






Abstract
MuV caused three epidemic waves in Spain since genotype G emerged in 2005, despite high vaccination coverage. SH gene sequencing according to WHO protocols allowed the identification of seven relevant variants and 88 haplotypes. While the originally imported MuVi/Sheffield.GBR/1.05/-variant prevailed during the first two waves, it was subsequently replaced by other variants originated by either local evolution or importation, according to the additional analysis of hypervariable NCRs. The time of emergence of the MRCA of each MuV variant clade was concordant with the data of the earliest sequence. The analysis of Shannon entropy showed an accumulation of variability on six particular positions as the cause of the increase on the number of circulating SH variants. Consequently, SH gene sequencing needs to be complemented with other more variable markers for mumps surveillance immediately after the emergence of a new genotype, but the subsequent emergence of new SH variants turns it unnecessary.
Mumps is a vaccine-preventable disease caused by mumps virus (MuV), which is a member of the Orthorubulavirusgenus (Paramyxoviridae family). The MuV virion is an enveloped particle containing a nonsegmented negative strand ribonucleic acid molecule of 15 384 nucleotides (nt) [1].
Mumps is a highly transmissible disease whose main clinical symptom is swelling of the parotid glands. Previously, nonspecific symptoms appeared such as fever, headache, malaise, and anorexia. Other less frequent symptoms include orchitis, mastitis, oophoritis, and pancreatitis. After infection, rare complications can arise, such as aseptic meningitis and encephalitis [2]. Infected people can transmit the virus through respiratory droplets from 2 days before to 5 days after onset of parotitis [2]. Asymptomatic and subclinical infected people can also transmit the virus.
Mumps vaccination was introduced into the Spanish childhood immunization schedule in combination with measles and rubella virus as a part of Measles Mumps Rubella Vaccine (MMR vaccine) in 1981 [3]. As a result of the high vaccine coverage, mumps incidence dropped from 211 cases per 100 000 inhabitants in 1982 to 3–31 cases per 100 000 inhabitants from 1997. However, in Spain, as in other countries, the virus still generates outbreaks among highly vaccinated population and causes cyclic epidemic waves that peak every 4–7 years, most recently in 1996, 2000, 2007, 2013, and 2019 [3, 4]. The origins of the capacity of MuV to circulate among immunized people are not well established. The use of the Rubini vaccine strain from 1992 to 1999 in Spain, which was later withdrawn due to reduced effectiveness, has been indicated as a major cause [4]. Additional hypothetical causes include waning of vaccine-induced immunity [2, 5], incomplete cross-neutralization between heterologous mumps virus genotypes [6], and a decreased capacity of the antibodies induced by the vaccine to protect against presently circulating wild-type MuV as a result of long-term antigenic drift [7]. Despite this continuous circulation, the prevalence of severe complications and hospitalizations due to MuV have decreased significantly [4].
The World Health Organization (WHO) recommends mumps genotyping by sequencing of the SH gene as a part of the epidemiologic surveillance system required to control the disease [8]. This molecular tool enables us to (1) identify the general patterns of viral circulation as well as to (2) trace the source of the outbreaks and the chains of transmission. According to this molecular method, 12 genotypes have been recognized: A, B, C (including former genotype E), D, F, G, H, I, J, K (including former genotype M), L and N [9].
The K genotype of mumps was the first reported to be circulating in Spain in the Basque country (Northern Spain) between 1987 and 1990 [10]. Genotype H was predominant from 1996 to 2003, until it was replaced by genotype G in 2005, whereas genotypes A, C, D, and J were reported in several isolated cases or limited outbreaks [11]. Genotype G is still predominant after 15 years. A single SH variant (MuVi/Sheffield.GBR/1.05/) of genotype G was predominant from 2005 to 2015, although cryptic strain replacement between epidemic waves was demonstrated by the use of additional molecular tools based on genomic noncoding regions (NCRs) [12].
In the present work, we show the implications for molecular surveillance of the emergence of new predominant SH variants after an increase of the genomic diversity of mumps virus circulating in Spain during the period of study from 2007 to 2019.
METHODS
Samples and Sequences
A total of 8.742 samples were tested from 2008 to 2019 at the National Center for Microbiology (CNM) as part of the activities of diagnosis and surveillance of mumps, 2464 of which were found to be positive by reverse-transcription polymerase chain reaction. In general, every positive case was genotyped, except during high incidence periods, and 1 case per location and epidemiological week was randomly selected. The MuV genotype was investigated in 1153 of the cases, and 1125 were genotype G, which constitutes the aim of this work, together with 121 Spanish sequences from 2007 to 2019 obtained from GenBank [13].
The first cases of relevant emerging and re-emerging variants were selected for sequencing of hypervariable NCRs together with concomitant cases of previously circulating variants of the same locations (see Results).
Ribonucleic Acid Extraction, Genetic Amplification, and Sequencing
Total nucleic acids were extracted with an automatic extractor (QIAsymphony, QIAGEN) using a commercial kit (QIAsymphony DSP Virus/ Pathogen Midi Kit (96 preps); QIAGEN). Four hundred microliters of sample were extracted to obtain 40 µL of final eluate. The whole SH gene sequence (316 nt) was obtained from all samples according to a published protocol [14]. Genomic fragments including the NCRs located between the nucleoprotein (N) and phosphoprotein (P) coding sequences (CDSs) (N-P, 768 nt) and the Matrix protein (M) and Fusion protein (F) CDSs (M-F, 452 nt), respectively, were amplified in a subset of samples (see below), as previously described [12]. Polymerase chain reaction-amplified products were purified by an enzymatic reaction using Illustra ExoProStar 1-Step (GE Health Care Life Science, Freiburg, Germany) and sequenced using the Sanger method with the ABI Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Branchburg, NJ) using the corresponding forward and reverse primers.
Phylogenetic Analysis, Identification of Mutations, and Variant Classification
Sequences were edited using BioEdit v.7.2.5 [15] and aligned with MAFFT v.7 [16]. Phylogenetic analyses were performed by the method of maximum likelihood (ML) using the IQ-TREE software via the webserver (W-IQ-TREE) [17], with the best evolutionary model previously selected in the model selection tool, based on the Akaike information criterion. The assessment of nodes was supported by ultrafast bootstrap approach (UFboot) [18]. Phylogenetic trees were edited using MEGA v.7 [19].
Every SH sequence was named in accordance with the WHO’s standard nomenclature [8]. Haplotypes (a set of identical sequences) were identified using the phylogenetic analysis and Basic Local Alignment Search Tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) [20] to identify any identical sequence in GenBank. Haplotypes were named using the name of the earliest sequence. Haplotypes that showed an extensive circulation were considered variants. The specific conditions for variant assignation were continuous detection for 6 months or more and/or spreading to 3 or more Spanish provinces.
Phylodynamic Analysis
The time of emergence of the most recent common ancestor (MRCA) of the identified variants were estimated using SH sequences with the Bayesian Markov chain Monte Carlo (MCMC) coalescent method implemented in BEAST v1.10.4 [21]. Regression of root-to-tip genetic distances against sampling time was estimated using Tempest [22]. BEAST analysis was carried out using a SRD06 codon-based evolutionary model and an uncorrelated relaxed molecular clock model with a lognormal rate distribution and the Bayesian Skyline Plot population growth model. The coalescent model was selected over other models because it assumes that a small random sample from a large population is included in the dataset. BEAST was running for at least 100 million MCMC steps, sampling every 5000 steps, and removing 10% as burn-in. The convergence of MCMC chains was checked using Tracer v.1.7.1 [23], ensuring that the effective sample size values were greater than 100 for each estimated parameter. The maximum clade credibility tree was obtained from the sampled trees using TreeAnnotator v1.10.4, then visualized and edited with FigTree v1.4.4 [24].
Shannon Entropy Analysis
Nucleotide variability was measured as entropy using the Shannon Entropy-Two tool (https://www.hiv.lanl.gov/content/sequence/ENTROPY/entropy.html) [25], which looked for the difference in entropies between 2 MuV SH gene alignments in each position. The first alignment included sequences from the 2010 to 2015 epidemic wave and was used as background. The second one collected sequences from the 2016 to 2019 epidemic wave and was used as query. Shannon entropy score was calculated by summing the individual values of each position.
GenBank Accession Numbers
The sequences obtained in this study have been deposited in GenBank under accession numbers MN436853-MN437318, MN308288-MN308363, MN489001-MN489004, MN567307-MN567478, MT906866-MT907264, MT919088-MT919099, MT858762-MT858792, MT670354-MT670376, MT26985-MT27001, MW044679-MW044695, and MW657773-MW657788.
Ethics Statement
The samples used in this work were obtained in the context of the Mumps Microbiological Surveillance Programme of the CNM and used in accordance with the requirements of Spanish biomedical research law (Ley 14/2007 de Investigación Biomédica). The protocol was approved by the Comité de Ética de la Investigación del Instituto de Salud Carlos III (approval no. reference code: CEI PI 35-2015).
RESULTS
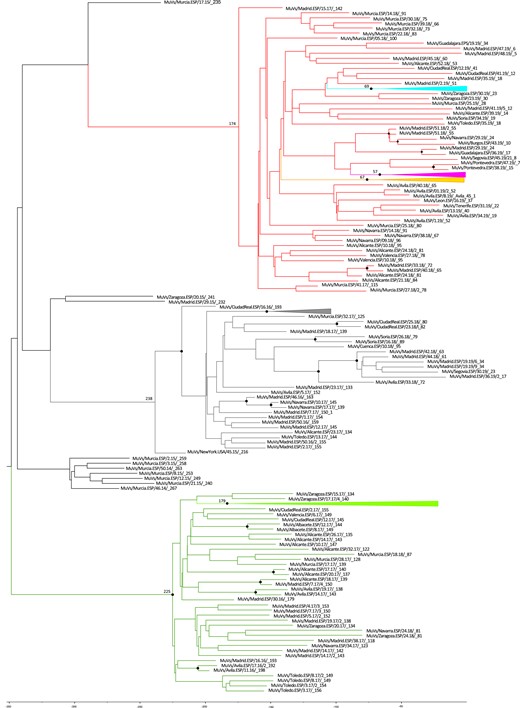
An ML phylogenetic analysis was made with all the available MuV SH Spanish sequences from genotype G (1246) during the period of study, using the generalized time-reversible (GTR) as an evolutionary model (data not shown). In total, 88 haplotypes were identified from 2007 to 2019 (Supplementary Table). Seven of these haplotypes, comprising 1072 sequences (86.0%), met the criteria to be considered variants. Four hundred thirty-seven sequences (35.0%) belonged to the previously described MuVi/Sheffield.GBR/1.05/variant (EU597478) [9].
The earliest sequence of each haplotype was used to construct an ML tree using the GTR as evolutionary model (Figure 1). Most of the haplotypes were from the same genetic lineage and related to MuVi/Sheffield.GBR/1.05/, according to the topology of the phylogenetic tree (Figure 1) and the nt differences (Supplementary Table). Six variants grouped into clades with their related haplotypes, sharing characteristic mutations in the SH sequences: MuVs/NewYork.USA/45.15/(C248T); MuVs/Avila.ESP/11.16/(A226T) and its derived variant MuVs/Madrid.ESP/50.16/2 subclade (A226T, T247C); MuVs/Avila.ESP/51.18/(C13A); MuVs/Salamanca.ESP/24.19/(T100C); and MuVs/New_Jersey.USA/20.10/(C67T). The only exception was a sequence (MuV/Segovia. ESP/39.19/2) of the MuVs/New_Jersey.USA/20.10/clade, which also showed the characteristic mutation of the MuVs/Tarragona.ESP/20.11/haplotype (G159T). Most of the haplotypes showed 1 to 3 mutations regarding the MuVi/Sheffield.GBR/1.05/variant. Nine haplotypes had 4 or more mutations, 2 of which were grouped into variant clades: MuVs/Guadalajara.ESP/22.19/ (11 nt differences), which shares the C13A mutation with MuVs/Avila.ESP/51.18/, and MuVs/Soria.ESP/48.19/ (4 nt differences), which shows the T100C mutation characteristic of the MuVs/Salamanca.ESP/24.19/variant.
![Phylogenetic tree of mumps virus (MuV) SH sequences haplotypes. Analysis used the maximum likelihood method in IQ-tree, using TNe as the evolutionary model. The MuV G genotype reference sequence (MuVi/Gloucester.GBR/32.96 [G], AF280799) was used as outgroup. Colored branches denote different variant clades, and the earliest SH variant sequence of each variant is indicated by colored dots. An asterisk next to the name of the SH sequences indicates those with ≥4 nucleotide differences with respect to the MuVi/Sheffield.GBR/1.05/variant. UltrafastBootstrap values >80 are shown.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/227/1/10.1093_infdis_jiac176/2/m_jiac176f1.jpeg?Expires=1750303690&Signature=kyb6ao6uFPkIo4itHJ1DHPKijBJz7vUPYHSWgkiOiaBgNiMMjy8zMoTrXnaKE0jTAYN9VkBKTpjD~uuHEoAVHI8od4~IWHzqYlbDCSYeabG7jdSGKUTB7hTnQ2SvnFvdd24sNTmjz7qjDPbzCwV-H2qJzJ0qYsUIP~p~O4JEvTu0soEM-3SHF8UhbVYcW09nybUZSCjl57hzwooO1XDqA6Ow-c1-KaULLeoIaLkUSmzxOKuOTAbMmjsAelobnQFTnunG-EbwXWaXSKPLMjnJMtNr2~wkplWf0o4EvgkeH02T~RfslQCSC2Q8F6Sxj9Rg6sQ9QLgMzdjnOtMRD08nCQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Phylogenetic tree of mumps virus (MuV) SH sequences haplotypes. Analysis used the maximum likelihood method in IQ-tree, using TNe as the evolutionary model. The MuV G genotype reference sequence (MuVi/Gloucester.GBR/32.96 [G], AF280799) was used as outgroup. Colored branches denote different variant clades, and the earliest SH variant sequence of each variant is indicated by colored dots. An asterisk next to the name of the SH sequences indicates those with ≥4 nucleotide differences with respect to the MuVi/Sheffield.GBR/1.05/variant. UltrafastBootstrap values >80 are shown.
A total of 44 of 88 (50%) of the haplotypes showed nonsynonymous changes. Most of them have single amino acid mutation; however, 9 of them accumulate more than 3. Three cases of 1246 (0.24%) contained sequences of abnormal length. Two of them lost a stop codon and extended 19 positions to reach a total of 76 amino acids instead of 57. Another one lost the final 13 amino acids showing an SH sequence of 44 amino acids.
The characteristic mutations of the variants are silent in most cases (Supplementary Table). However, MuVs/Salamanca.ESP/24.19/ presents a L17P substitution and MuVs/NewJersey.USA/20.10/has a P6L mutation.
An increase in the diversity of MuV SH variants and their derived haplotypes was observed throughout successive epidemic waves (Figure 2). The MuVi/Sheffield.GBR/1.05/variant was the only one identified in the 2005–2009 and 2010–2015 epidemic waves. However, in the last epidemic wave (2016–2019), several new circulating MuV variants were identified. The MuVs/Avila.ESP/11.16/variant, was the first one, replacing the MuVi/Sheffield.GBR/1.05/variant, which became undetectable by mid-2016 and disappeared for one year. Meanwhile, 2 new MuV variants (MuVs/NewYork.USA/45.15/ and MuVs/Madrid.ESP/50.16/2) cocirculated with MuVs/Avila.ESP/11.16/. In the mid-2017, the MuVi/Sheffield.GBR/1.05/variant reappeared, coexisting with the latter. During 2018 and 2019, the dominant variants, MuVs/NewYork.USA/45.15/ and MuVs/Avila.ESP/11.16/, were substituted by 3 new ones: MuVs/Avila.ESP/51.18/, MuVs/Salamanca.ESP/24.19/, and MuVs/New_Jersey.USA/20.10/ (Figure 2).
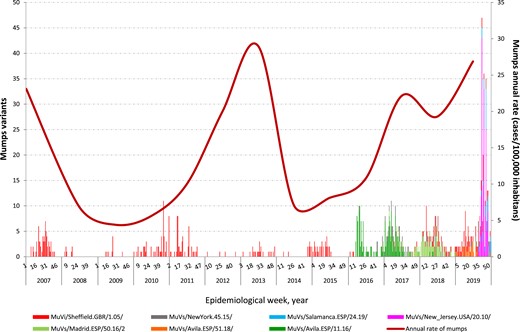
Mumps virus (MuV) genotype G SH variants with relevant circulation in Spain from 2007 to 2019.
A phylodynamic analysis was implemented in BEAST to estimate the time to the MRCA of each identified MuV variant clade. The estimated time of emergence of each clade was consistent with the epidemiological data (Figure 3).
Time-scale tree of mumps virus (MuV) SH haplotype and variant sequences. The time of emergence of the most recent common ancestor (MRCA) is estimated in weeks, taking the last week of 2019 as the temporal reference. The week of each haplotype is included at the end of the name. The week of the MRCA of each variant is shown at the corresponding node. The MuV variant clades are identified by the previously used color code. Posterior probabilities ≥0.8 are shown as black spots.
Entropy Shannon analysis revealed an entropy score along the SH gene of 6.83 for 2010–2015, whereas 4.72 for 2016–2019. The difference in entropy was more concentrated in some particular nucleotide positions in the second period, which correspond to the characteristic mutations of the new variants (C13A, C67T, T100C, A226T, T247C, and C248T) (Figure 4).
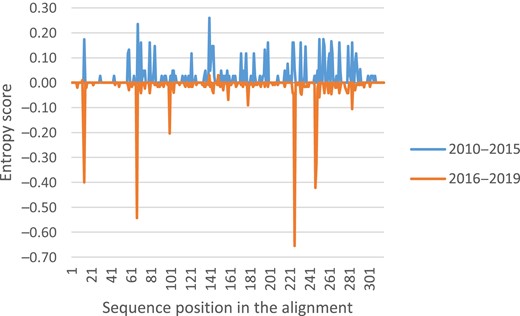
Shannon entropy differences between 2010–2015 and 2016–2019 epidemic waves for mumps virus (MuV) SH sequence alignments. The MuV SH sequences belonging to the 2010–2015 epidemic waves were used as background (blue) and were represented at the positive y-axis, whereas sequences belonging to 2016–2019 were used as query (orange) and were represented at the negative y-axis.
Additional molecular tools based on sequencing of MuV genomic fragments including NCR were used to increase the phylogenetic resolution [12], to establish whether the new MuV variants evolved from those circulating previously or were introduced by importation events. With this purpose, a phylogenetic tree was derived using the concatenated N-P NCR, M-F NCR, and SH sequences from the first 5 cases of the emerging MuVs/Avila.ESP/11.16/variant and the last 6 cases of the previously dominant MuVi/Sheffield.GBR/1.05/variant (Figure 5, Panel A). Sequences from cases of the MuVs/Avila.ESP/11.16/variant grouped in a separate/different branch of cases of the MuVi/Sheffield.GBR/1.05/variant, suggesting an importation event. To study the re-emergence of the MuVi/Sheffield.GBR/1.05/variant in 2017, a phylogenetic tree was derived using the concatenated N-P NCR, M-F NCR, and SH sequences from 13 cases of the MuVi/Sheffield.GBR/1.05/variant identified in 2017 and 2018 and simultaneous circulating variants. MuVi/Sheffield.GBR/1.05/variant sequences grouped in a different clade from MuVs/Avila.ESP/11.16/ and MuVs/NewYork.USA/45.15/ (Figure 5, Panel B), suggesting importation. Finally, sequences of the MuVs/New_Jersey.USA/20.10/variant, which produced a big outbreak in Segovia in mid-2019, were analyzed. According to the topology of the phylogenetic tree (Figure 5, Panel C), the concatenated N-P NCR, M-F NCR, and SH sequences from cases of the MuVs/New_Jersey.USA/20.10/variant grouped in a clade. MuVi/Sheffield.GBR/1.05/variant cases, which circulated earlier in 2019, were part of a different clade, but 1 sequence was at the base of the clade, suggesting that this variant evolved from existing one.
![Phylogenetic tree of mumps virus (MuV) genome regions including N-P NCR, M-F NCR, and SH concatenated sequences. Analysis involved the maximum likelihood method in W-IQ-TREE, using JC as evolutionary model. The MuV G genotype reference sequence (MuVi/Gloucester.GBR/32.96 [G], AF280799) was used as outgroup. (Panel A) Analysis to study the origin of the MuVs/Avila.ESP/11.16/ variant. Green spots correspond to the MuVs/Avila.ESP/11.16/variant and red spots correspond to the MuVi/Sheffield.GBR/1.05/variant. (Panel B) The study of the origin of the re-emergence of the MuVi/Sheffield.GBR/1.05/variant. Green dots correspond to the MuVs/Avila.ESP/11.16/variant, gray dots correspond to the MuVs/NewYork.USA/45.15/variant, and uncolored spots correspond to the re-emerged MuVi/Sheffield.GBR/1.05/variant. (Panel C) Analysis to study the origin of the MuVs/New_Jersey.USA/20.10/variant. Pink dots correspond to the MuVs/New_Jersey.USA/20.10/variant, whereas derived haplotypes (MuVs/Segovia.ESP/39.19/2, MuVs/Segovia.ESP/39.19/12, and MuVs/Segovia.ESP/39.19/32) are indicated by empty circles. Red samples correspond to the MuVi/Sheffield.GBR/1.05/variant.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/227/1/10.1093_infdis_jiac176/2/m_jiac176f5.jpeg?Expires=1750303690&Signature=meY1c0yaUyY5XCdCx1McigFtAPSBhFL9OQD5e4ApMXMig2cdZOYB4~PIQdzBhfCmXbUkV7DLlGMjoc8nSbWfBugyHjkWirMFAjUlVi1gDZl2uh9BDuK4lFEhvcukQeWEmwsfqEu4srqQ7yomzOnBl3Sc4oHkJiY4yWMlbk50I~NxD3XKQHVQgLBLfZCNJTyjZzrG607ZigenE8~kST~KCC0v-MsJwr-8vz25GL~67JBO18UJ97qii0mvbNV-c-CKQMvJ52x8d~ufcmrRHfM3A3PO2LynMLo9RrRtv8AQWe3VBREVe3tVs~wqZYON5uCLyI9YoDlwJ5x9CPbYXEw6gQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Phylogenetic tree of mumps virus (MuV) genome regions including N-P NCR, M-F NCR, and SH concatenated sequences. Analysis involved the maximum likelihood method in W-IQ-TREE, using JC as evolutionary model. The MuV G genotype reference sequence (MuVi/Gloucester.GBR/32.96 [G], AF280799) was used as outgroup. (Panel A) Analysis to study the origin of the MuVs/Avila.ESP/11.16/ variant. Green spots correspond to the MuVs/Avila.ESP/11.16/variant and red spots correspond to the MuVi/Sheffield.GBR/1.05/variant. (Panel B) The study of the origin of the re-emergence of the MuVi/Sheffield.GBR/1.05/variant. Green dots correspond to the MuVs/Avila.ESP/11.16/variant, gray dots correspond to the MuVs/NewYork.USA/45.15/variant, and uncolored spots correspond to the re-emerged MuVi/Sheffield.GBR/1.05/variant. (Panel C) Analysis to study the origin of the MuVs/New_Jersey.USA/20.10/variant. Pink dots correspond to the MuVs/New_Jersey.USA/20.10/variant, whereas derived haplotypes (MuVs/Segovia.ESP/39.19/2, MuVs/Segovia.ESP/39.19/12, and MuVs/Segovia.ESP/39.19/32) are indicated by empty circles. Red samples correspond to the MuVi/Sheffield.GBR/1.05/variant.
DISCUSSION
Genotype G was the most frequently detected in Spain during the period of the study, as well as in other Western countries and Japan [9, 11]. The MuVi/Sheffield.GBR.1.05/variant seemed to be dominant, based on the sequences available in GenBank as previously described for the United States of America [26]. In Spain, MuV was able to circulate among the highly vaccinated population and generated successive epidemic waves [4, 10, 13]. It might be expected that periods of lower incidence could act as evolutionary bottlenecks in which some strains could disappear. The replacement of genotype H by the MuVi/Sheffield.GBR/1.05/variant of genotype G in 2005 is an example of this [11]. However, according to the SH gene analysis, the MuVi/Sheffield.GBR/1.05/variant was able to remain the dominant strain throughout 2 subsequent epidemic waves (2005–2009, 2010–2015) with no apparent replacement. Additional molecular tools, based on sequencing of MuV genomic fragments including NCRs, were necessary to reveal the hidden strain replacement between the 2 epidemic waves, which remained unnoticed with the WHO-recommended genotyping tool [12]. However, in the course of the last epidemic wave (2016–2019), SH gene analysis was sufficient to reveal an increase of circulating variants of the G genotype mumps virus (Figure 2). During this period, 7 variants were identified, based on criteria detailed in the Materials and Methods, all of which belonged to the MuVi/Sheffield.GBR/1.05/lineage. Haplotypes related to each variant grouped into the same clades of the ML phylogenetic tree (Figure 1) and shared 1 or 2 specific mutations (Supplementary Table), suggesting that most of them were derived from each variant by genetic evolution. The number of mutations identified were concordant with the evolution rate previously estimated for the MuV SH gene (1.71 × 10−3 substitutions/site/year) [27].
The increase of new variants seems to be related to the emergence of new strains of MuV by local evolution over the time, which can eventually establish a sustained circulation. These variants subsequently generate derived haplotypes that are generally unable to spread through the population. However, one of them, MuVs/Madrid.ESP/50.16/2, succeeded in establish circulation as a new variant. Nevertheless, an importation event was probably the origin of the MuVs/Avila.ESP/11.16/variant circulation in Spain, as suggested by the phylogenetic analysis of the concatenated SH, N-P NCR, and M-F NCR regions (Figure 5A). This could also be the case for MuVs/NewYork/45.15/variant, which was circulating extensively during the previous weeks in other countries, such as the United States of America [26]. This variant is characterized by mutation C248T and was previously named as Sheffield-C248 T by McNall et al [26]. As suggested by the NCR analysis (Figure 5B), the re-emergence of the MuVi/Sheffield.GBR/1.05/variant in 2017 was probably due to an importation event, rather than local evolution by reversion of the characteristic mutation from the cocirculating variants. In addition to importation events, local evolution was implied in the emergence of MuVs/New_Jersey.USA/20.10/variant and its derived haplotypes, which were responsible for causing an important outbreak in Segovia in 2019 (Figure 5C). The high number of mutations identified in 9 haplotypes, many of them nonsynonymous (Supplementary Table), could not be explained by the local evolution of sequences, according to the evolution rate estimated for the MuV SH gene, and probably corresponded to distinct self-limited importation events. Two of them (MuVs/Guadalajara.ESP/22.19/ and MuVs/Soria.ESP/48.19/) grouped into variant clades because they shared the specific characteristic mutations.
The topology of the phylogenetic tree of MuV variants and haplotypes circulating in Spain is consistent, with the temporal data of the sequences by the epidemiological week and year in which they were detected (Figure 2), and shows their sequential emergence over the time. These results were confirmed, using the MCMC coalescent method implemented in BEAST, which estimated the times of emergence of the MRCA of each identified variant, (Figure 3), which always occurred before the first week of detection of each variant.
Although the total Shannon entropy score obtained along the SH sequence was higher for the 2010–2015 epidemic wave than the 2016–2019 epidemic wave, suggesting more genetic diversity in the first wave, an unique variant (MuVi/Sheffield.GBR/1.05/) was the predominant variant, and most of the haplotypes detected were not successful. The total Shannon entropy score during the 2016–2019 epidemic wave was lower, but the difference in the entropy value was concentrated in 6 nucleotide positions (Figure 4), concordant with the specific variant point mutations (C13A, C67T, T100C, A226T, T247C, and C248T), resulting in an increase in the number of circulating variants. Cocirculation of different haplotypes has been previously described [26]. However, the number of circulating SH variants associated to characteristic mutations described here was not found in the United States of America, where C248T seemed to be the only one selected [26]. The longer period of study of the present work could explain this difference. Surprisingly, we found a lower frequency (0.24% versus 1.79%) of SH protein sequences with modified length [26], all 3 from different locations and years.
The cocirculation of several variants of one predominant lineage that emerged by local evolution, and from multiple independent introductions, has previously been reported in the United States of America, from combining molecular tools based on the full-genome and epidemiological analysis [28]. Full-genome sequencing is replacing partial sequencing for molecular epidemiology studies of mumps [28–30] and other viral diseases. However, the recommendation of full-genome sequencing as a global tool for laboratory surveillance of mumps is not feasible at present, due to the lack of availability of deep-sequencing technology in many countries. As a result, current WHO protocols for mumps genotyping will continue to be used in the immediate future. In a previous study [12], we showed that additional markers based on the sequencing of genomic regions including NCRs are required for the laboratory surveillance of mumps, in addition to the WHO protocols based on the SH sequence, after the emergence of a new dominant genotype. Other authors have used these new molecular markers to improve the resolution in molecular epidemiology studies [31], although they recommend the use of the complete genome to track chains of transmission [29].
Consequently, our results show that the WHO-recommended protocol for mumps genotyping based on the sequencing of the SH gene could not be discriminative enough to trace the circulation patterns of MuV in the years after the emergence of a new dominant genotype, as suggested in a previous work [12]. Under these circumstances, additional auxiliary markers such as NCR sequences are needed. However, as genetic variability accumulated, SH sequence became discriminative enough for this purpose, although additional markers are still needed to establish the origin of the new variants as the result of local evolution or importation. Genetic variation initially seems to spread randomly through the SH sequence while the original imported variant persists as the dominant variant. After several years of continuous circulation, variability seems to concentrate on some particular positions that seems to be fixed to become new variants that are able to establish circulation, until they are replaced by new ones. However, such mutations are synonymous in most cases. On the contrary, mutants in other positions fail to spread and are detected as sporadic haplotypes, even when some changes are not synonymous. This fact suggests that the SH gene would not be the target for positive selection. The comparative study of the full genomes of SH MuV haplotypes that were or were not able to establish circulation could allow to identify features that could eventually facilitate virus-spreading among highly immunized populations.
Study Limitations
This study has 2 main limitations. The work made use of the samples received at the CNM through the specific surveillance mumps program and the Spanish MuV SH sequences available in GenBank. Although most Spanish provinces are represented in the dataset, they are not proportionally representative of their population. Fewer samples were received in 2012 and 2013 than expected, given the incidence of mumps (Figure 2), probably due to the coexistence of large measles outbreaks that placed great pressure on the work of the surveillance system.
CONCLUSIONS
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Sara Ruiz and the Genomics Unit (Instituto de Salud Carlos III) for technical assistance with sequencing.
Authors contributions. A. M. G. contributed to technical work, data analysis, and writing as main author. A. F.-G. and J. E. E. contributed to design of the study, data analysis, and writing as main authors. F. D.-F. contributed to technical bioinformatics, data analysis, and drafting and revision of the manuscript. N. L.-P. contributed to data analysis and drafting and revision of the manuscript. J. C. S., S. M. J., C. R.-S., and L. G.-C. contributed to identification and confirmation of mumps cases and review and assistance in the editing the final version of the manuscript. J. M.-C., M. P.-O., and F. d. O. contributed to review and assistance in editing the final version of the manuscript.
Financial support. This work was funded by the instituto de Salud Carlos III (ISCIII) (Grants PI15CIII/00023 and PI19ICIII/0041). A. M. G. was funded by Consorcio de Investigación Biomédica en Red de Epidemiología y Salud Pública, ISCIII.
References
Author notes
A. F.-G. and J. E. E. contributed equally as cosenior authors.
Presented in part: 23rd Annual Conference of the European Society for Clinical Virology, Manchester, UK, 15th-18th September 2021.
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}