






Abstract
This study describes the genotypic and phenotypic characterization of novel human cytomegalovirus (HCMV) genetic variants of a cohort of 94 clinically resistant HCMV patients.
Antiviral-resistant mutations were detected in the UL97, UL54, and UL56 target genes of 25 of 94 (26.6%) patients. The genotype-phenotype correlation study resolved the status of 5 uncharacterized UL54 deoxyribonucleic acid polymerase (G441S, A543V, F460S, R512C, A928T) and 2 UL56 terminase (F345L, P800L) mutations found in clinical isolates. A928T conferred high, triple resistance to ganciclovir, foscarnet, and cidofovir, and A543V had 10-fold reduced susceptibility to cidofovir. Viral growth assays showed G441S, A543V, F345L, and P800L impaired viral growth capacities compared with wild-type AD169 HCMV. Three-dimensional modeling predicted A543V and A928T phenotypes but not R512C, reinforcing the need for individual characterization of mutations by recombinant phenotyping.
Extending mutation databases is crucial to optimize treatments and to improve the assessment of patients with resistant/refractory HCMV infection.
Characterization of human cytomegalovirus (HCMV) antiviral drug resistance mutations has contributed to improving HCMV therapy and to the knowledge of viral proteins that serve as new antiviral targets. Ganciclovir (GCV), its oral prodrug valganciclovir (VGCV), foscarnet (FOS), and cidofovir (CDV) target the viral UL54 deoxyribonucleic acid (DNA) polymerase and are currently licensed for treatment of HCMV infection [1, 2]. Maribavir (MBV) is an inhibitor of UL97 phosphokinase undergoing a phase 3 clinical trial; however, the US Food and Drug Administration has not yet approved MBV for patients with post-transplant HCMV infection who do not respond to the antivirals available [3]. Letermovir (LMV) targets the viral terminase complex (UL51, UL56, UL89) and has recently been approved for primary prophylaxis of HCMV infections in allogenic hematopoietic stem cell transplant (HSCT) recipients [4]. Human cytomegalovirus (HCMV) antiviral resistance is an underestimated emergent problem, especially in transplant recipients, presenting an incidence of 5%–12% [5].
Resistance to VGCV/GCV, as first-line therapy, is most commonly due to mutations in UL97 involved in the initial phosphorylation of GCV [6]. Maribavir resistance mutations have primarily been mapped to this gene and show compensatory mutations in UL27 [7]; however, only few mutations in UL97 cause MBV-GCV cross-resistance [8, 9]. Mutations in UL54 are associated with resistance to FOS and CDV and can also appear after prolonged GCV therapy, contributing to a high level of resistance to GCV and inducing cross-resistance to FOS or/and CDV [10]. Resistance mutations are mainly located in the conserved domains of the viral DNA polymerase, whereas sensitive polymorphisms appear in the highly variable regions located between domains delta-C and II and between domains III and I [11]. In contrast, LMV resistance mutations are mainly described in the UL56 terminase subunit and rarely in UL89 and UL51 [12].
Confirmation of antiviral resistance is based on the detection of viral mutations that confer drug resistance by genotypic antiviral resistance testing, providing timely data to facilitate clinical decision making. However, the accuracy of genotypic antiviral resistance testing depends on validated databases linking specific mutations with levels of drug resistance. Three-dimensional (3D) protein modeling provides useful prediction of crucial residues for protein-antiviral molecules interaction [13]. Nevertheless, recombinant phenotyping is the reference method to confirm the level of antiviral resistance and the impact of individual mutations on viral growth.
Therefore, the aim of this study is to describe the incidence of mutations under GCV, FOS, CDV, LMV, and MBV therapy in patients with refractory HCMV infection; and to phenotype previously uncharacterized HCMV genetic variants by 3D protein modeling and recombinant phenotyping.
METHODS
Study Specimens and Population
Positive samples from patients with refractory HCMV infection (n = 94) who fulfilled criteria of suspicion of resistance to antiviral treatment (VGCV/GCV, FOS, CDV, LMV, MBV) were included from April 2012 until September 2021 [1]. Antiviral treatment was administered according to the clinical judgment of the attending physician. The study population has been enlarged from a cohort previously published for a different purpose [14]. The patients belonged to the hospitals included in the Spanish Network for Research in Infectious Diseases (REIPI) and the Group for the Study of Infection in Transplantation (GESITRA). All samples were collected on suspicion of antiviral resistance, frozen at −80°C, and sent to the coordinating center (Hospital Clinic of Barcelona, Barcelona, Spain) so that genotypic antiviral resistance testing could be performed.
Human Cytomegalovirus Load Quantification
The HCMV load was measured in liquid samples by quantitative real-time polymerase chain reaction (qPCR) in a Cobas 6800 (Roche, Basel, Switzerland) according to the manufacturer’s instructions. Viral load of gastrointestinal biopsies was quantified by qPCR (Q-CMV Real Time Complete Kit; Nanogen Advanced Diagnostics, Buttigliera, Italy) using a 7300 Real-Time PCR System (Applied Biosystems).
Genotypic Antiviral Resistance Testing
Extraction of total nucleic acids from liquid samples was performed (1) in MagNA Pure Compact (Roche, Switzerland) and (2) with the EZ1 DNA Tissue Kit (QIAGEN, Hilden, Germany) in paraffin-embedded tissue and fresh tissue according to the manufacturer’s specifications and using the Bio-Robot EZ1 (QIAGEN).
Genotypic testing was done by Sanger sequencing based on PCR amplification of HCMV UL97 (residues 270–670), UL54 (300–1000), and UL56 (180–395) regions. These regions correspond to resistance-associated domains and were sequenced using previously described primers and procedures [14, 15]. Each amplicon was bidirectionally sequenced to avoid artifacts. Sequences were analyzed and aligned using the MEGA v.7. software [16] and were compared with the HCMV TB40 strain (GenBank accession number MF871618.1) using the MRA-Mutation Resistance Analyzer tool provided by the University of Ulm [17].
Prioritization of Sequence Variants for Phenotyping
The gene position of the variants found was used to establish priority for phenotyping. Mutations located at the 2 previously characterized hypervariable, nonconserved regions located at residues 614–697 and 874–898 of UL54 [10] are shown in Supplementary Table 1 and did not undergo phenotyping.
Phenotypic Assay by Recombinant Bacterial Artificial Chromosome Technology
Mutations with previously uncharacterized phenotypes at the time of genotypic detection were individually tested at the French National Reference Centre for Herpesviruses (Limoges, France) and the Hospital Clinic of Barcelona using a phenotypic assay with recombinant bacterial artificial chromosome (BAC) technology as described previously [18]. Each mutation was introduced by “en passant” mutagenesis into an HCMV BAC [19] containing an enhanced green fluorescent protein (EGFP) gene in the unique short region derived from the AD169 laboratory strain (provided by M. Messerle, Hannover Medical School, Hannover, Germany). The recombinant BAC was transfected into MRC-5 cells (bioMérieux, Lyon, France) using the liposomal reagent Transfast (Promega, Madison, WI) following the manufacturer’s instructions. The presence of the desired mutation was confirmed by Sanger sequencing.
A focus reduction assay in a 48-well MRC-5 culture plate with a multiplicity of infection (MOI) of 0.01 was used to assess antiviral susceptibility in triplicate to GCV, FOS, CDV, and LMV for mutations found in UL56. The half-maximal effective concentration (EC50) of the mutant was compared to that obtained for the wild-type control HCMV BAC.
To estimate the impact of each mutation on viral growth, the recombinant strain and the AD169-EGFP control were inoculated into 48-well MRC-5 culture with an MOI of 0.01. The number of fluorescent plaque-forming units (PFUs) was counted from days 1 to 4 and on day 7 postinoculation to establish viral growth curves for each recombinant.
Structure of the Protein 3D Model
The theoretical structure of the UL54 DNA polymerase was built by homology modeling with the standalone version of MODELLER 9.9 [20]. The UL54 sequence was aligned with 3 templates as described previously [21]. Sequence alignment included primary structures of the UL52 homolog from herpes simplex virus-1 (HSV-1) (protein data bank [PDB] ID 2GV9), the C-terminal part of UL54 taken from PDB ID 1YYP (ie, complex of UL44 with fragment 1223–1242 of UL54), and the exonuclease domain (ie, amino acid sequence 109–342), metal ions, and DNA duplex of PDB ID 1CLQ. UL54 moieties aligned to nonresolved loops of UL52 were retrieved from sequence alignment for calculations. A fast molecular dynamic optimization implemented in MODELLER was applied to each 100 calculated structures. Quality structures were assessed by calculating their Q-mean score on a dedicated web server [22]. For UL56 mutations, the model with an HSV-1 homolog did not allow localizing the mutations concerned because there was no correspondence with the amino acid sequence.
Ethical Approval
This study was approved by the Ethical Committee of the Hospital Clínic of Barcelona (ref. no. HCB/2018/0634) as the reference committee for all the participating hospitals and endorsed by GESITRA according to CPMP/ICH/135/95 regulations. All of the patients included in the study provided signed informed consent.
RESULTS
Overview of UL54, UL56, and UL97 Sequence Variation Under Treatment
This study comprised a cohort of 94 patients with refractory HCMV infection who fulfilled the criteria of suspicion of HCMV resistance to standard antiviral treatment (GCV, FOS, CDV). Seven patients additionally received LMV and 4 MBV as salvage therapy. One clinical sample from each patient was collected at the time of suspicion to perform genotypic antiviral resistance testing, except for patients with examined mutations, from which multiple samples were evaluated retrospectively when possible. Subjects with previously characterized mutations associated with either resistance or sensitive response to antivirals located in the target UL97, UL54, and UL56 genes were classified according to their clinical history and sample type (Table 1). All resistant mutations were detected in plasma samples despite the high viral loads detected in the rest of clinical isolates.
Clinical History of the Study Populationa
| Clinical | Resistance Mutation | Sensitive Polymorphisms | |||||
|---|---|---|---|---|---|---|---|
| History | Nb | UL97 | UL54 | UL56 | UL97 | UL54 | UL56 |
| Congenital HCMV | 6 | 0 | 0 | 0 | 0 | 5 | 0 |
| HIV | 2 | 0 | 0 | 0 | 1 | 2 | 0 |
| IBD | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| CVID | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Transplant Recipients | 83 | 21 | 5 | 1 | 4 | 58 | 4 |
| HSCT | 30 | 3 | 3 | 1 | 2 | 23 | 2 |
| SOT | 53 | 18 | 2 | 0 | 2 | 35 | 2 |
| Heart | 11 | 4 | 1 | 0 | 0 | 5 | 1 |
| Lung | 6 | 4 | 1 | 0 | 0 | 5 | 0 |
| Liver | 8 | 3 | 0 | 0 | 0 | 6 | 0 |
| Kidney | 25 | 7 | 0 | 0 | 2 | 17 | 1 |
| Liver-kidney | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Pancreas-kidney | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| Sample Type | |||||||
| Plasma | 84 | 20 | 4 | 1 | 5 | 58 | 4 |
| Whole blood | 4 | 1 | 1 | 0 | 0 | 4 | 0 |
| GI biopsy | 5 | 0 | 0 | 0 | 0 | 4 | 0 |
| Aqueous humour | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Total | 94 | 21 | 5 | 1 | 5 | 67 | 4 |
| Clinical | Resistance Mutation | Sensitive Polymorphisms | |||||
|---|---|---|---|---|---|---|---|
| History | Nb | UL97 | UL54 | UL56 | UL97 | UL54 | UL56 |
| Congenital HCMV | 6 | 0 | 0 | 0 | 0 | 5 | 0 |
| HIV | 2 | 0 | 0 | 0 | 1 | 2 | 0 |
| IBD | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| CVID | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Transplant Recipients | 83 | 21 | 5 | 1 | 4 | 58 | 4 |
| HSCT | 30 | 3 | 3 | 1 | 2 | 23 | 2 |
| SOT | 53 | 18 | 2 | 0 | 2 | 35 | 2 |
| Heart | 11 | 4 | 1 | 0 | 0 | 5 | 1 |
| Lung | 6 | 4 | 1 | 0 | 0 | 5 | 0 |
| Liver | 8 | 3 | 0 | 0 | 0 | 6 | 0 |
| Kidney | 25 | 7 | 0 | 0 | 2 | 17 | 1 |
| Liver-kidney | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Pancreas-kidney | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| Sample Type | |||||||
| Plasma | 84 | 20 | 4 | 1 | 5 | 58 | 4 |
| Whole blood | 4 | 1 | 1 | 0 | 0 | 4 | 0 |
| GI biopsy | 5 | 0 | 0 | 0 | 0 | 4 | 0 |
| Aqueous humour | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Total | 94 | 21 | 5 | 1 | 5 | 67 | 4 |
Abbreviations: CVID, common variable immunodeficiency; GI, gastrointestinal; HCMV, human cytomegalovirus; HIV, human immunodeficiency virus; HSCT, hematopoietic stem cell transplant; IBD, inflammatory bowel disease; SNP, single-nucleotide polymorphism; SOT, solid organ transplant.
Number of subjects with resistance mutations and sensitive polymorphisms in the antiviral target genes are indicated according to the clinical history and the type of sample.
The number of subjects.
Clinical History of the Study Populationa
| Clinical | Resistance Mutation | Sensitive Polymorphisms | |||||
|---|---|---|---|---|---|---|---|
| History | Nb | UL97 | UL54 | UL56 | UL97 | UL54 | UL56 |
| Congenital HCMV | 6 | 0 | 0 | 0 | 0 | 5 | 0 |
| HIV | 2 | 0 | 0 | 0 | 1 | 2 | 0 |
| IBD | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| CVID | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Transplant Recipients | 83 | 21 | 5 | 1 | 4 | 58 | 4 |
| HSCT | 30 | 3 | 3 | 1 | 2 | 23 | 2 |
| SOT | 53 | 18 | 2 | 0 | 2 | 35 | 2 |
| Heart | 11 | 4 | 1 | 0 | 0 | 5 | 1 |
| Lung | 6 | 4 | 1 | 0 | 0 | 5 | 0 |
| Liver | 8 | 3 | 0 | 0 | 0 | 6 | 0 |
| Kidney | 25 | 7 | 0 | 0 | 2 | 17 | 1 |
| Liver-kidney | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Pancreas-kidney | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| Sample Type | |||||||
| Plasma | 84 | 20 | 4 | 1 | 5 | 58 | 4 |
| Whole blood | 4 | 1 | 1 | 0 | 0 | 4 | 0 |
| GI biopsy | 5 | 0 | 0 | 0 | 0 | 4 | 0 |
| Aqueous humour | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Total | 94 | 21 | 5 | 1 | 5 | 67 | 4 |
| Clinical | Resistance Mutation | Sensitive Polymorphisms | |||||
|---|---|---|---|---|---|---|---|
| History | Nb | UL97 | UL54 | UL56 | UL97 | UL54 | UL56 |
| Congenital HCMV | 6 | 0 | 0 | 0 | 0 | 5 | 0 |
| HIV | 2 | 0 | 0 | 0 | 1 | 2 | 0 |
| IBD | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| CVID | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Transplant Recipients | 83 | 21 | 5 | 1 | 4 | 58 | 4 |
| HSCT | 30 | 3 | 3 | 1 | 2 | 23 | 2 |
| SOT | 53 | 18 | 2 | 0 | 2 | 35 | 2 |
| Heart | 11 | 4 | 1 | 0 | 0 | 5 | 1 |
| Lung | 6 | 4 | 1 | 0 | 0 | 5 | 0 |
| Liver | 8 | 3 | 0 | 0 | 0 | 6 | 0 |
| Kidney | 25 | 7 | 0 | 0 | 2 | 17 | 1 |
| Liver-kidney | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Pancreas-kidney | 2 | 0 | 0 | 0 | 0 | 1 | 0 |
| Sample Type | |||||||
| Plasma | 84 | 20 | 4 | 1 | 5 | 58 | 4 |
| Whole blood | 4 | 1 | 1 | 0 | 0 | 4 | 0 |
| GI biopsy | 5 | 0 | 0 | 0 | 0 | 4 | 0 |
| Aqueous humour | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Total | 94 | 21 | 5 | 1 | 5 | 67 | 4 |
Abbreviations: CVID, common variable immunodeficiency; GI, gastrointestinal; HCMV, human cytomegalovirus; HIV, human immunodeficiency virus; HSCT, hematopoietic stem cell transplant; IBD, inflammatory bowel disease; SNP, single-nucleotide polymorphism; SOT, solid organ transplant.
Number of subjects with resistance mutations and sensitive polymorphisms in the antiviral target genes are indicated according to the clinical history and the type of sample.
The number of subjects.
Previously described resistant mutations were detected in 24 of 94 (25.5%) patients, all of whom were transplant recipients (6 HSCT, 18 solid organ transplant [SOT]): 19 patients had resistant mutations in UL97 (1 presenting 2 mutations), 2 in UL54 (1 presenting 2 mutations), 1 patient with mutations in UL54 and UL56, and 2 in UL54 and UL97. These mutations conferred resistance to GCV, FOS, CDV, LMV, or MBV as well as multiple resistance or cross-resistance (Table 2). Data of cumulative treatment, time from transplantation until the detection of the variant, viral loads of the detection sample, and level of resistance to each antiviral are shown in Table 2.
Resistance Mutations Detected in Association With Current Antiviral Drugs in the Overall Study Population
| Gene | Mutation | Na | Transplanta | Days of Cumulative Treatmenta,b | Days Until Detectionc | Viral Load at the Detection (IU/mL)d | Resistance Levele | Reference |
|---|---|---|---|---|---|---|---|---|
| UL97 | M460V | 2 | HSCT, liver | 19d VGCV/GCV (n = 2) | 493 | 5.82E+03 | 5–10× GCV | [24] |
| UL97 | C480F | 2 | HSCT, kidney | 60d MBV (n = 2), 67 GCV (n = 2) | 222 | 9.97E+03 | 2–5× GCV, 223× MBV | [8, 9] |
| UL97 | C592G | 1 | Kidney | 138d VGCV | 235 | 9.87E+03 | 2–5× GCV, 2–5× FCV | [25] |
| UL97 | A594V | 5 | Kidney (n = 2), liver (n = 2), lung | 56d (14; 121) VGCV/GCV (n = 5), 52d FOS + 51d IgG (n = 1) | 137 (100; 156) | 2.03E+04 (1.20E+04; 3.68E+04) | 5–10× GCV | [26] |
| UL97 | A594P | 1 | Heart | 24d GCV | 534 | 1.84E+04 | 5–20× GCV | [25] |
| UL97 | L595S | 5 | HSCT, heart, lung (n = 2), kidney | 38d (20; 54) VGCV/GCV (n = 5), 36d FOS + 16d IgG (n = 1) | 158 (66; 357) | 3.87E+04 (2.29E+04; 5.80E+04) | 5× GCV | [27] |
| UL97 | L595W + A594V | 1 | Heart | 14d VGCV | 110 | 4.12E+03 | 5.1× GCV | [27] |
| UL54 | V781I | 1 | HSCT | 38d GCV + 79d FOS + LT | 153 | 1.63E+03 | 1–4× GCV, 4–5.2× FOS | [28] |
| UL54 | A928T | 1 | cCMV + 2 HSCT | 43d GCV + 23d FOS | 145 | 2.17E+04 | NP | … |
| UL54 | L773V + G841A | 1 | HSCT | 35d VGCV/GCV + 45d FOS + 10d ACV | 133 | 3.55E+03 | 2× GCV, 5× FOS/3.2× GCV, 2.6× CDV, 4.3× FOS | [29, 30] |
| UL54 + UL97 | D413N + M460I | 1 | Lung | 24d GCV | 668 | 6.20E+04 | 6.5× GCV, 11× CDV | [31] |
| UL54 + UL97 | A543V + M460I | 2 | Kidney (n = 2) | 64d VGCV/GCV + 9d FOS | 185 | 7.01E+04 | NP/5–10× GCV | [32] |
| UL54 + UL97 | A987G + C603W | 1 | Heart | 150d VGCV prophylaxis + 84d VGCV | 238 | 1.90E+04 | 6.8× GCV, 5.3× CDV/8.3× GCV | [28, 33] |
| UL54 + UL56 | T700A + C325F | 1 | HSCT | 33d VGCV/GCV + 74d FOS + 4d CDV + 27d LMV + LT | 198 | 3.41E+03 | 4.7× FOS/>3000× LMV | [30, 34] |
| Gene | Mutation | Na | Transplanta | Days of Cumulative Treatmenta,b | Days Until Detectionc | Viral Load at the Detection (IU/mL)d | Resistance Levele | Reference |
|---|---|---|---|---|---|---|---|---|
| UL97 | M460V | 2 | HSCT, liver | 19d VGCV/GCV (n = 2) | 493 | 5.82E+03 | 5–10× GCV | [24] |
| UL97 | C480F | 2 | HSCT, kidney | 60d MBV (n = 2), 67 GCV (n = 2) | 222 | 9.97E+03 | 2–5× GCV, 223× MBV | [8, 9] |
| UL97 | C592G | 1 | Kidney | 138d VGCV | 235 | 9.87E+03 | 2–5× GCV, 2–5× FCV | [25] |
| UL97 | A594V | 5 | Kidney (n = 2), liver (n = 2), lung | 56d (14; 121) VGCV/GCV (n = 5), 52d FOS + 51d IgG (n = 1) | 137 (100; 156) | 2.03E+04 (1.20E+04; 3.68E+04) | 5–10× GCV | [26] |
| UL97 | A594P | 1 | Heart | 24d GCV | 534 | 1.84E+04 | 5–20× GCV | [25] |
| UL97 | L595S | 5 | HSCT, heart, lung (n = 2), kidney | 38d (20; 54) VGCV/GCV (n = 5), 36d FOS + 16d IgG (n = 1) | 158 (66; 357) | 3.87E+04 (2.29E+04; 5.80E+04) | 5× GCV | [27] |
| UL97 | L595W + A594V | 1 | Heart | 14d VGCV | 110 | 4.12E+03 | 5.1× GCV | [27] |
| UL54 | V781I | 1 | HSCT | 38d GCV + 79d FOS + LT | 153 | 1.63E+03 | 1–4× GCV, 4–5.2× FOS | [28] |
| UL54 | A928T | 1 | cCMV + 2 HSCT | 43d GCV + 23d FOS | 145 | 2.17E+04 | NP | … |
| UL54 | L773V + G841A | 1 | HSCT | 35d VGCV/GCV + 45d FOS + 10d ACV | 133 | 3.55E+03 | 2× GCV, 5× FOS/3.2× GCV, 2.6× CDV, 4.3× FOS | [29, 30] |
| UL54 + UL97 | D413N + M460I | 1 | Lung | 24d GCV | 668 | 6.20E+04 | 6.5× GCV, 11× CDV | [31] |
| UL54 + UL97 | A543V + M460I | 2 | Kidney (n = 2) | 64d VGCV/GCV + 9d FOS | 185 | 7.01E+04 | NP/5–10× GCV | [32] |
| UL54 + UL97 | A987G + C603W | 1 | Heart | 150d VGCV prophylaxis + 84d VGCV | 238 | 1.90E+04 | 6.8× GCV, 5.3× CDV/8.3× GCV | [28, 33] |
| UL54 + UL56 | T700A + C325F | 1 | HSCT | 33d VGCV/GCV + 74d FOS + 4d CDV + 27d LMV + LT | 198 | 3.41E+03 | 4.7× FOS/>3000× LMV | [30, 34] |
Abbreviations: ACV, acyclovir; cCMV, congenital cytomegalovirus infection; CDV, cidofovir; CMV, cytomegalovirus; d, days; FCV, faldaprevir; FOS, foscarnet; HSCT, hematopoietic stem cell transplant; IgG, CMV-specific immunoglobulin G; LMV, letermovir; LT, CMV-specific lymphocytes T infusion; MBV, maribavir; NP, new phenotype; VGCV/GCV, valganciclovir/ganciclovir.
NOTE: New phenotype mutation in this study are indicated in bold and by NP resistance level.
Number of patients infected with the human cytomegalovirus mutant indicated, receiving the transplant type, receiving the treatment indicated.
Days of cumulative treatment until the time of mutation detection are indicated as the median when n ≥ 2 and the (Q1; Q3) when n > 2.
Days until detection are calculated from the transplant date until the detection of the variant in the clinical sample by sequencing are indicated as the median when n ≥ 2 and the (Q1; Q3) when n > 2.
Viral loads of the clinical sample in which the variant was detected are indicated as the median when n ≥ 2 and the (Q1; Q3) when n > 2 in IU/mL.
Level of resistance is indicated as fold-shift increase of the effective concentration 50% (EC50) of the mutant compared with the CMV control strain.
Resistance Mutations Detected in Association With Current Antiviral Drugs in the Overall Study Population
| Gene | Mutation | Na | Transplanta | Days of Cumulative Treatmenta,b | Days Until Detectionc | Viral Load at the Detection (IU/mL)d | Resistance Levele | Reference |
|---|---|---|---|---|---|---|---|---|
| UL97 | M460V | 2 | HSCT, liver | 19d VGCV/GCV (n = 2) | 493 | 5.82E+03 | 5–10× GCV | [24] |
| UL97 | C480F | 2 | HSCT, kidney | 60d MBV (n = 2), 67 GCV (n = 2) | 222 | 9.97E+03 | 2–5× GCV, 223× MBV | [8, 9] |
| UL97 | C592G | 1 | Kidney | 138d VGCV | 235 | 9.87E+03 | 2–5× GCV, 2–5× FCV | [25] |
| UL97 | A594V | 5 | Kidney (n = 2), liver (n = 2), lung | 56d (14; 121) VGCV/GCV (n = 5), 52d FOS + 51d IgG (n = 1) | 137 (100; 156) | 2.03E+04 (1.20E+04; 3.68E+04) | 5–10× GCV | [26] |
| UL97 | A594P | 1 | Heart | 24d GCV | 534 | 1.84E+04 | 5–20× GCV | [25] |
| UL97 | L595S | 5 | HSCT, heart, lung (n = 2), kidney | 38d (20; 54) VGCV/GCV (n = 5), 36d FOS + 16d IgG (n = 1) | 158 (66; 357) | 3.87E+04 (2.29E+04; 5.80E+04) | 5× GCV | [27] |
| UL97 | L595W + A594V | 1 | Heart | 14d VGCV | 110 | 4.12E+03 | 5.1× GCV | [27] |
| UL54 | V781I | 1 | HSCT | 38d GCV + 79d FOS + LT | 153 | 1.63E+03 | 1–4× GCV, 4–5.2× FOS | [28] |
| UL54 | A928T | 1 | cCMV + 2 HSCT | 43d GCV + 23d FOS | 145 | 2.17E+04 | NP | … |
| UL54 | L773V + G841A | 1 | HSCT | 35d VGCV/GCV + 45d FOS + 10d ACV | 133 | 3.55E+03 | 2× GCV, 5× FOS/3.2× GCV, 2.6× CDV, 4.3× FOS | [29, 30] |
| UL54 + UL97 | D413N + M460I | 1 | Lung | 24d GCV | 668 | 6.20E+04 | 6.5× GCV, 11× CDV | [31] |
| UL54 + UL97 | A543V + M460I | 2 | Kidney (n = 2) | 64d VGCV/GCV + 9d FOS | 185 | 7.01E+04 | NP/5–10× GCV | [32] |
| UL54 + UL97 | A987G + C603W | 1 | Heart | 150d VGCV prophylaxis + 84d VGCV | 238 | 1.90E+04 | 6.8× GCV, 5.3× CDV/8.3× GCV | [28, 33] |
| UL54 + UL56 | T700A + C325F | 1 | HSCT | 33d VGCV/GCV + 74d FOS + 4d CDV + 27d LMV + LT | 198 | 3.41E+03 | 4.7× FOS/>3000× LMV | [30, 34] |
| Gene | Mutation | Na | Transplanta | Days of Cumulative Treatmenta,b | Days Until Detectionc | Viral Load at the Detection (IU/mL)d | Resistance Levele | Reference |
|---|---|---|---|---|---|---|---|---|
| UL97 | M460V | 2 | HSCT, liver | 19d VGCV/GCV (n = 2) | 493 | 5.82E+03 | 5–10× GCV | [24] |
| UL97 | C480F | 2 | HSCT, kidney | 60d MBV (n = 2), 67 GCV (n = 2) | 222 | 9.97E+03 | 2–5× GCV, 223× MBV | [8, 9] |
| UL97 | C592G | 1 | Kidney | 138d VGCV | 235 | 9.87E+03 | 2–5× GCV, 2–5× FCV | [25] |
| UL97 | A594V | 5 | Kidney (n = 2), liver (n = 2), lung | 56d (14; 121) VGCV/GCV (n = 5), 52d FOS + 51d IgG (n = 1) | 137 (100; 156) | 2.03E+04 (1.20E+04; 3.68E+04) | 5–10× GCV | [26] |
| UL97 | A594P | 1 | Heart | 24d GCV | 534 | 1.84E+04 | 5–20× GCV | [25] |
| UL97 | L595S | 5 | HSCT, heart, lung (n = 2), kidney | 38d (20; 54) VGCV/GCV (n = 5), 36d FOS + 16d IgG (n = 1) | 158 (66; 357) | 3.87E+04 (2.29E+04; 5.80E+04) | 5× GCV | [27] |
| UL97 | L595W + A594V | 1 | Heart | 14d VGCV | 110 | 4.12E+03 | 5.1× GCV | [27] |
| UL54 | V781I | 1 | HSCT | 38d GCV + 79d FOS + LT | 153 | 1.63E+03 | 1–4× GCV, 4–5.2× FOS | [28] |
| UL54 | A928T | 1 | cCMV + 2 HSCT | 43d GCV + 23d FOS | 145 | 2.17E+04 | NP | … |
| UL54 | L773V + G841A | 1 | HSCT | 35d VGCV/GCV + 45d FOS + 10d ACV | 133 | 3.55E+03 | 2× GCV, 5× FOS/3.2× GCV, 2.6× CDV, 4.3× FOS | [29, 30] |
| UL54 + UL97 | D413N + M460I | 1 | Lung | 24d GCV | 668 | 6.20E+04 | 6.5× GCV, 11× CDV | [31] |
| UL54 + UL97 | A543V + M460I | 2 | Kidney (n = 2) | 64d VGCV/GCV + 9d FOS | 185 | 7.01E+04 | NP/5–10× GCV | [32] |
| UL54 + UL97 | A987G + C603W | 1 | Heart | 150d VGCV prophylaxis + 84d VGCV | 238 | 1.90E+04 | 6.8× GCV, 5.3× CDV/8.3× GCV | [28, 33] |
| UL54 + UL56 | T700A + C325F | 1 | HSCT | 33d VGCV/GCV + 74d FOS + 4d CDV + 27d LMV + LT | 198 | 3.41E+03 | 4.7× FOS/>3000× LMV | [30, 34] |
Abbreviations: ACV, acyclovir; cCMV, congenital cytomegalovirus infection; CDV, cidofovir; CMV, cytomegalovirus; d, days; FCV, faldaprevir; FOS, foscarnet; HSCT, hematopoietic stem cell transplant; IgG, CMV-specific immunoglobulin G; LMV, letermovir; LT, CMV-specific lymphocytes T infusion; MBV, maribavir; NP, new phenotype; VGCV/GCV, valganciclovir/ganciclovir.
NOTE: New phenotype mutation in this study are indicated in bold and by NP resistance level.
Number of patients infected with the human cytomegalovirus mutant indicated, receiving the transplant type, receiving the treatment indicated.
Days of cumulative treatment until the time of mutation detection are indicated as the median when n ≥ 2 and the (Q1; Q3) when n > 2.
Days until detection are calculated from the transplant date until the detection of the variant in the clinical sample by sequencing are indicated as the median when n ≥ 2 and the (Q1; Q3) when n > 2.
Viral loads of the clinical sample in which the variant was detected are indicated as the median when n ≥ 2 and the (Q1; Q3) when n > 2 in IU/mL.
Level of resistance is indicated as fold-shift increase of the effective concentration 50% (EC50) of the mutant compared with the CMV control strain.
The incidence of sensitive polymorphisms, previously described to be sensitive to antivirals, was 76 of 94 (80.9%): 5 (6.6%) patients presented them in UL97, 67 (88.2%) in UL54, and 4 (5.3%) in UL56 (Table 1, Supplementary Table 1).
Fourteen previously undescribed genetic variants were found in 16 of 94 (17.0%) patients, all located in UL54, but 1 was in UL56 after LMV therapy (Table 3, Supplementary Table 1). Each genetic variant was detected in 1 patient, except for UL54 A543V and S883I, which were detected in 2 different individuals. Five UL54 and 1 UL56 variants located outside hypervariable regions were selected for recombinant phenotyping and 3D protein modeling. The UL56 P800L substitution was previously described by Champier et al [23] in 1 LMV-naive patient. However, because it was detected in association with a LMV resistance mutation in this study, we wanted to measure its potential impact on decreasing sensitivity to LMV and on viral replicative capacity.
Results of Antiviral Susceptibility and Replicative Capacity Assays of Novel HCMV Genetic Variants
| Gene | Mutation | Na | Patient History | Antiviral Susceptibility (RI)b | |||
|---|---|---|---|---|---|---|---|
| GCV | CDV | FOS | LMV | ||||
| UL54 | G441S | 1 | CVID | 0.67 (±0.24) | 1 (±0.75) | 1.13 (±0.8) | … |
| UL54 | F460S | 1 | HSCT | 0.86 (±0.45) | 0.88 (±0.78) | 1.11 (±0.8) | … |
| UL54 | A543V | 2 | Kidney (2) | 1.5 (±0.1) | 10 (±5.37) | 1.21 (±0.27) | … |
| UL54 | R512C | 1 | Kidney | 1 | 1 | 0.74 | … |
| UL54 | A928T | 1 | cCMV + 2 HSCT | >3 | >3 | >3 | … |
| UL56 | F345L | 1 | HSCT | 1.42 (±1.07) | 1.67 (±46) | 0.98 (±0.04) | 0.99 (±0.04) |
| UL56 | P800L | 1 | HSCT | 0.23 | 0.13 | 1 | 1.11 |
| Gene | Mutation | Na | Patient History | Antiviral Susceptibility (RI)b | |||
|---|---|---|---|---|---|---|---|
| GCV | CDV | FOS | LMV | ||||
| UL54 | G441S | 1 | CVID | 0.67 (±0.24) | 1 (±0.75) | 1.13 (±0.8) | … |
| UL54 | F460S | 1 | HSCT | 0.86 (±0.45) | 0.88 (±0.78) | 1.11 (±0.8) | … |
| UL54 | A543V | 2 | Kidney (2) | 1.5 (±0.1) | 10 (±5.37) | 1.21 (±0.27) | … |
| UL54 | R512C | 1 | Kidney | 1 | 1 | 0.74 | … |
| UL54 | A928T | 1 | cCMV + 2 HSCT | >3 | >3 | >3 | … |
| UL56 | F345L | 1 | HSCT | 1.42 (±1.07) | 1.67 (±46) | 0.98 (±0.04) | 0.99 (±0.04) |
| UL56 | P800L | 1 | HSCT | 0.23 | 0.13 | 1 | 1.11 |
Abbreviations: cCMV, congenital cytomegalovirus infection; CDV, cidofovir; CVID, common variable immunodeficiency; FOS, foscarnet; GCV, ganciclovir; HCMV, human cytomegalovirus; HSCT, hematopoietic stem cell transplant; LMV, letermovir; RI, resistance index.
NOTE: RI >3 is considered drug resistant and is indicated in bold.
Number of patients in whom the mutation was detected.
The RI is the half-maximal effective concentration ([EC50] µM) value for the mutant strain divided by the EC50 of the AD169 HCMV control strain. Each mutant was checked against different antiviral concentrations in triplicate. Each individual experiment was repeated 3 times for each mutant, except for R512C and P800L, which could only be performed once. Data are indicated as the mean of the 3 technical replicates.
Results of Antiviral Susceptibility and Replicative Capacity Assays of Novel HCMV Genetic Variants
| Gene | Mutation | Na | Patient History | Antiviral Susceptibility (RI)b | |||
|---|---|---|---|---|---|---|---|
| GCV | CDV | FOS | LMV | ||||
| UL54 | G441S | 1 | CVID | 0.67 (±0.24) | 1 (±0.75) | 1.13 (±0.8) | … |
| UL54 | F460S | 1 | HSCT | 0.86 (±0.45) | 0.88 (±0.78) | 1.11 (±0.8) | … |
| UL54 | A543V | 2 | Kidney (2) | 1.5 (±0.1) | 10 (±5.37) | 1.21 (±0.27) | … |
| UL54 | R512C | 1 | Kidney | 1 | 1 | 0.74 | … |
| UL54 | A928T | 1 | cCMV + 2 HSCT | >3 | >3 | >3 | … |
| UL56 | F345L | 1 | HSCT | 1.42 (±1.07) | 1.67 (±46) | 0.98 (±0.04) | 0.99 (±0.04) |
| UL56 | P800L | 1 | HSCT | 0.23 | 0.13 | 1 | 1.11 |
| Gene | Mutation | Na | Patient History | Antiviral Susceptibility (RI)b | |||
|---|---|---|---|---|---|---|---|
| GCV | CDV | FOS | LMV | ||||
| UL54 | G441S | 1 | CVID | 0.67 (±0.24) | 1 (±0.75) | 1.13 (±0.8) | … |
| UL54 | F460S | 1 | HSCT | 0.86 (±0.45) | 0.88 (±0.78) | 1.11 (±0.8) | … |
| UL54 | A543V | 2 | Kidney (2) | 1.5 (±0.1) | 10 (±5.37) | 1.21 (±0.27) | … |
| UL54 | R512C | 1 | Kidney | 1 | 1 | 0.74 | … |
| UL54 | A928T | 1 | cCMV + 2 HSCT | >3 | >3 | >3 | … |
| UL56 | F345L | 1 | HSCT | 1.42 (±1.07) | 1.67 (±46) | 0.98 (±0.04) | 0.99 (±0.04) |
| UL56 | P800L | 1 | HSCT | 0.23 | 0.13 | 1 | 1.11 |
Abbreviations: cCMV, congenital cytomegalovirus infection; CDV, cidofovir; CVID, common variable immunodeficiency; FOS, foscarnet; GCV, ganciclovir; HCMV, human cytomegalovirus; HSCT, hematopoietic stem cell transplant; LMV, letermovir; RI, resistance index.
NOTE: RI >3 is considered drug resistant and is indicated in bold.
Number of patients in whom the mutation was detected.
The RI is the half-maximal effective concentration ([EC50] µM) value for the mutant strain divided by the EC50 of the AD169 HCMV control strain. Each mutant was checked against different antiviral concentrations in triplicate. Each individual experiment was repeated 3 times for each mutant, except for R512C and P800L, which could only be performed once. Data are indicated as the mean of the 3 technical replicates.
Phenotypic Results
For antiviral drug susceptibility assays, the resistance index of each mutant was calculated by dividing the EC50 value (µM) of the mutant strain by the EC50 of the AD169 HCMV control strain (Table 3). Replicative capacity assays were performed for each mutant and in parallel with the AD169 for each repetition (Table 3, Figure 1).
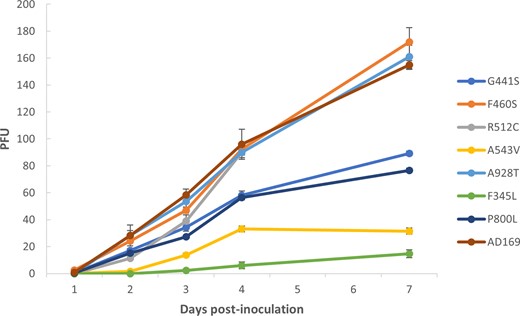
Growth capacity of newly characterized human cytomegalovirus variants. Each recombinant cytomegalovirus (CMV) strain was compared with an AD169 CMV control strain. Each strain was inoculated at an equal multiplicity of infection of 0.01. The plaque-forming units (PFUs) were counted from days 1 to 4 and on day 7 postinoculation. Data shown are the mean of 3 replicates set up simultaneously, with the corresponding standard deviation. The AD169 curve is the mean of 7 different experiments (1 repetition per variant).
UL54 A928T conferred high triple-resistance to GCV, FOS, and CDV at a level of resistance that any antiviral concentration could not inhibit 50% of the replication of the A928T mutant. UL54 A543V conferred 10-fold decreased susceptibility to CDV, and it impaired growth capacity. The 3 remaining UL54 (G441S, F460S, and R512C) were sensitive to GCV, FOS, and CDV, but G441S involved a defective replicative capacity (Figure 1).
None of the phenotypes of the UL56 mutations (F345L, P800L) conferred reduced response to LMV or cross-resistance to the DNA polymerase inhibitors tested. However, both mutations involved lower viral kinetics of replication (Figure 1).
Correlation of Newly Characterized Phenotype Mutations With Clinical Outcomes
UL54 A543V was detected in 2 kidney transplant recipients in combination with the known GCV-resistant mutation UL97 M460I (Figure 2A). In one patient, this mutation was observed after 90 days of GCV and 9 days of FOS treatment, reaching viral loads of 137 000 IU/mL. In the other patient, the mutation was detected after 37 days of prophylaxis with VGCV, with a viral load of 3100 IU/mL. The phenotypic results showed A543V conferred a defective replicative capacity and resistance to CDV (Table 3), which was not administered to either subject.
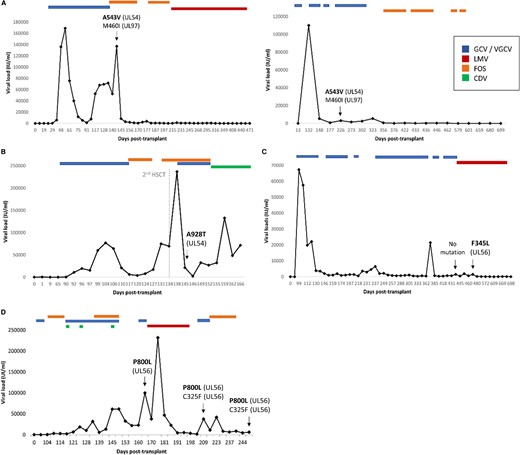
Clinical follow-up of patients with newly characterized phenotype mutations. Viral loads in IU/mL were tracked against days after transplantation. Anti-human cytomegalovirus treatment outset and end are indicated. Genotypic assays were performed in the clinical samples indicated with an arrow. New phenotype mutations in UL54 (A and B) and UL56 (C and D) are indicated in bold, together with previously characterized resistant mutations. CDV, cidofovir; FOS, foscarnet; GCV, ganciclovir; LMV, letermovir; VGCV, valganciclovir.
UL54 A928T was detected in a plasma sample with a viral load of 21 732 IU/mL in a congenital HCMV individual after 2 HSCTs (145 days after the first intervention, and 11 after the second) (Figure 2B). This mutation was detected after cumulative treatment of 43 days of GCV and 23 days of FOS. Cidofovir was administered afterwards because of adenovirus reactivation and was maintained until limitation of therapeutic efforts; however, HCMV DNA clearance was not achieved until its death. This clinical unresponsiveness correlated with the in vitro phenotypic results of GCV, CDV, and FOS multi-resistance.
A UL56 F345L missense mutation was located within the LMV resistance region (residues 230–370) (Supplementary Figure 1) in a HSCT recipient after 209 days of VGCV/GCV and 20 days of LMV but not before LMV therapy. Viral loads were 3 log10 IU/mL and were well controlled until HCMV DNA cleared after 148 days with LMV (Figure 2C). This mutation conferred a highly impaired replicative capacity and was sensitive to GCV, FOS, CDV, and LMV in concordance with the clinical antiviral response and clearance after the detection of F345L (Table 3).
P800L was detected in an HSCT recipient at day 163 post-transplant, 7 days before LMV therapy onset (Figure 2D). After LMV was administered for 28 days, the high level LMV-resistant mutation C325F emerged in combination with the previously detected P800L polymorphism. Despite discontinuation of LMV, both mutations persisted until the patient developed multiorgan failure.
Follow-up of viral loads and antiviral treatment could not be retrieved for the subject with the UL54 G441S variant. The clinical data of patients infected with wild-type phenotypes characterized in this study are not presented.
Predicting the Phenotype of New UL54 Variants by 3D Protein Modeling
UL54 3D protein modeling showed that the amino acids G441S (Figure 3A) and F460S (Figure 3B) were localized in nonstructured regions of UL54 DNA polymerase. These domains are outside conserved regions, which agrees with the absence of impact on antiviral resistance obtained by phenotypic methods. A543V (Figure 3C) and R512C (Figure 3D) are part of a helix. Removing 1 amino acid of a helix can change repartition of amino acids exposed to solvent or the interior of the protein. Both are localized in the Exo-III/delta-C region but are associated with a completely different impact on antiviral resistance as shown by recombinant phenotyping. A928T (Figure 3E, Supplementary Figure 2) is part of a helix between region I and VII of the palm domain. The change of an alanine to a threonine impacts both the polarity and the size of the amino acid, modifying the potential interaction between UL54 and antivirals.
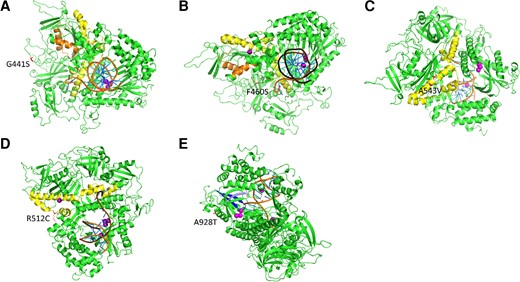
Three-dimensional protein models showing the location of newly characterized UL54 mutations. Theoretical structures of UL54 calculated with MODELLER are represented in cartoon mode. The different domains close to the new mutations are colored as follows: residues 379–421 of region IV/Exo-II in orange, residues 492–588 of region delta-C/Exo-III in yellow, residues 905–919 of region I in blue, and residues 962–970 of region VII in gray. Purple dots in active sites represent metal ions. The DNA duplex is colored in orange and blue. (A) The G441S mutation in red between the Exo-II and Exo-III regions. (B) The A543V mutation in red in the Exo-III region. (C) The F460S mutation in red between the Exo-II and the Exo-III regions. (D) The R512C mutation in red in the Exo-III region. (E) The A928T mutation in red between regions I and VII.
DISCUSSION
The results of this study demonstrate a high incidence of antiviral-resistant mutations in patients with refractory HCMV infection (n = 25 of 94, 26.6%). Considering all mutations emerged in transplant patients, the resulting incidence in this subpopulation is higher (n = 25 of 83, 30.1%). Recombinant phenotyping of previously uncharacterized mutations showed that the UL54 A928T mutation conferred high-level resistance to GCV, FOS, and CDV, and UL54 A543V conferred intermediate resistance to CDV. In addition, G441S and A543V located in UL54, and F345L and P800L in UL56 variants showed lower growth capacities than wild-type AD169 HCMV.
Uncharacterized mutations were selected for the correlation of their gene position to antiviral resistance. G441S and F460S were located in the region associated with GCV and CDV cross-resistance (Supplementary Figure 2), but neither conferred resistance to these antivirals, as predicted in silico. In agreement, the F460L variant, located in the same residue, was described to be sensitive (1.2-fold shift) to DNA-polymerase inhibitors and did not show a slow-growth phenotype [4].
A543V and R512C were mutations located in the Exo-III/delta-C domain. This domain has been described as being involved in exonuclease and polymerase catalytic function, which are well conserved among mammals and yeast, and was associated with resistance to GCV, FOS, and CDV (Supplementary Figure 2) [24]. Our study showed A543V conferred CDV resistance and impaired its viral growth, whereas a previously reported A543S mutation had a wild-type phenotype [4], suggesting that when alanine is substituted by valine, its bulky lateral aliphatic chain could block its polymerase function, but the small hydroxyl group of the serine does not. Therefore, residue A543 seems to play an important role in viral growth. However, this variant clearly replicated in the 2 clinical cases presented (Figure 2A), suggesting an A543V growth defect could be compensated by other mutations, such as UL97 M460I.
The A928T mutation, located just outside the catalytic polymerase function in a region supposedly associated with antiviral susceptibility, conferred a high level of resistance to 3 DNA polymerase inhibitors (Supplementary Figure 2). Previous reports showed certain triple-resistance mutations in the surroundings, such as A836P and del981-2, detected in patients receiving prolonged therapy and causing loss of growth fitness [33]. In contrast, many variants in the nearby amino acids (V902G, E903G, K947E, M959T) were susceptible to GCV, FOS, and CDV [4]. This result also strengthens the importance of discerning high from intermediate-low levels of resistance, because it influences the dosages and antiviral molecules chosen in clinical practice.
UL56 mutations emerged in the context of infections unresponsive to standard therapy and the use of LMV as salvage therapy. F345L was located inside the LMV resistance region (Supplementary Figure 1) and strongly impaired viral replication, but surprisingly it did not involve a loss of response to the different antiviral molecules tested. This suggests that the F345 residue is critical for biological terminase function but not for interaction with LMV, as has been described for R215, because R215C amino acid substitution increased growth capacity but did not confer loss of response to LMV [14].
The P800L polymorphism emerged before LMV therapy and remained detectable until the patient’s death after 28 days of LMV in association with the C325F resistance mutation (Figure 2D), suggesting the need to measure its potential impact on decreasing sensitivity to LMV and on viral replicative capacity. Phenotypic assays showed this mutation impaired virus growth and was confirmed as having no effect on virus sensitivity to LMV. As the P800L + C325F variant continued replicating, the defective growth associated with P800L alone seemed to be compensated by the emergence of C325F, the residue of which appeared to be critical for LMV binding but unimportant for HCMV terminase function.
It has been described that UL54 mutations can confer multiresistance and may emerge after a previous UL97 mutation, enhancing the level of resistance [18], as shown in 4 SOT recipients in our cohort. However, the order of emergence of mutations could not be determined because only 1 sample was collected. In addition, this study presents the uncommon detection of 2 UL54 resistance mutations with no previous change in UL97 and the combination of mutations in UL54 together with UL56 after GCV + LMV therapy.
Fewer sensitive polymorphisms arose in UL97 than in UL54 during standard treatment, supporting the existing literature [6, 11], and barely any were detected in UL56, in agreement with the previously described UL56 genetic conservation among herpesvirus before LMV therapy [14, 35]. However, the prevalence of sensitive polymorphisms in UL56 and UL97 was understated because full-length genes were not amplified and only the regions associated with drug-resistance were sequenced. This study was limited by the incapacity to recover previous isolates of the patients infected with the newly characterized variants to determine the timing of the earliest emergence of the respective mutations. Phenotyping assays involve long, laborious work, and results were obtained months after the genotypic resistance testing was requested. Phenotyping the novel mutations found in the UL54 would be interesting to confirm the absence of resistant mutations in highly variable regions.
Three-dimensional protein modeling is a useful and fast tool to predict the impact of genetic variants in silico. It correctly predicted the effect of A928T, A543V, G441S, and F460S mutations on antiviral susceptibility, but not with the R512C mutation, highlighting its limitations and the necessity of confirmation by recombinant phenotyping, especially when it concerns clinical practice.
CONCLUSIONS
This study reinforces that genotype-phenotype correlation studies not only serve to define the level of antiviral resistance and viral kinetics associated with each genetic variant, but they also determine the biological role of each residue and allow constructing comprehensive and reliable mutation maps to help develop new anti-HCMV therapies. The high incidence of HCMV resistance mutations in transplant recipients presents a worrisome scenario for their clinical management. However, early genotypic testing and increasing databases of mutations by phenotyping can optimize treatments and improve the assessment of patients with refractory/resistant HCMV infection.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. We thank all the participating hospitals endorsed by the Spanish Network for Research in Infectious Diseases (REIPI) and the Group for the Study of Infection in Transplantation (GESITRA) and the work of the investigators of these groups: Francisco López-Medrano, Jose María Agüado, Cecilia Martin-Gandul, Jordi Carratalá, Jordí Niubó, Carlos Cervera, Patricia Muñoz, María Carmen Fariñas, Andrés Antón, Miguel Montejo, Pilar Pérez-Romero, and Julián Torres-Cisneros. We also thank Donna Pringle for her contribution to English language editing. Finally, we thank the Merck Sharp & Dohme (MSD) pharmaceutical company for providing Letermovir and Montserrat Tuset from the Hospital Pharmacy Unit of the Hospital Clinic of Barcelona for providing ganciclovir, cidofovir, and foscarnet.
Financial support. The research was carried out in the Hospital Clinic of Barcelona, Spain and was funded by “Fondo de Investigación en Salud (FIS) PI 17/0150 of the Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación del Gobierno de España”, the Agency for Health Technology Assessment, and “Ministerio de Economía y Competitividad”, by “Fundació La Marató” 201824 (21/267), by “Centro de Investigación Biomédica en Red” (CIBER) CB21/13/0081 and “Agencia de Gestión de Ayudas Universitarias y de Investigación” (AGAUR). The work performed in the French National Center in Limoges, France, was funded by the “Santé Publique France” and Inserm for the UMR1092.
References
Author notes
Potential conflict of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Conflicts of Interest.


{kind=link}
{kind=link}
{kind=link}