming that the main ancestral groups of the Brazilian population are Europeans, Africans, and Native Americans [27].
Locus ancestry was inferred with RFmix version 1.5.4 software [28] by using autosomal variants shared by the BR-MIS-C samples and reference populations with low admixture rates: Iberian in Spain, Toscani in Italy, Yoruba in Nigeria, Luhya in Kenya, and Peruvians (from 1000 Genomes Project); Surui and Karitiana in Brazil and Maya and Pima in Mexico (from the Human Genome Diversity Project). The analysis was carried out via the software's default parameters, including a window length of 0.2 cm. Only the windows with a posterior probability of ancestry >0.90, inferred by the forward-backward algorithm, were retained in the study. The results shown here are focused on IL17RC (3:9 917 074–9 933 630), IFNA10 (9:21 206 181–21 207 143), and NLRP12 (19:53 793 584–53 824 403) (version GRCh38).
Brazilian Reference Populations
The frequencies of candidate MIS-C–predisposing variations in the Brazilian population were investigated in 2 cohorts: SABE [29], which includes 1171 unrelated individuals from the city of São Paulo and 61 174 462 variants, and EPIGEN [27, 30], which genotyped/imputed 30 055 724 variants in 6487 admixed persons from the northeast, southeast, and south of Brazil.
Evolutionary Conservation of Amino Acids
Amino acid sequences of IL-17RC (ENST00000295981.7), IFNA10 (ENST00000357374.2), and NLRP12 (ENST00000324134.11) were obtained from the Ensembl genome browser 107 (ensembl.org/). The structure and functional domains of IL-17RC (Q8NAC3), IFNA10 (P01566), and NLRP12 (P59046) were obtained from the UniProt database (uniprot.org/). Evolutionary conservation of amino acid positions was predicted with the ConSurf server (consurf.tau.ac.il/) [31].
NLRP12 In Vitro Functional Analysis
HEK 293T–NF-κB reporter cells for luciferase were plated at a density of 2.5 × 104 and transfected with lipofectamine with 100 ng of expression plasmids (Vector Builder) for wild type NLRP12 or its variants (R284X, D142N+P319R, R1016X, V901L) in addition to the empty vector. After 48 hours, the cells were stimulated with interleukin 1β (IL-1β; 1 ng/mL) or tumor necrosis factor α (10 ng/mL). Twelve hours later, luciferase activity was assessed by the Luciferase Assay Kit (Promega), following the manufacturer's instructions. Luciferase activity was normalized by total protein concentration (bicinchoninic acid assay; Merck Millipore Sigma). The results of nuclear factor–κB (NF-κB) activation indicated by the reaction of luciferase with its substrate were expressed as mean ± SD from 3 independent tests (each experimental condition in triplicate).
To Western blotting analysis, cells were collected in RIPA Buffer (Merck Millipore Sigma) with a protease/phosphatase inhibitor cocktail (Cell Signaling Technology). Protein content was quantified by bicinchoninic acid assay. Extracts were separated by 4%–20% precast gels (Bio-Rad) by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with 5% bovine serum albumin solution for 2 hours at room temperature. Then, membranes were incubated with monoclonal antibody anti-NLRP12 (1:1000; Tebu-Bio) overnight at 4 °C. After washes, the membranes were incubated with horseradish peroxidase–conjugated anti-mouse antibody (1:3000; Cell Signaling Technology) for 2 hours, and fluorescence was detected. Furthermore, β-actin expression was used as loading control, and the membranes were incubated with anti-β-actin antibody (1:1000; Cell Signaling Technology) for 2 hours at room temperature. Fluorescence was detected by an ECL system (GE Healthcare) and Chemidoc imaging system (Bio-Rad).
Statistical Analysis
Associations were evaluated by a median test for continuous variables or Fisher exact test for categorical variables. The differences in NF-κB activation (luciferase expression) among the experimental conditions were evaluated through the Kruskal-Wallis test, followed by Dunn posttest. P < .05 was considered statistically significant.
RESULTS
Characteristics of Patients With MIS-C
The median age of BR-MIS-C was 7 years (IQR, 4–11) and 62% was male (Supplementary Figure 1A). Most patients (71%) were declared ethnically as pardos (mixed; Supplementary Figure 1B). SARS-CoV-2 recent or current infection was confirmed for all (Supplementary Figure 1C). The symptoms found in more than half of the MIS-C cases were fever, abdominal pain, rash, dyspnea, and vomiting (Supplementary Figure 1D). All were discharged from the hospital, with a median length of stay of 10 days (IQR, 8–13; Supplementary Figure 1E). Most patients (86%) were admitted to the pediatric intensive care unit. Sepsis occurred in 48% of the cases, while shock affected 38% (Supplementary Figure 1F). As shown in Supplementary Figure 1G, most patients underwent treatment with intravenous immunoglobulin (86%) and corticosteroids (76%).
The laboratory findings during hospitalization are shown in Supplementary Figure 2. Remarkably, most MIS-C cases developed leukocytosis and neutrophilia. D-dimer levels were substantially increased in the patients' plasma, such as ferritin and C-reactive protein.
Sequencing Data and BR-MIS-C Ancestry
The median read count for the exomes of the 21 patients with MIS-C was 75 million, with a median read depth of 126× (Supplementary Figure 3). A summary of statistics of the detected SNVs is described in Supplementary Table 1.
Principal component analysis (Figure 1A) and admixture analysis (Figure 1B) revealed the admixed patterns of the patients. The average global ancestries for BR-MIS-C are 0.49 (IQR, 0.37–0.56) European, 0.38 (IQR, 0.30–0.54) African, and 0.12 (IQR, 0.08–0.14) Native American. There is significant variation in ancestries, ranging from 0.77 European (patient 7) to 0.78 African (patient 1).
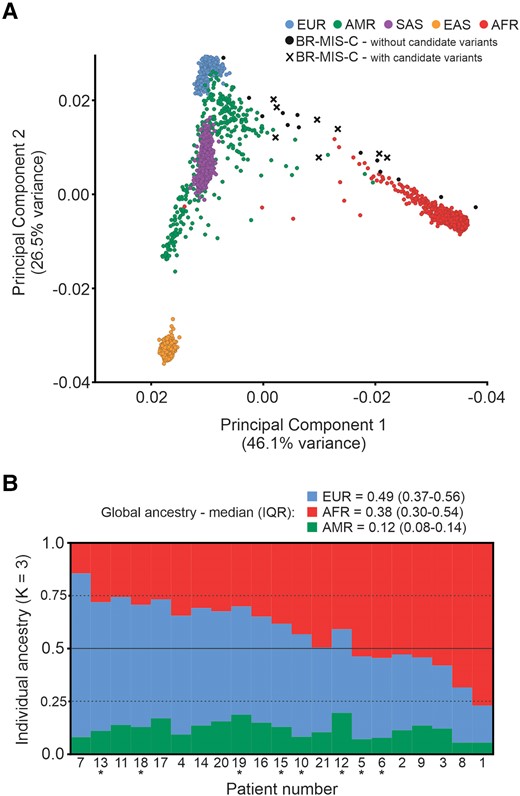
Ancestry analyses of the MIS-C cases. A, Principal component analysis of 21 patients (with or without variants potentially implicated in MIS-C) and samples from the 1000 Genomes Project. B, Individual ancestry bar plot of BR-MIS-C via unsupervised ADMIXTURE analysis. Patients with candidate MIS-C–predisposing variations variants are marked with an asterisk. AFR, Africans; AMR, Native Americans; BR-MIS-C, current cohort with MIS-C; EAS, East Asians; EUR, Europeans; MIS-C, multisystem inflammatory syndrome in children; SAS, South Asians.
Variant Prioritization and Association Analysis
As shown in Figure 2, the first step in the identification of candidate MIS-C–predisposing variations was the selection of variants in exons, resulting in 65 286 observations in at least 1 individual. Only variants located in 316 human protein-coding genes related to SARS-CoV-2 infection were selected (Reactome R-HSA-9694516, n = 574 variants; Supplementary Table 2). Given the assumption that these genetic factors may be rare, only variants with MAF ≤1% in the global databases of the 1000 Genomes Project and gnomAD were selected (n = 86 variants). Then, a filter was applied to remove synonymous variants (n = 82 variants). Additional filtering was performed according to a predicted combined annotation–dependent depletion score ≥15, which is consistent with deleteriousness. This approach revealed 9 variants in 3 genes. Notably, these genes have a predicted gene damage index <13.84, indicating that their variants may lead to altered phenotypes.
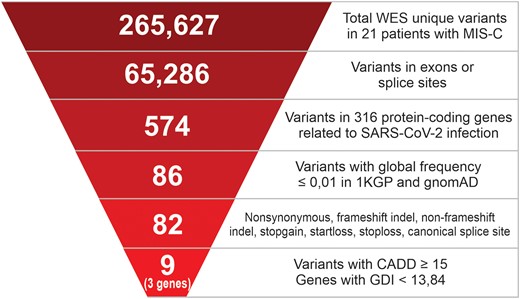
Strategy for prioritizing functional variants in MIS-C cases. The numbers indicated at each step are the remaining unique small-nucleotide variants found after filter application. The list of 316 human protein-coding genes related to SARS-CoV-2 infection was obtained from the Reactome database (R-HSA-9694516). 1KGP, 1000 Genomes Project, phase 3; CADD, combined annotation–dependent depletion algorithm; GDI, gene damage index; gnomAD, Genome Aggregation Database version 2.1.1; MIS-C, multisystem inflammatory syndrome in children; WES, whole exome sequencing.
This filtering strategy revealed the presence of rare, potentially pathogenic variants that may be implicated in MIS-C in 8 patients (∼38%; Table 1). All variants are in heterozygous state. Five of these SNVs—rs34330210 (D142N), rs104895564 (R284X), rs35401786 (P319R), rs104895568 (V901L), and rs35064500 (R1016X)—are located in the NLRP12 gene (NLR family pyrin domain containing 12). The variant rs10485564 (R284X), resulting in a premature stop codon of the NLRP12 transcript with an MAF ≤0.002 in the reference populations, was found in 2 unrelated MIS-C cases.
Candidate MIS-C–Predisposing Variants
| MAF a | |||||||
|---|---|---|---|---|---|---|---|
| Gene: Patient | SNV-IDconsequence | Genotype | Protein Change | GDI | CADD Score | 1KGP | gnomAD |
| IL17RC | 6.6 | ||||||
| P6 | rs115505677missense | G/C | Q329H | 20 | 0.005 | 0.001 | |
| P18 | rs148575246splice site | G/C | … | 33 | 0.004 | 0.006 | |
| P19 | rs115461448missense | G/A | A395T | 23 | 0.009 | 0.01 | |
| IFNA10 | 5.8 | ||||||
| P15 | rs145785282stopgain | G/A | Q125X | 36 | 0.01 | 0.002 | |
| NLRP12 | 11.1 | ||||||
| P5 | rs104895568missense | G/C | V901L | 15 | 0.002 | 0.0006 | |
| P6 | rs35401786missense | C/G | P319R | 23 | 0.009 | 0.002 | |
| P6 | rs34330210missense | G/A | D142N | 24 | 0.009 | 0.002 | |
| P10, P12 | rs104895564stopgain | C/T | R284X | 34 | 0.002 | 0.0002 | |
| P13 | rs35064500stopgain | C/T | R1016X | 37 | 0.002 | 0.0002 | |
| MAF a | |||||||
|---|---|---|---|---|---|---|---|
| Gene: Patient | SNV-IDconsequence | Genotype | Protein Change | GDI | CADD Score | 1KGP | gnomAD |
| IL17RC | 6.6 | ||||||
| P6 | rs115505677missense | G/C | Q329H | 20 | 0.005 | 0.001 | |
| P18 | rs148575246splice site | G/C | … | 33 | 0.004 | 0.006 | |
| P19 | rs115461448missense | G/A | A395T | 23 | 0.009 | 0.01 | |
| IFNA10 | 5.8 | ||||||
| P15 | rs145785282stopgain | G/A | Q125X | 36 | 0.01 | 0.002 | |
| NLRP12 | 11.1 | ||||||
| P5 | rs104895568missense | G/C | V901L | 15 | 0.002 | 0.0006 | |
| P6 | rs35401786missense | C/G | P319R | 23 | 0.009 | 0.002 | |
| P6 | rs34330210missense | G/A | D142N | 24 | 0.009 | 0.002 | |
| P10, P12 | rs104895564stopgain | C/T | R284X | 34 | 0.002 | 0.0002 | |
| P13 | rs35064500stopgain | C/T | R1016X | 37 | 0.002 | 0.0002 | |
Abbreviations: CADD, combined annotation–dependent depletion; GDI, gene damage index; MAF, minor allele frequency; MIS-C, multisystem inflammatory syndrome in children; SNV-ID, single-nucleotide variant identifier.
aGlobal databases from 1KGP (1000 Genomes Project phase 3) and gnomAD (Genome Aggregation Database version 2.1.1).
Candidate MIS-C–Predisposing Variants
| MAF a | |||||||
|---|---|---|---|---|---|---|---|
| Gene: Patient | SNV-IDconsequence | Genotype | Protein Change | GDI | CADD Score | 1KGP | gnomAD |
| IL17RC | 6.6 | ||||||
| P6 | rs115505677missense | G/C | Q329H | 20 | 0.005 | 0.001 | |
| P18 | rs148575246splice site | G/C | … | 33 | 0.004 | 0.006 | |
| P19 | rs115461448missense | G/A | A395T | 23 | 0.009 | 0.01 | |
| IFNA10 | 5.8 | ||||||
| P15 | rs145785282stopgain | G/A | Q125X | 36 | 0.01 | 0.002 | |
| NLRP12 | 11.1 | ||||||
| P5 | rs104895568missense | G/C | V901L | 15 | 0.002 | 0.0006 | |
| P6 | rs35401786missense | C/G | P319R | 23 | 0.009 | 0.002 | |
| P6 | rs34330210missense | G/A | D142N | 24 | 0.009 | 0.002 | |
| P10, P12 | rs104895564stopgain | C/T | R284X | 34 | 0.002 | 0.0002 | |
| P13 | rs35064500stopgain | C/T | R1016X | 37 | 0.002 | 0.0002 | |
| MAF a | |||||||
|---|---|---|---|---|---|---|---|
| Gene: Patient | SNV-IDconsequence | Genotype | Protein Change | GDI | CADD Score | 1KGP | gnomAD |
| IL17RC | 6.6 | ||||||
| P6 | rs115505677missense | G/C | Q329H | 20 | 0.005 | 0.001 | |
| P18 | rs148575246splice site | G/C | … | 33 | 0.004 | 0.006 | |
| P19 | rs115461448missense | G/A | A395T | 23 | 0.009 | 0.01 | |
| IFNA10 | 5.8 | ||||||
| P15 | rs145785282stopgain | G/A | Q125X | 36 | 0.01 | 0.002 | |
| NLRP12 | 11.1 | ||||||
| P5 | rs104895568missense | G/C | V901L | 15 | 0.002 | 0.0006 | |
| P6 | rs35401786missense | C/G | P319R | 23 | 0.009 | 0.002 | |
| P6 | rs34330210missense | G/A | D142N | 24 | 0.009 | 0.002 | |
| P10, P12 | rs104895564stopgain | C/T | R284X | 34 | 0.002 | 0.0002 | |
| P13 | rs35064500stopgain | C/T | R1016X | 37 | 0.002 | 0.0002 | |
Abbreviations: CADD, combined annotation–dependent depletion; GDI, gene damage index; MAF, minor allele frequency; MIS-C, multisystem inflammatory syndrome in children; SNV-ID, single-nucleotide variant identifier.
aGlobal databases from 1KGP (1000 Genomes Project phase 3) and gnomAD (Genome Aggregation Database version 2.1.1).
Three other SNVs predicted to be highly pathogenic—rs115505677 (Q329H), rs115461448 (A395T), and rs148575246 (splice site)—were identified in the IL17RC coding sequence (interleukin-17 receptor C). An additional nonsense variant, rs145785282 (Q125X) in the IFNA10 gene (interferon alpha 10), was identified in a single patient. Notably, patient 6, who presented a severe condition, accumulates 3 of the 9 candidate MIS-C–predisposing variations identified here.
Finally, patients carrying the candidate MIS-C–predisposing variations had a significantly higher median fever duration than the group of patients without these variants (P = .03; Figure 3).
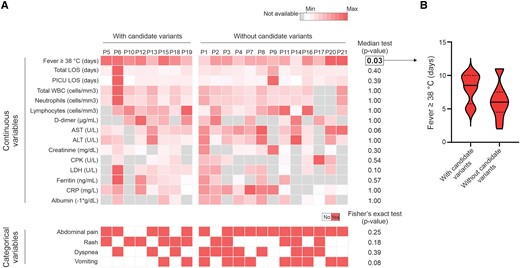
Association between the occurrence of candidate MIS-C–predisposing variations and clinical/laboratory parameters. A, Heat maps representing clinical and laboratory results from patients with or without the prioritized variants. Continuous variables: only parameters available for >50% of the individuals are displayed. The color scales consider the minimum and maximum values found for each variable. Categorical variables: only symptoms reported by at least 50% of the MIS-C cases were analyzed. P < .05 was considered statistically significant. B, Violin plot showing the distribution of fever duration in the groups of patients with or without candidate MIS-C–predisposing variations. Continuous and dashed lines represent the medians and first/third quartiles, respectively. ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; CRP, C-reactive protein; LDH, lactate dehydrogenase; LOS, length of stay (in hospital); MIS-C, multisystem inflammatory syndrome in children; PICU, pediatric intensive care unit; WBC, white blood cell.
Local Ancestry Estimation and Frequency of Variants in Brazilian Reference Populations
Local ancestry inference data show that in persons carrying variants potentially implicated in MIS-C, the IL17RC, IFNA10, and NLPR12 loci were from European or African origins (Supplementary Figure 4).
The frequencies of the target variants in the Brazilian population were estimated in 2 cohorts: SABE [29] and EPIGEN [27] (Supplementary Table 3). Except for rs115461448 in IL17RC (MAF, 0.02 in both cohorts), the prioritized variants are rare (MAF ≤0.01) in the SABE cohort (European ancestry, 0.73) and in the EPIGEN subpopulations from southeast or south Brazil (both with European ancestry >0.76). A similar pattern was found in the EPIGEN subpopulation from northeast Brazil, which has the lowest European ancestry (0.43) among the EPIGEN subpopulations. In this last sample, rs35401786 and rs34330210 in NLRP12 have an MAF of 0.02, while the other target variants show an MAF ≤0.01.
Amino Acid Conservation and the Effects of Variants on NLRP12 Function
Evolutionary conservation of amino acids, which reflects its structural and/or functional importance, was evaluated. Positions of amino acids in IL-17RC (Supplementary Figure 5), IFNA10 (Supplementary Figure 6), and NLRP12 (Supplementary Figure 7) that are affected by the variants prioritized here were assigned intermediate to high conservation grades (4–8).
As described earlier, 5 of the 21 patients of the BR-MIS-C cohort carry heterozygous missense or truncating variants in NLRP12. This gene encodes the cytosolic innate immune receptor NLRP12 [32], which functions as an essential regulator of inflammation, mitigating canonical and noncanonical NF-κB activation in myeloid cells [33, 34]. Additionally, NLRP12 mutant proteins may exhibit a diminished capacity to regulate NF-κB activation, resulting in systemic inflammatory disorders [35, 36]. These evidences led us to hypothesize that the identified NLRP12 variants have reduced inhibitory properties, potentially contributing to MIS-C pathogenesis in certain children. As no fresh blood sample was available, this hypothesis was tested by carrying out in vitro functional experiments with HEK 293T–NF-κB luciferase reporter cells. These HEK cells, which exhibit limited expression of endogenous NLRP12 [35], were transfected with expression plasmids containing either wild type NLRP12 or its variants: R284X, D142N+P319R, R1016X, and V901L (Figure 4A). Notably, our results confirmed that wild type NLRP12 significantly (P < .05) inhibits NF-κB activation induced by IL-1β, in contrast to all evaluated NLRP12 protein variants, which exhibited reduced inhibitory properties (Figure 4B). Similar results were obtained when the transfected cells were stimulated with tumor necrosis factor α (Figure 4C).
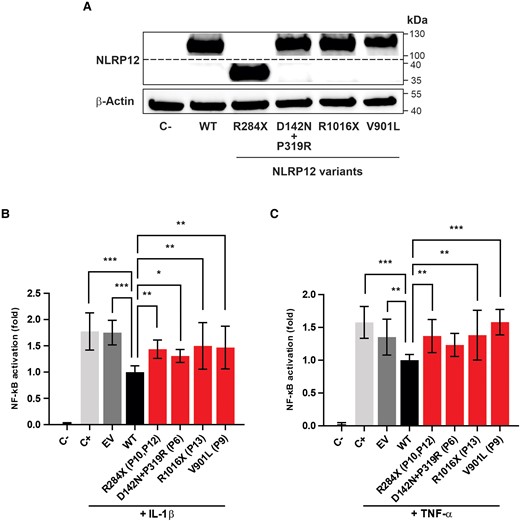
Inhibitory properties of mutant NLRP2 proteins on nuclear factor κB (NF-κB) activation. A, HEK 293T–NF-κB reporter cells were transfected with expression plasmids for wild type (WT) NLRP12 or its variants (R284X, D142N+P319R, R1016X, V901L). NLRP12 proteins were detected by Western blot analysis. The expression of β-actin was evaluated in the same samples as an endogenous control. Impact of NLRP12 WT or NLRP12 mutated proteins on NF-κB activation from HEK 293T–NF-κB reporter cells stimulated with (B) interleukin 1β (IL-1β; 1 ng/mL) or (C) tumor necrosis factor α (TNF-α; 10 ng/mL). NF-κB activation, expressed by the ratio (fold) between WT and other tested conditions, is presented as the mean ± SD derived from 3 independent experiments, with each experimental condition tested in triplicate. Statistical analyses were performed by Kruskal-Wallis test with Dunn correction. *P < .05. **P < .001. ***P < .0001. C−, unstimulated cells; C+, stimulated cells; EV, empty vector; P, patient number in which the variant was found.
DISCUSSION
Whole exome sequencing was performed on Brazilian patients with MIS-C to identify genetic factors explaining individual predisposition to this syndrome. Given that MIS-C incidence rates are significantly higher among Black, Latino, and Asian persons as compared with White individuals [4], studying diverse populations is crucial for identifying population-specific variations and understanding the genetic architecture of this syndrome.
It should be noted that no one in the BR-MIS-C cohort died during the period of this study. Nonetheless, most patients developed severe conditions and were admitted to the pediatric intensive care unit. This panorama highlights the importance of identifying the genetic factors underling MIS-C to define strategies for prevention and treatment. Notably, patient 6, a 6-year-old child, developed a persistent systemic inflammatory disorder with severe progression and difficult management. A myelogram confirmed that the patient had hemophagocytic lymphohistiocytosis secondary to MIS-C. This patient is currently undergoing chemotherapy with etoposide, and bone marrow transplant is under consideration.
Three variants in IL17RC were identified as potentially implicated in MIS-C in 3 unrelated individuals. This gene, located at 3p25.3, encodes one of the IL-17 receptor chains [37]. The IL-17 signaling pathway, which has been implicated in the progression of several inflammatory and autoimmune diseases [38], leads to the activation of NF-κB and MAPK, increasing the production of proinflammatory factors [39, 40]. Therefore, variants in this pathway could also explain the occurrence of MIS-C.
In line with previous findings [16, 20], our data indicate that a dysregulation of a type I interferon response may contribute to the occurrence of MIS-C. Notably, patient 15 carries a nonsense variant in IFNA10. This gene, at the 9p21.3 locus, encodes a type I interferon. Although this SNV in IFNA10 has never been related to susceptibility or severity in SARS-CoV-2 infection, another missense variant (rs28368148) has been implicated in life-threatening COVID-19 [41].
Approximately 24% of the BR-MIS-C cohort carries potentially deleterious variants in NLRP12. The NLRP12 gene, located at 19q13.42, encodes a cytoplasmic protein involved in microbial recognition by dendritic cells and macrophages [42]. This molecule is a potent inflammation reliever and inhibits the NFκB and ERK activation pathways [43]. Recent findings indicate that NLRP12 inhibits caspase recruitment domain (ASC) inflammasome assembly mediated by NLRP3 [44].
In silico analysis conducted here evidenced that the amino acid positions affected by candidate MIS-C–predisposing variations in NLRP12 show intermediate to high evolutionary conservation grades, highlighting their functional relevance. Additionally, all these variants were classified as probably damaging by the LoFtool algorithm (data not shown), a widely used method for predicting loss-of-function variants [45]. Indeed, our in vitro analysis confirmed that the studied NLRP12 variants affect its function. The wild type NLRP12 protein inhibits NF-κB activation in HEK 293T cells, whereas the mutant proteins show significantly reduced inhibitory properties.
The present study is the first to investigate the potential impact of the variants D142N+P319R (combined), R1016X, and V901L on NLRP12 function. Yet, the truncating variant R284X, found in a heterozygous state in 2 unrelated cases of the BR-MIS-C cohort, has been implicated in autosomal dominant autoinflammatory syndromes [35]. Additionally, the NRLP12 D142N was previously described as a candidate variant for cryopyrinopathies [46]. NLRP12-associated systemic autoinflammatory disorders are characterized by an altered innate immune response, with increased NF-κB and caspase 1 activities [36], leading to excessive production of inflammatory cytokines such as IL-1β [47].
Even though there is no unified treatment for autoinflammatory conditions, the use of monoclonal antibodies targeting IL-1–related inflammatory pathways results in a sustained reduction of disease severity [48]. Interestingly, activation of the IL-1 signaling pathway has been observed in MIS-C [12], and the effectiveness of IL-1 blockade for the treatment of this potentially severe illness is currently under discussion [49, 50]. To our knowledge, the present study is the first to provide a plausible molecular mechanism for MIS-C involving variants in NLRP12. In this context, our results suggest that some patients with MIS-C may benefit from IL-1 pathway blockade treatments, which are generally safe and well tolerated.
Some limitations should be considered. Although we identified MIS-C–predisposing variations in some patients, genetic variants potentially involved in MIS-C were not detected in 13 individuals. This observation may suggest that only some patients with MIS-C exhibit functional variants in IL17RC, IFNA10, and NLRP12 or that the potential genetic contributions may arise from unexplored loci or polygenic small-effect variants. Furthermore, additional experiments involving cells from persons carrying the prioritized variants should be considered in future studies. Finally, while our results highlight the differential activation of the NF-κB pathway by NRLP12 variants in HEK 293T cells, the absence of some components of the inflammasome complex in this model represents a limitation. Another system with an intact inflammasome would allow for a more comprehensive evaluation of the impact of NLRP12 variants on ASC polymerization induced by NLRP3. Notably, Coombs et al [44] found that monocytes from patients with periodic fever syndrome carrying the NLRP12 R284X mutation (heterozygous state) display enhanced NLRP3 activity.
With previous findings, our results indicate that rare deleterious variants in genes encoding immune molecules, particularly of the innate immunity, potentially increase the susceptibility to MIS-C and reinforce the genetic heterogeneity of this condition. Furthermore, the MIS-C–predisposing genetic variants discovered here provide new insights for the appropriate management of this severe condition.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgment. We thank the medical staffs of the Hospital Martagão Gesteira, Instituto Couto Maia, and Hospital Universitário Oswaldo Cruz, whose support and collaboration made this study possible. We extend our thanks to the ABraOM team, for publicly sharing genomic data from the SABE cohorts, and to the EPIGEN consortium, for providing information on the frequencies of target variants in Brazilian populations.
Author contributions. P. R. S. O. and L. R. S. V. contributed to the conception and design of the study. T. M. M. B., A. P. N. B., N. L. S. D., R. C. F. R., N. M. T., S. N., R. B. S. P., A. S. S., A. L. N., M. V. B. O.-S., and R. S. S. contributed to biological sample collection and acquisition of medical data. P. R. S. O., L. R. S. V., and T. L. C. contributed to sequencing data acquisition. P. R. S. O., E. T.-S., C. S.-C., F. S. G. K., and R. L. contributed with ancestry analysis. T. M. C. and I. M. P. conducted the in vitro functional analysis. P. R. S. O., L. R. S. V., R. F. C., R. C. F. R., T. M. M. B., R. B. S. P., G. S. G. B., E. H. B. A., P. M. M. F. M., and N. M. T. were responsible for data analysis and interpretation. P. R. S. O., L. R. S. V., R. F. C., G. D. R., R. C. F. R., T. M. M. B., G. D. R., and R. B. S. P. wrote the original draft. All authors were involved in the reviewing and editing of the manuscript and approved the final version.
Financial support. This work was supported by the Fundação de Apoio à Fundação Oswaldo Cruz (FIOTEC), INOVA/FIOCRUZ Program (project VPPCB-005-FIO-20-2-57-30); and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (project 311048/2022-6). ET-S and the EPIGEN group received funding from the Ministério da Saúde do Brasil-DECIT (Genomas-SUS), CNPq, and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG).
References
Author notes
T. M. M. B. and R. S. S. contributed equally to this work.
L. R. S. V. and P. R. S. O. contributed equally to this work.
Potential conflict of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}