






Abstract
Infection with varicella zoster virus (VZV) may involve different central nervous system (CNS) manifestations, including meningitis, encephalitis, and vasculitis. In cases in which otherwise healthy individuals are affected, an inborn error of immunity may underlie increased susceptibility or severity of infection.
We collected a cohort of 17 adults who experienced VZV encephalitis and performed whole exome sequencing. Patient peripheral blood mononuclear cells were infected with VZV, and innate antiviral interferon (IFN) and cytokine responses as well as viral replication were evaluated. Data were analyzed by Mann-Whitney U test.
We identified a total of 21 different potentially disease-causing variants in a total of 13 of the 17 patients included. These gene variants were within 2 major functional clusters: (1) innate viral sensors and immune pathways and (2) autophagy pathways. Antiviral IFN and cytokine responses were abnormal in the majority of patients, whereas viral replication was increased in only 2 of 17 patients.
This study identifies a list of variants of pathogenic potential, which may serve as a platform for generating hypotheses for future studies addressing genetic and immunological factors associated with susceptibility to VZV encephalitis. These data, taken together, suggest that disturbances in innate sensing and autophagy pathways may predispose to VZV encephalitis.
Varicella zoster virus (VZV) is a human (deoxyribonucleic acid (DNA) virus of the alphaherpesvirus subfamily and the causative agent of varicella (chickenpox) [1]. Primary infection with VZV is followed by latency in peripheral ganglia from where the virus may reactivate decades later and cause herpes zoster (shingles) [2]. Varicella zoster virus reactivation is estimated to occur in 1 of 4 seropositive individuals and is associated with immunosuppression and age above 65 years [2]. In rare cases, VZV may spread to the central nervous system (CNS) and cause neurological complications, including meningitis, encephalitis, meningoencephalitis, or vasculitis [3]. Varicella zoster virus encephalitis can occur both during primary infection or reactivation, it is observed in approximately 1 in 10 000 of all VZV infections, and it is associated with serious neurological sequelae, such as stroke, hemiparesis, and cognitive impairment, including attention and memory deficits years after the infection [3]. Whereas immunosenescence and other causes of immunosuppression from malignancy or certain medications are well recognized, VZV infection in the CNS is also sometimes observed in younger, otherwise apparently healthy individuals and children, suggesting a role for inborn errors of immunity (IEI) in the pathogenesis in these cases [4, 5].
The pathogenesis of VZV CNS invasion and infection remains incompletely understood. In principle, factors in both host and virus are likely of importance to the quite rare involvement of the CNS. However, despite sequencing of the complete genomes of a large number of clinical VZV strains and the finding of increased viral diversity among CNS strains, no specific viral mutations have been found that can explain CNS engagement. On the other hand, several host factors are known to play a role in conferring protective immunity to VZV. First, it is well established that cellular immunity plays an essential role, because individuals with inborn errors affecting natural killer cells and T cells may experience severe and disseminated infection with VZV [5, 6]. This includes, but is not limited to, genetic defects in DOCK2, DOCK8, CORO1A, MCM4, and STK4 [7–12]. Because cellular immunity plays a fundamental role in the host, patients with defects in these genes often present with a wide variety of viral and bacterial infections in addition to VZV. Moreover, defects in IFNGR1, STAT1, and TYK2 lead to severe and disseminated VZV infection together with mycobacterial disease by disrupting macrophage responses against intracellular pathogens [13–15].
More recently, it has been acknowledged that innate immunity also plays a key role in restricting and controlling VZV infection, through early immune recognition of VZV and subsequent production of antiviral interferons (IFNs) [16]. The discovery of defects in the innate DNA sensor Ribonucleic acid (RNA) polymerase III (POL III) in the genes POLR3A and POLR3C in children with severe VZV pneumonitis and CNS infection demonstrated an important role of innate recognition of POL III and induction of type I IFN in protection against severe VZV disease [17]. Ribonucleic acid POL III recognizes AT-rich DNA and produces 5’ppp RNA species that serve as a substrate for RIG-I, which induces transcription of type I and III IFNs [18]. Subsequently, a genetic variant in another POL III component, POLR3F, was described in monozygotic twins with VZV reactivation and recurrent VZV CNS infection and vasculitis [19], followed by the description of POL III variants in adults with VZV encephalitis [20]. These findings together suggested a role of POL III in sensing of VZV DNA and triggering antiviral responses to VZV, not only during primary infection in children but also in viral reactivation from latency in adults [21]. Finally, a recent study described a heterozygous variant in TLR3 in a patient with recurrent herpes zoster ophthalmicus [22].
The majority of these abnormalities in innate immunity has been described in single patients or family studies. The proportion of genetic variants in otherwise healthy individuals with VZV encephalitis is unknown. To further explore the genetic and immunological basis of susceptibility to VZV CNS infection, in this study we collected a cohort of 17 younger or middle-aged patients with VZV encephalitis, some of whom also presented with vasculitis or Ramsay Hunt syndrome, and performed whole exome sequencing (WES). Based on current knowledge on VZV immunity the whole exome dataset was analyzed, leading to the identification of several potentially deleterious variants in genes related to innate immune responses and autophagy pathways. Finally, these data were combined with VZV infection of patient peripheral blood mononuclear cells (PBMCs) and examination of antiviral responses and VZV replication.
METHODS
Patient Cohort
The patient cohort consisted of 17 patients of Caucasian origin with clinical symptoms of CNS infection and documented pleocytosis and were polymerase chain reaction positive for VZV DNA or VZV immunoglobulin G index in the cerebrospinal fluid (CSF). Whole blood was collected by ambulatory contact after months to years after recovery or in the case of P13 at the end of hospitalization after having recovered from acute infection. (See Supplementary Methods for further details.)
Whole Exome Sequencing and Bioinformatics
Whole exome sequencing (WES) was conducted on genomic DNA from all study participants using KAPA HTP library preparation and Nimblegen SeqCap EZ MedExome Plus kits and analyzed with NextSeq version 2 chemistry (2 × 150 base pairs) (Ilumina). See Supplementary Methods for details.
Varicella Zoster Virus Infection of Peripheral Blood Mononuclear Cells
Patient and control PBMCs were thawed and seeded in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin (complete RPMI [cRPMI]) at a density of 5 × 105 cells/well in 96-well plates and left to incubate overnight at 37°C and 5% CO2. Viability was approximately 80%. Varicella zoster virus-infected MeWo cells (VZV recombinant Oka strain, ROka strain; no. VR-1832, American Type Culture Collection [ATCC]) or uninfected MeWo cells were thawed in cRPMI and spun down at 500 ×g for 5 minutes and resuspended in 1 mL cRPMI, after which they were added in a ratio of 1:0.4 PBMC/MeWo and spun down at 250 ×g for 10 minutes. Cells were incubated for 48 hours after harvest of supernatants for Meso Scale and cell lysis for RNA isolation.
Meso Scale Measurements of Supernatants
Expression of IFNs (IFNα2a, IFNβ, IFNλ1, and IFNγ), proinflammatory cytokines (interleukin [IL]-6, IL-1β, and tumor necrosis factor [TNF]α), and the IFN inducible protein CXCL10 were measured in cell culture supernatants using U-PLEX assays (Meso Scale Diagnostics) according to manufacturer’s protocols, on a Meso Quickplex SQ 120 instrument.
STRING Analysis
All final variant-containing genes were analyzed for known and predicted associations using the STRING analysis software (version 11.0; STRING Consortium 2020), which identifies physical and functional protein-protein associations [23].
Statistics
Experiments were performed in biological triplicates. The Mann-Whitney rank-sum test (GraphPad Prism 9) was used to determine whether there was a statistical significant difference between patient and pooled controls, and this approach was chosen, because the need for a non-parametric test for comparison of unpaired data.
Ethics
The patients and healthy controls were included after oral and written consent in accordance with The Helsinki Declaration and national ethics guidelines and after approval from the Danish National Committee on Health Ethics (no. 1-10-72-275-15), the Data Protection Agency, and Institutional Review Board and the Swedish Ethical Review Authority (no. 2019-02544).
Data Availability Statement
All data related to this study will be shared at the request of other investigators for purposes of replicating procedures and results, according to national and international General Data Protection Regulation (GDPR) rules and after individual Data Transfer Agreement (DTA) and Material Transfer Agreement (MTA) rules with relevant investigators.
RESULTS
Identification of Rare Genetic Variants by Whole Exome Sequencing in Patients With Varicella Zoster Virus Central Nervous System Infection
In this study, we included 17 patients of Caucasian ethnicity from Denmark and Sweden who had previously suffered from VZV encephalitis or meningoencephalitis. The patients were selected based on well defined VZV neuroinfection and for some of them the presence of additional complications, such as vasculitis or cranial nerve palsy. An overview of the demographic, clinical, and paraclinical findings in the patient cohort is presented in Table 1.
Demographics and Clinical Characteristics of the Cohort of Patients With VZV Encephalitis or Meningoencephalitis
| Patient Number | Agea, Sex | Country | Clinical Diagnosis | Medical History | MRI/CT-C Findings | CSF Findings | Date of Admission | Date of Sample Collection |
|---|---|---|---|---|---|---|---|---|
| P1 | 56, male | Sweden | Meningoencephalitis | Deep venous thrombosis | MRI and CT-C with one-sided prominent temporal horn. No signs of encephalitis | Cell count: 58/mm3 | August 16, 2007 | October 25, 2019 |
| P2 | 20, female | Sweden | Meningoencephalitis and Ramsay Hunt syndrome | Healthy, recent child delivery | CT-angiography normal, MRI ND | Cell count: 728/mm3 | June 29, 2011 | October 29, 2019 |
| P3 | 38, male | Sweden | Meningoencephalitis | Healthy | CT-C normal, MRI ND | Cell count: 594/mm3 | January 20, 2016 | November 1, 2019 |
| P4 | 22, male | Sweden | Meningoencephalitis, vasculitis with ischemic stroke | Migraine, asthma | CT-angiography with left-sided temporal infarction, MRI normal | Cell count: 716/mm3 | September 07, 2016 | November 8, 2019 |
| P5 | 58, male | Sweden | Vasculitis with subarachnoid hemorrhage | Healthy | MRI and CT-C with one-sided prominent temporal horn. No signs of encephalitis | Cell count: 181/mm3 | January 26, 2004 | November 8, 2019 |
| P6 | 28, male | Sweden | Meningoencephalitis | Healthy | CT-angiography normal, MRI ND | Cell count: 274/mm3 | February 22, 2015 | November 13, 2019 |
| P7 | 37, female | Sweden | Meningoencephalitis | Chronic posttraumatic neck and back pain, migraine, chronic fatigue syndrome, earlier motoric function loss diagnosed as dissociative motor disorder | CT-C normal, MRI ND | Cell count: 376/mm3 | September 15, 2015 | November 13, 2019 |
| P8 | 74, female | Sweden | Meningoencephalitis, Ramsay Hunt syndrome and vasculitis | Migraine, anxiety | CT-angiography with left-sided temporal infarction, MRI normal | Cell count: 297/mm3 | May 10, 2019 | November 13, 2019 |
| P9 | 4, female | Sweden | Encephalitis | Asthma | MRI and CT-C with one-sided prominent temporal horn | Cell count: 102/mm3 | April 21, 2016 | November 19, 2019 |
| P10 | 32, female | Sweden | Meningoencephalitis | Healthy | CT-angiography normal MRI ND | Cell count: 266/mm3 | January 30, 2016 | November 29, 2019 |
| P11 | 58, male | Denmark | Meningoencephalitis | Healthy | MRI and CT-C normal | Cell count: 142/mm3 | February 8, 2019 | June 12, 2020 |
| P12 | 21, female | Denmark | Encephalitis | Healthy | MRI (+angiography) normal | Cell count: 585/mm3 | June 14, 2016 | July 29, 2019 |
| P13 | 36, female | Denmark | Meningoencephalitis | Polycystic ovary syndrome | White matter lesions | Cell count: 264/mm3 | October 23, 2019 | November 14, 2019 |
| P14 | 56, male | Denmark | Encephalitis | Rheumatoid arthritis treated with Mtx | MRI normal | Cell count: 115/mm3 | June 10, 2016 | June 30, 2019 |
| P15 | 45, male | Sweden | Meningoencephalitis, vasculitis with ischemic stroke | Depression | MRI with left-sided thalamic and occipital ischemic lesions, angiography with no signs of vasculitis, but a narrow arteria cerebri posterior sin, CT-C normal | Cell count: 75/mm3 | May 18, 2007 | October 25, 2019 |
| P16 | 25, male | Sweden | Meningoencephalitis | Delusional disorder, anxiety, recurrent depressive disorder | CT-C and MRI normal | Cell count: 446/mm3 | May 4, 2011 | November 8, 2019 |
| P17 | 42, female | Denmark | Encephalitis | Benign intracraniel hypertension | CT-C (+angiography): normal3 | Cell count: 107/mm3 | March 3, 2014 | January 20, 2020 |
| One prior episode of serous meningitis without microbial findings |
| Patient Number | Agea, Sex | Country | Clinical Diagnosis | Medical History | MRI/CT-C Findings | CSF Findings | Date of Admission | Date of Sample Collection |
|---|---|---|---|---|---|---|---|---|
| P1 | 56, male | Sweden | Meningoencephalitis | Deep venous thrombosis | MRI and CT-C with one-sided prominent temporal horn. No signs of encephalitis | Cell count: 58/mm3 | August 16, 2007 | October 25, 2019 |
| P2 | 20, female | Sweden | Meningoencephalitis and Ramsay Hunt syndrome | Healthy, recent child delivery | CT-angiography normal, MRI ND | Cell count: 728/mm3 | June 29, 2011 | October 29, 2019 |
| P3 | 38, male | Sweden | Meningoencephalitis | Healthy | CT-C normal, MRI ND | Cell count: 594/mm3 | January 20, 2016 | November 1, 2019 |
| P4 | 22, male | Sweden | Meningoencephalitis, vasculitis with ischemic stroke | Migraine, asthma | CT-angiography with left-sided temporal infarction, MRI normal | Cell count: 716/mm3 | September 07, 2016 | November 8, 2019 |
| P5 | 58, male | Sweden | Vasculitis with subarachnoid hemorrhage | Healthy | MRI and CT-C with one-sided prominent temporal horn. No signs of encephalitis | Cell count: 181/mm3 | January 26, 2004 | November 8, 2019 |
| P6 | 28, male | Sweden | Meningoencephalitis | Healthy | CT-angiography normal, MRI ND | Cell count: 274/mm3 | February 22, 2015 | November 13, 2019 |
| P7 | 37, female | Sweden | Meningoencephalitis | Chronic posttraumatic neck and back pain, migraine, chronic fatigue syndrome, earlier motoric function loss diagnosed as dissociative motor disorder | CT-C normal, MRI ND | Cell count: 376/mm3 | September 15, 2015 | November 13, 2019 |
| P8 | 74, female | Sweden | Meningoencephalitis, Ramsay Hunt syndrome and vasculitis | Migraine, anxiety | CT-angiography with left-sided temporal infarction, MRI normal | Cell count: 297/mm3 | May 10, 2019 | November 13, 2019 |
| P9 | 4, female | Sweden | Encephalitis | Asthma | MRI and CT-C with one-sided prominent temporal horn | Cell count: 102/mm3 | April 21, 2016 | November 19, 2019 |
| P10 | 32, female | Sweden | Meningoencephalitis | Healthy | CT-angiography normal MRI ND | Cell count: 266/mm3 | January 30, 2016 | November 29, 2019 |
| P11 | 58, male | Denmark | Meningoencephalitis | Healthy | MRI and CT-C normal | Cell count: 142/mm3 | February 8, 2019 | June 12, 2020 |
| P12 | 21, female | Denmark | Encephalitis | Healthy | MRI (+angiography) normal | Cell count: 585/mm3 | June 14, 2016 | July 29, 2019 |
| P13 | 36, female | Denmark | Meningoencephalitis | Polycystic ovary syndrome | White matter lesions | Cell count: 264/mm3 | October 23, 2019 | November 14, 2019 |
| P14 | 56, male | Denmark | Encephalitis | Rheumatoid arthritis treated with Mtx | MRI normal | Cell count: 115/mm3 | June 10, 2016 | June 30, 2019 |
| P15 | 45, male | Sweden | Meningoencephalitis, vasculitis with ischemic stroke | Depression | MRI with left-sided thalamic and occipital ischemic lesions, angiography with no signs of vasculitis, but a narrow arteria cerebri posterior sin, CT-C normal | Cell count: 75/mm3 | May 18, 2007 | October 25, 2019 |
| P16 | 25, male | Sweden | Meningoencephalitis | Delusional disorder, anxiety, recurrent depressive disorder | CT-C and MRI normal | Cell count: 446/mm3 | May 4, 2011 | November 8, 2019 |
| P17 | 42, female | Denmark | Encephalitis | Benign intracraniel hypertension | CT-C (+angiography): normal3 | Cell count: 107/mm3 | March 3, 2014 | January 20, 2020 |
| One prior episode of serous meningitis without microbial findings |
Abbreviations: CSF, cerebrospinal fluid; CT-C CAT, scan of the cerebrum; MRI, magnetic resonance imagining; Mtx, methotrexate; N/A, not available; ND, not done; VZV, varicella zoster virus.
aAge stated is the age of each patient upon hospital admission. Date of admission and sample collection are presented as month, day, and year.
Demographics and Clinical Characteristics of the Cohort of Patients With VZV Encephalitis or Meningoencephalitis
| Patient Number | Agea, Sex | Country | Clinical Diagnosis | Medical History | MRI/CT-C Findings | CSF Findings | Date of Admission | Date of Sample Collection |
|---|---|---|---|---|---|---|---|---|
| P1 | 56, male | Sweden | Meningoencephalitis | Deep venous thrombosis | MRI and CT-C with one-sided prominent temporal horn. No signs of encephalitis | Cell count: 58/mm3 | August 16, 2007 | October 25, 2019 |
| P2 | 20, female | Sweden | Meningoencephalitis and Ramsay Hunt syndrome | Healthy, recent child delivery | CT-angiography normal, MRI ND | Cell count: 728/mm3 | June 29, 2011 | October 29, 2019 |
| P3 | 38, male | Sweden | Meningoencephalitis | Healthy | CT-C normal, MRI ND | Cell count: 594/mm3 | January 20, 2016 | November 1, 2019 |
| P4 | 22, male | Sweden | Meningoencephalitis, vasculitis with ischemic stroke | Migraine, asthma | CT-angiography with left-sided temporal infarction, MRI normal | Cell count: 716/mm3 | September 07, 2016 | November 8, 2019 |
| P5 | 58, male | Sweden | Vasculitis with subarachnoid hemorrhage | Healthy | MRI and CT-C with one-sided prominent temporal horn. No signs of encephalitis | Cell count: 181/mm3 | January 26, 2004 | November 8, 2019 |
| P6 | 28, male | Sweden | Meningoencephalitis | Healthy | CT-angiography normal, MRI ND | Cell count: 274/mm3 | February 22, 2015 | November 13, 2019 |
| P7 | 37, female | Sweden | Meningoencephalitis | Chronic posttraumatic neck and back pain, migraine, chronic fatigue syndrome, earlier motoric function loss diagnosed as dissociative motor disorder | CT-C normal, MRI ND | Cell count: 376/mm3 | September 15, 2015 | November 13, 2019 |
| P8 | 74, female | Sweden | Meningoencephalitis, Ramsay Hunt syndrome and vasculitis | Migraine, anxiety | CT-angiography with left-sided temporal infarction, MRI normal | Cell count: 297/mm3 | May 10, 2019 | November 13, 2019 |
| P9 | 4, female | Sweden | Encephalitis | Asthma | MRI and CT-C with one-sided prominent temporal horn | Cell count: 102/mm3 | April 21, 2016 | November 19, 2019 |
| P10 | 32, female | Sweden | Meningoencephalitis | Healthy | CT-angiography normal MRI ND | Cell count: 266/mm3 | January 30, 2016 | November 29, 2019 |
| P11 | 58, male | Denmark | Meningoencephalitis | Healthy | MRI and CT-C normal | Cell count: 142/mm3 | February 8, 2019 | June 12, 2020 |
| P12 | 21, female | Denmark | Encephalitis | Healthy | MRI (+angiography) normal | Cell count: 585/mm3 | June 14, 2016 | July 29, 2019 |
| P13 | 36, female | Denmark | Meningoencephalitis | Polycystic ovary syndrome | White matter lesions | Cell count: 264/mm3 | October 23, 2019 | November 14, 2019 |
| P14 | 56, male | Denmark | Encephalitis | Rheumatoid arthritis treated with Mtx | MRI normal | Cell count: 115/mm3 | June 10, 2016 | June 30, 2019 |
| P15 | 45, male | Sweden | Meningoencephalitis, vasculitis with ischemic stroke | Depression | MRI with left-sided thalamic and occipital ischemic lesions, angiography with no signs of vasculitis, but a narrow arteria cerebri posterior sin, CT-C normal | Cell count: 75/mm3 | May 18, 2007 | October 25, 2019 |
| P16 | 25, male | Sweden | Meningoencephalitis | Delusional disorder, anxiety, recurrent depressive disorder | CT-C and MRI normal | Cell count: 446/mm3 | May 4, 2011 | November 8, 2019 |
| P17 | 42, female | Denmark | Encephalitis | Benign intracraniel hypertension | CT-C (+angiography): normal3 | Cell count: 107/mm3 | March 3, 2014 | January 20, 2020 |
| One prior episode of serous meningitis without microbial findings |
| Patient Number | Agea, Sex | Country | Clinical Diagnosis | Medical History | MRI/CT-C Findings | CSF Findings | Date of Admission | Date of Sample Collection |
|---|---|---|---|---|---|---|---|---|
| P1 | 56, male | Sweden | Meningoencephalitis | Deep venous thrombosis | MRI and CT-C with one-sided prominent temporal horn. No signs of encephalitis | Cell count: 58/mm3 | August 16, 2007 | October 25, 2019 |
| P2 | 20, female | Sweden | Meningoencephalitis and Ramsay Hunt syndrome | Healthy, recent child delivery | CT-angiography normal, MRI ND | Cell count: 728/mm3 | June 29, 2011 | October 29, 2019 |
| P3 | 38, male | Sweden | Meningoencephalitis | Healthy | CT-C normal, MRI ND | Cell count: 594/mm3 | January 20, 2016 | November 1, 2019 |
| P4 | 22, male | Sweden | Meningoencephalitis, vasculitis with ischemic stroke | Migraine, asthma | CT-angiography with left-sided temporal infarction, MRI normal | Cell count: 716/mm3 | September 07, 2016 | November 8, 2019 |
| P5 | 58, male | Sweden | Vasculitis with subarachnoid hemorrhage | Healthy | MRI and CT-C with one-sided prominent temporal horn. No signs of encephalitis | Cell count: 181/mm3 | January 26, 2004 | November 8, 2019 |
| P6 | 28, male | Sweden | Meningoencephalitis | Healthy | CT-angiography normal, MRI ND | Cell count: 274/mm3 | February 22, 2015 | November 13, 2019 |
| P7 | 37, female | Sweden | Meningoencephalitis | Chronic posttraumatic neck and back pain, migraine, chronic fatigue syndrome, earlier motoric function loss diagnosed as dissociative motor disorder | CT-C normal, MRI ND | Cell count: 376/mm3 | September 15, 2015 | November 13, 2019 |
| P8 | 74, female | Sweden | Meningoencephalitis, Ramsay Hunt syndrome and vasculitis | Migraine, anxiety | CT-angiography with left-sided temporal infarction, MRI normal | Cell count: 297/mm3 | May 10, 2019 | November 13, 2019 |
| P9 | 4, female | Sweden | Encephalitis | Asthma | MRI and CT-C with one-sided prominent temporal horn | Cell count: 102/mm3 | April 21, 2016 | November 19, 2019 |
| P10 | 32, female | Sweden | Meningoencephalitis | Healthy | CT-angiography normal MRI ND | Cell count: 266/mm3 | January 30, 2016 | November 29, 2019 |
| P11 | 58, male | Denmark | Meningoencephalitis | Healthy | MRI and CT-C normal | Cell count: 142/mm3 | February 8, 2019 | June 12, 2020 |
| P12 | 21, female | Denmark | Encephalitis | Healthy | MRI (+angiography) normal | Cell count: 585/mm3 | June 14, 2016 | July 29, 2019 |
| P13 | 36, female | Denmark | Meningoencephalitis | Polycystic ovary syndrome | White matter lesions | Cell count: 264/mm3 | October 23, 2019 | November 14, 2019 |
| P14 | 56, male | Denmark | Encephalitis | Rheumatoid arthritis treated with Mtx | MRI normal | Cell count: 115/mm3 | June 10, 2016 | June 30, 2019 |
| P15 | 45, male | Sweden | Meningoencephalitis, vasculitis with ischemic stroke | Depression | MRI with left-sided thalamic and occipital ischemic lesions, angiography with no signs of vasculitis, but a narrow arteria cerebri posterior sin, CT-C normal | Cell count: 75/mm3 | May 18, 2007 | October 25, 2019 |
| P16 | 25, male | Sweden | Meningoencephalitis | Delusional disorder, anxiety, recurrent depressive disorder | CT-C and MRI normal | Cell count: 446/mm3 | May 4, 2011 | November 8, 2019 |
| P17 | 42, female | Denmark | Encephalitis | Benign intracraniel hypertension | CT-C (+angiography): normal3 | Cell count: 107/mm3 | March 3, 2014 | January 20, 2020 |
| One prior episode of serous meningitis without microbial findings |
Abbreviations: CSF, cerebrospinal fluid; CT-C CAT, scan of the cerebrum; MRI, magnetic resonance imagining; Mtx, methotrexate; N/A, not available; ND, not done; VZV, varicella zoster virus.
aAge stated is the age of each patient upon hospital admission. Date of admission and sample collection are presented as month, day, and year.
To explore the genetics underlying severe VZV CNS infection in these individuals, we performed WES, and we filtered data in Ingenuity Variant Analysis (IVA) as described in Supplementary Methods to identify the variants most likely to have contributed to disease in the cohort. The analysis was based on a hypothesis of autosomal dominant inheritance with possible incomplete penetrance. In brief, variants were filtered out if they were frequent (frequency >0.1%), not predicted to be pathogenic as defined by a combined annotation dependent depletion (CADD) score <15, or lacking biological significance. Variants in genes without a substantial role in the immune system or in autophagy pathways (Supplementary Table 1) or variants with uncertain relevance to VZV infection (Supplementary Table 2) were excluded. Variants in genes previously described to cause severe or disseminated VZV infection but not passing filters are listed in Supplementary Table 3. The final list of variants, shown in Table 2, thus comprises 21 heterozygous variants in 21 different genes. All variants were missense variants except for a stop gain mutation in DDX60 in P11 and a frameshift in NLRC5 in P13. In addition, variants were only present in single patients beside a variant in ATG2B that was common to P1 and P7. Finally, genes described in the literature to be associated with disseminated or severe VZV CNS infection [6, 17, 19, 20] but present with higher frequency than cutoff are shown in Supplementary Table 3. Altogether, in the 17 VZV CNS infection patients included, we identified 21 potentially disease-causing variants in 13 (76%) of the patients.
Genetic Variants Identified in Patients With VZV Encephalitis and Meningoencephalitis
| Patient ID | Gene Symbol | Biological Category | Transcript ID | Transcript Variant; Protein Variant | Translation Impact | CADD; MSC Scores | SIFT Function Prediction | gnomAD Frequency (%) |
|---|---|---|---|---|---|---|---|---|
| P1 | ULK1 | Autophagy | NM_003565.4 | c.1699C > T; p.R567C | Missense | 22.5; 3.313 | N/A | 0.012 |
| ATG2B | Autophagy | NM_018036.7 | c.4706C > T; p.P1569L | Missense | 23.2; 3.313 | N/A | 0.030 | |
| P2 | ATG2A | Autophagy | NM_015104.3 | c.3152G > A; p.R1051H | Missense | 26.9; 3.313 | Damaging | -a |
| TGM2 | Autophagy | NM_004613.4 | c.2005G > A; p.E669K | Missense | 23.9; 23.1 | Tolerated | 0.023 | |
| P3 | IRF1 | Innate immune system | NM_002198.3 | c.524T > C; p.V175A | Missense | 21.5; 16.232 | Benign | 0.012 |
| P4 | RELB | Innate immune system | NM_006509.4 | c.847C > T; p.R283W | Missense | 23.8; 3.313 | Damaging | 0.003 |
| P5 | POLR3E | Innate immune system | NM_018119.4 | c.1559A > G; p.D520G | Missense | 26; 3.313 | Damaging | 0.007 |
| LAMP2 | Autophagy | NM_001122606.1 | c.1078G > A; p.G360R | Missense | 24.5; 0.001 | Damaging | 0.002 | |
| IRAK1 | Innate immune system | NM_001025242.2 | c.1096C > T; p.R366C | Missense | 28.2; 3.313 | Damaging | 0.084 | |
| P6 | TRIM38 | Innate immune system | NM_006355.5 | c.1060G > C; p.G354R | Missense | 23; 3.313 | Tolerated | -a |
| P7 | ATG2B | Autophagy | NM_018036.7 | c.4706C > T; p.P1569L | Missense | 23.2; 3.313 | Benign | 0.030 |
| P8 | DHX36 | Innate immune system | NM_020865.3 | c.1539G > C; p.M513I | Missense | 22.9; 3.313 | Tolerated | -a |
| CGAS | Innate immune system | NM_138441.3 | c.986G > C; p.S329T | Missense | 22.7; NA | Damaging | 0.001 | |
| P9 | TANK | Innate immune system | NM_004180.3 | c.902A > G; p.Q301R | Missense | 22.0; 3.313 | Damaging | 0.044 |
| P10 | POLR3A | Innate immune system | NM_007055.4 | c.1729G > A; p.E577K | Missense | 23.5; 3.313 | Tolerated | 0.049 |
| P11 | TLR9 | Innate immune system | NM_017442.3 | c.853C > T; p.R285C | Missense | 23.4; 3.313 | Damaging | 0.003 |
| NFKB1 | Innate immune system | NM_003998.4 | c.1601G > A; p.R534H | Missense | 25.6; 3.313 | Damaging | 0.053 | |
| DDX60 | Innate immune system | NM_017631.6 | c.4681A > T; p.K1561* | Stop gain | 33; 3.313 | N/A | 0.000 | |
| TSC1 | Autophagy | NM_000368.5 | c.2285A > G; p.N762S | Missense | 15.31; 9.086 | Tolerated | 0.039 | |
| P12 | ATG9A | Autophagy | NM_024085.5 | c.1688C > G; p.T563S | Missense | 22.6; 313 | Tolerated | 0.028 |
| RIOK3 | Innate immune system | NM_003831.5 | c.890A > G; p.N297S | Missense | 23.9; 3.313 | Tolerated | 0.001 | |
| P13 | NLRC5 | Innate immune system | NM_032206.4 | c.5292delC; p.F1764fs*9 | Frameshift | 26.1; | N/A | 0.000 |
| Patient ID | Gene Symbol | Biological Category | Transcript ID | Transcript Variant; Protein Variant | Translation Impact | CADD; MSC Scores | SIFT Function Prediction | gnomAD Frequency (%) |
|---|---|---|---|---|---|---|---|---|
| P1 | ULK1 | Autophagy | NM_003565.4 | c.1699C > T; p.R567C | Missense | 22.5; 3.313 | N/A | 0.012 |
| ATG2B | Autophagy | NM_018036.7 | c.4706C > T; p.P1569L | Missense | 23.2; 3.313 | N/A | 0.030 | |
| P2 | ATG2A | Autophagy | NM_015104.3 | c.3152G > A; p.R1051H | Missense | 26.9; 3.313 | Damaging | -a |
| TGM2 | Autophagy | NM_004613.4 | c.2005G > A; p.E669K | Missense | 23.9; 23.1 | Tolerated | 0.023 | |
| P3 | IRF1 | Innate immune system | NM_002198.3 | c.524T > C; p.V175A | Missense | 21.5; 16.232 | Benign | 0.012 |
| P4 | RELB | Innate immune system | NM_006509.4 | c.847C > T; p.R283W | Missense | 23.8; 3.313 | Damaging | 0.003 |
| P5 | POLR3E | Innate immune system | NM_018119.4 | c.1559A > G; p.D520G | Missense | 26; 3.313 | Damaging | 0.007 |
| LAMP2 | Autophagy | NM_001122606.1 | c.1078G > A; p.G360R | Missense | 24.5; 0.001 | Damaging | 0.002 | |
| IRAK1 | Innate immune system | NM_001025242.2 | c.1096C > T; p.R366C | Missense | 28.2; 3.313 | Damaging | 0.084 | |
| P6 | TRIM38 | Innate immune system | NM_006355.5 | c.1060G > C; p.G354R | Missense | 23; 3.313 | Tolerated | -a |
| P7 | ATG2B | Autophagy | NM_018036.7 | c.4706C > T; p.P1569L | Missense | 23.2; 3.313 | Benign | 0.030 |
| P8 | DHX36 | Innate immune system | NM_020865.3 | c.1539G > C; p.M513I | Missense | 22.9; 3.313 | Tolerated | -a |
| CGAS | Innate immune system | NM_138441.3 | c.986G > C; p.S329T | Missense | 22.7; NA | Damaging | 0.001 | |
| P9 | TANK | Innate immune system | NM_004180.3 | c.902A > G; p.Q301R | Missense | 22.0; 3.313 | Damaging | 0.044 |
| P10 | POLR3A | Innate immune system | NM_007055.4 | c.1729G > A; p.E577K | Missense | 23.5; 3.313 | Tolerated | 0.049 |
| P11 | TLR9 | Innate immune system | NM_017442.3 | c.853C > T; p.R285C | Missense | 23.4; 3.313 | Damaging | 0.003 |
| NFKB1 | Innate immune system | NM_003998.4 | c.1601G > A; p.R534H | Missense | 25.6; 3.313 | Damaging | 0.053 | |
| DDX60 | Innate immune system | NM_017631.6 | c.4681A > T; p.K1561* | Stop gain | 33; 3.313 | N/A | 0.000 | |
| TSC1 | Autophagy | NM_000368.5 | c.2285A > G; p.N762S | Missense | 15.31; 9.086 | Tolerated | 0.039 | |
| P12 | ATG9A | Autophagy | NM_024085.5 | c.1688C > G; p.T563S | Missense | 22.6; 313 | Tolerated | 0.028 |
| RIOK3 | Innate immune system | NM_003831.5 | c.890A > G; p.N297S | Missense | 23.9; 3.313 | Tolerated | 0.001 | |
| P13 | NLRC5 | Innate immune system | NM_032206.4 | c.5292delC; p.F1764fs*9 | Frameshift | 26.1; | N/A | 0.000 |
Abbreviations: CADD, combined annotation dependent depletion; gnomAD, genome aggregation database; ID, identification; MSC, mutation significance cutoff; N/A, not applicable; SIFT, sorting intolerant from tolerant; VZV, varicella zoster virus.
aVariant not present in gnomAD database.
NOTE: See Supplementary Table 2 for additional variants identified in innate adaptive immune signaling pathways and autophagy-related genes.
Genetic Variants Identified in Patients With VZV Encephalitis and Meningoencephalitis
| Patient ID | Gene Symbol | Biological Category | Transcript ID | Transcript Variant; Protein Variant | Translation Impact | CADD; MSC Scores | SIFT Function Prediction | gnomAD Frequency (%) |
|---|---|---|---|---|---|---|---|---|
| P1 | ULK1 | Autophagy | NM_003565.4 | c.1699C > T; p.R567C | Missense | 22.5; 3.313 | N/A | 0.012 |
| ATG2B | Autophagy | NM_018036.7 | c.4706C > T; p.P1569L | Missense | 23.2; 3.313 | N/A | 0.030 | |
| P2 | ATG2A | Autophagy | NM_015104.3 | c.3152G > A; p.R1051H | Missense | 26.9; 3.313 | Damaging | -a |
| TGM2 | Autophagy | NM_004613.4 | c.2005G > A; p.E669K | Missense | 23.9; 23.1 | Tolerated | 0.023 | |
| P3 | IRF1 | Innate immune system | NM_002198.3 | c.524T > C; p.V175A | Missense | 21.5; 16.232 | Benign | 0.012 |
| P4 | RELB | Innate immune system | NM_006509.4 | c.847C > T; p.R283W | Missense | 23.8; 3.313 | Damaging | 0.003 |
| P5 | POLR3E | Innate immune system | NM_018119.4 | c.1559A > G; p.D520G | Missense | 26; 3.313 | Damaging | 0.007 |
| LAMP2 | Autophagy | NM_001122606.1 | c.1078G > A; p.G360R | Missense | 24.5; 0.001 | Damaging | 0.002 | |
| IRAK1 | Innate immune system | NM_001025242.2 | c.1096C > T; p.R366C | Missense | 28.2; 3.313 | Damaging | 0.084 | |
| P6 | TRIM38 | Innate immune system | NM_006355.5 | c.1060G > C; p.G354R | Missense | 23; 3.313 | Tolerated | -a |
| P7 | ATG2B | Autophagy | NM_018036.7 | c.4706C > T; p.P1569L | Missense | 23.2; 3.313 | Benign | 0.030 |
| P8 | DHX36 | Innate immune system | NM_020865.3 | c.1539G > C; p.M513I | Missense | 22.9; 3.313 | Tolerated | -a |
| CGAS | Innate immune system | NM_138441.3 | c.986G > C; p.S329T | Missense | 22.7; NA | Damaging | 0.001 | |
| P9 | TANK | Innate immune system | NM_004180.3 | c.902A > G; p.Q301R | Missense | 22.0; 3.313 | Damaging | 0.044 |
| P10 | POLR3A | Innate immune system | NM_007055.4 | c.1729G > A; p.E577K | Missense | 23.5; 3.313 | Tolerated | 0.049 |
| P11 | TLR9 | Innate immune system | NM_017442.3 | c.853C > T; p.R285C | Missense | 23.4; 3.313 | Damaging | 0.003 |
| NFKB1 | Innate immune system | NM_003998.4 | c.1601G > A; p.R534H | Missense | 25.6; 3.313 | Damaging | 0.053 | |
| DDX60 | Innate immune system | NM_017631.6 | c.4681A > T; p.K1561* | Stop gain | 33; 3.313 | N/A | 0.000 | |
| TSC1 | Autophagy | NM_000368.5 | c.2285A > G; p.N762S | Missense | 15.31; 9.086 | Tolerated | 0.039 | |
| P12 | ATG9A | Autophagy | NM_024085.5 | c.1688C > G; p.T563S | Missense | 22.6; 313 | Tolerated | 0.028 |
| RIOK3 | Innate immune system | NM_003831.5 | c.890A > G; p.N297S | Missense | 23.9; 3.313 | Tolerated | 0.001 | |
| P13 | NLRC5 | Innate immune system | NM_032206.4 | c.5292delC; p.F1764fs*9 | Frameshift | 26.1; | N/A | 0.000 |
| Patient ID | Gene Symbol | Biological Category | Transcript ID | Transcript Variant; Protein Variant | Translation Impact | CADD; MSC Scores | SIFT Function Prediction | gnomAD Frequency (%) |
|---|---|---|---|---|---|---|---|---|
| P1 | ULK1 | Autophagy | NM_003565.4 | c.1699C > T; p.R567C | Missense | 22.5; 3.313 | N/A | 0.012 |
| ATG2B | Autophagy | NM_018036.7 | c.4706C > T; p.P1569L | Missense | 23.2; 3.313 | N/A | 0.030 | |
| P2 | ATG2A | Autophagy | NM_015104.3 | c.3152G > A; p.R1051H | Missense | 26.9; 3.313 | Damaging | -a |
| TGM2 | Autophagy | NM_004613.4 | c.2005G > A; p.E669K | Missense | 23.9; 23.1 | Tolerated | 0.023 | |
| P3 | IRF1 | Innate immune system | NM_002198.3 | c.524T > C; p.V175A | Missense | 21.5; 16.232 | Benign | 0.012 |
| P4 | RELB | Innate immune system | NM_006509.4 | c.847C > T; p.R283W | Missense | 23.8; 3.313 | Damaging | 0.003 |
| P5 | POLR3E | Innate immune system | NM_018119.4 | c.1559A > G; p.D520G | Missense | 26; 3.313 | Damaging | 0.007 |
| LAMP2 | Autophagy | NM_001122606.1 | c.1078G > A; p.G360R | Missense | 24.5; 0.001 | Damaging | 0.002 | |
| IRAK1 | Innate immune system | NM_001025242.2 | c.1096C > T; p.R366C | Missense | 28.2; 3.313 | Damaging | 0.084 | |
| P6 | TRIM38 | Innate immune system | NM_006355.5 | c.1060G > C; p.G354R | Missense | 23; 3.313 | Tolerated | -a |
| P7 | ATG2B | Autophagy | NM_018036.7 | c.4706C > T; p.P1569L | Missense | 23.2; 3.313 | Benign | 0.030 |
| P8 | DHX36 | Innate immune system | NM_020865.3 | c.1539G > C; p.M513I | Missense | 22.9; 3.313 | Tolerated | -a |
| CGAS | Innate immune system | NM_138441.3 | c.986G > C; p.S329T | Missense | 22.7; NA | Damaging | 0.001 | |
| P9 | TANK | Innate immune system | NM_004180.3 | c.902A > G; p.Q301R | Missense | 22.0; 3.313 | Damaging | 0.044 |
| P10 | POLR3A | Innate immune system | NM_007055.4 | c.1729G > A; p.E577K | Missense | 23.5; 3.313 | Tolerated | 0.049 |
| P11 | TLR9 | Innate immune system | NM_017442.3 | c.853C > T; p.R285C | Missense | 23.4; 3.313 | Damaging | 0.003 |
| NFKB1 | Innate immune system | NM_003998.4 | c.1601G > A; p.R534H | Missense | 25.6; 3.313 | Damaging | 0.053 | |
| DDX60 | Innate immune system | NM_017631.6 | c.4681A > T; p.K1561* | Stop gain | 33; 3.313 | N/A | 0.000 | |
| TSC1 | Autophagy | NM_000368.5 | c.2285A > G; p.N762S | Missense | 15.31; 9.086 | Tolerated | 0.039 | |
| P12 | ATG9A | Autophagy | NM_024085.5 | c.1688C > G; p.T563S | Missense | 22.6; 313 | Tolerated | 0.028 |
| RIOK3 | Innate immune system | NM_003831.5 | c.890A > G; p.N297S | Missense | 23.9; 3.313 | Tolerated | 0.001 | |
| P13 | NLRC5 | Innate immune system | NM_032206.4 | c.5292delC; p.F1764fs*9 | Frameshift | 26.1; | N/A | 0.000 |
Abbreviations: CADD, combined annotation dependent depletion; gnomAD, genome aggregation database; ID, identification; MSC, mutation significance cutoff; N/A, not applicable; SIFT, sorting intolerant from tolerant; VZV, varicella zoster virus.
aVariant not present in gnomAD database.
NOTE: See Supplementary Table 2 for additional variants identified in innate adaptive immune signaling pathways and autophagy-related genes.
Presence of Variants in Genes Within Innate Immune Responses and Autophagy Pathways
The final variants identified in the patient cohort (Table 2) were analyzed for protein-protein interactions using STRING association analysis software [23]. Within the STRING protein-protein association network depicted in Figure 1, variants could be separated into 2 major groups consisting of proteins participating in innate immune responses and proteins in autophagy-related processes. Proteins within each group were strongly associated with each other, and, moreover, the 2 groups were also interconnected (Figure 1).
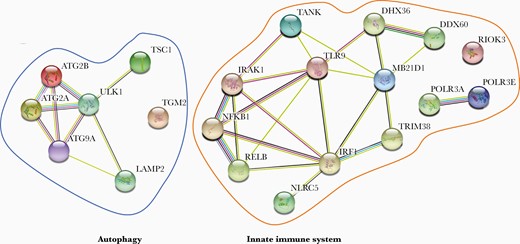
STRING protein-protein association network for proteins identified in the varicella zoster virus encephalitis cohort. All 21 proteins identified in the final whole exome sequencing analysis were subjected to STRING software analysis. Each circle represents a protein affected by a variant and lines denote predicted or known interactions.
Within the group of innate immune response genes, variants can be divided into a group of genes participating in virus recognition and/or sensing and another group of genes directly involved in induction of IFNs and proinflammatory cytokines. In the former category, variants were found in DNA sensors recognizing double-stranded DNA (dsDNA) (cGAS in P8), AT_rich dsDNA (POLR3A in P10, POLR3E in P5), and unmethylated DNA (TLR9 in P11). In addition, in P12 a variant was found in RIO kinase (RIOK)3, which acts as an adaptor protein important for cytosolic DNA- and RNA-induced type I IFN induction [24]. Moreover, 3 different variants were found in DDX60 in P11, DHX36 in P8 and tripartite motif containing (TRIM)38 in P6, which all participate in RIG-I-like receptor signaling [25–27]. A variant was also found in P13 in NOD-like receptor 5 (NLRC5), which has been implicated in IFNγ-dependent antiviral immune response [28]. In the group of genes encoding innate immune signaling molecules that participate in IFN induction, a variant in P3 was identified in IRF1, a transcription factor that together with IRF3 and IRF7 contributes to type I IFN induction [29]. In addition, in P5 we noted a variant in interleukin-1 receptor associated kinase (IRAK)1, an important kinase that activates IRF7 downstream of TLR7 and TLR9 leading to induction of IFNα [30]. P9 harbored a variant in TANK, which plays a role in type I IFN induction downstream of TLR3, -4, -7, -9, and RIG-I, as well as regulating nuclear factor (NF)-κB signaling [31, 32]. Finally, in P4 and P11, 2 variants were discovered in RELB and NFKB1, respectively, both of which participate in induction of proinflammatory cytokines through activation of the NF-κB transcription complex [33].
In processes related to the autophagy pathway, variants were found in genes essential for initiation of autophagy (ULK1 in P1) [34], for the formation and maturation of the autophagosome (ATG2A in P2, ATG2B in P1 and P7, and transglutaminase (TGM)2 in P2) [35–37], as well as expansion of the phagophore (ATG9A in P12) [38]. In addition, a variant was present instead of a variant was found in the lysosome-associated membrane protein (LAMP)2 gene in P5, which is important for lysosomal stability and maturation of the autophagosome [39]. Finally, in P11 a variant was identified in tuberous sclerosis complex (TSC)1, which together with TSC2 have been implicated in regulating autophagy through ULK1 in neurons [40].
Antiviral Immune Responses and Varicella Zoster Virus Gene Expression/Replication in Patient Peripheral Blood Mononuclear Cells
To functionally investigate the impact of the identified variants in the patient cohort, we examined innate immune responses in patient cells infected with VZV. Patient PBMCs from P1 - P13 as well as 6 healthy controls were infected with VZV-MeWo cells for 48 hours, after which IFNs and proinflammatory cytokines in cell culture supernatants (Figures 2 and 3) as well as viral gene expression in PBMC-VZV-MeWo coculture lysates were measured (Figure 4). We found that P1, P3, P4, P5, P6, and P7 showed impaired production of IFNα2a, IFNβ, IFNλ, and/or CXCL10 in response to VZV-MeWo infection compared with pooled healthy controls (Figure 2A–D and Figure 3D), and additionally, P1, P5, P7, and P9 also exhibited decreased expression of 1 or more of the proinflammatory cytokines measured (IL-6, IL-1β, and TNFα) (Figure 3A–C). Of these, P3, P4, P5, P6, and P9 harbored variants in innate immune signaling molecules, whereas P1 and P7 had autophagy-related variants. Induction of IFNγ was decreased in P1 and P9 (Figure 2D). On the other hand, VZV infection resulted in increased expression of IFNα2a and IFNβ in P11, and IFNλ1 in P8 (Figure 2A–C), as well as CXCL10 in P6, P8, P11, and P12, compared with healthy controls (Figure 3D). Moreover, IFNγ protein expression was significantly increased in P6, P8, P11, and P12 (Figure 2D). When measuring the expression of viral open reading frame (ORF) genes as an indirect measure of VZV replication in the VZV-MeWo-PBMC coculture, we found that P1 showed a significant increase in the expression of ORF40 and ORF63 messenger RNA (mRNA), whereas P11 demonstrated significantly increased expression of ORF40 mRNA (Figure 4A–C). In addition, P3, P9, and P11 also exhibited higher induction of ORF9, ORF40, and/or ORF63, although not reaching statistical significance (Figure 4A–C).
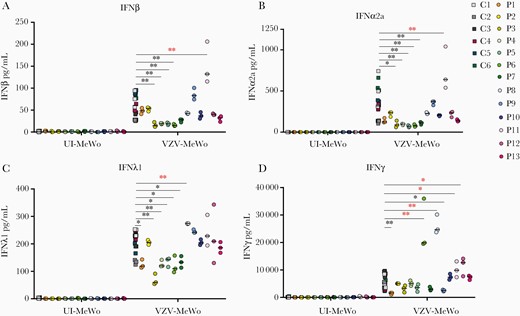
Antiviral immune responses to varicella zoster virus (VZV) infection in patient and control cells. Peripheral blood mononuclear cells (PBMCs) from 13 patients from the VZV encephalitis cohort and 6 healthy controls were cocultured with uninfected MeWo cells (UI-MeWo) or VZV-ROka-infected MeWo cells (VZV-MeWo) at a ratio of 1:0.4 (PBMC/MeWo) for 48 hours, and the production of interferon (IFN)β (A), IFNα2a (B), IFNλ1 (C), and IFNγ (D) was measured in culture supernatants by Meso Scale. Statistics were calculated using the Mann-Whitney rank-sum test. *, P ≤ .05; **, P ≤ .01. Line with black asterisks indicates decreased expression, and line with red asterisks indicates increased expression. No line and asterisk = nonsignificant.
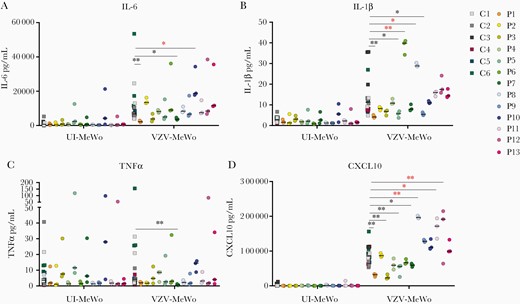
Production of proinflammatory and interferon-stimulated cytokines in patient and control cells infected with varicella zoster virus (VZV). Patient and control peripheral blood mononuclear cells (PBMCs) were cocultured with uninfected MeWo cells (UI-MeWo) or VZV-ROKa infected MeWo cells (VZV-MeWo) at a ratio of 1:0.4 (PBMC/MeWo) for 48 hours, and the induction of interleukin (IL)-6 (A), IL-1β (B), tumor necrosis factor (TNF)α (C), and CXCL10 (D) were measured by Meso Scale technology in the coculture supernatants. Statistics were calculated using the Mann-Whitney rank-sum test. Line with black asterisks indicates decreased expression, and line with red asterisks indicates increased expression. No line and asterisk, nonsignificant. *, P ≤ .05; **, P ≤ .01. No line and asterisk, nonsignificant.
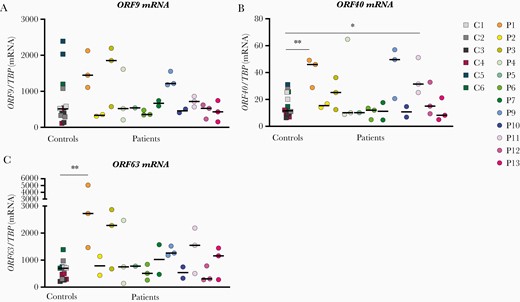
Expression of viral genes in peripheral blood mononuclear cell (PBMC)-varicella zoster virus (VZV)-MeWo cocultures. Patient and control PBMCs were cocultured with uninfected MeWo cells (UI-MeWo) or VZV-ROKa-infected MeWo cells (VZV-MeWo) at a ratio of 1:0.4 (PBMC/MeWo) and lysed 48 hours postinfection. Total ribonucleic acid (RNA) was purified from cell lysates and subjected to quantitative reverse-transcription polymerase chain reaction for expression of the viral open reading frame (ORF) genes ORF9 (A), ORF40 (B), and ORF63 (C). Messenger RNA (mRNA) levels were normalized to the expression of TATA binding protein (TBP). Statistics were calculated using the Mann-Whitney rank-sum test.*, P ≤ .05; **, P ≤ .01. Line with black asterisks indicates decreased expression, and line with red asterisks indicates increased expression. No line and asterisk, nonsignificant.
DISCUSSION
In this study, we performed WES combined with functional immunological analyses on material from patients with VZV encephalitis to gain insights into the pathogenesis of this severe neuroinfectious disease. We investigated genetic variants that were rare and predicted to be damaging by different in silico tools and with a known role in viral sensing or induction of innate immune circuits in antiviral defense against viruses. By this approach we identified 21 potentially disease-causing variants in 13 of the 17 patients, participating in innate antiviral immune responses or representing different components of the autophagy pathway, that we suggest may have contributed to neuroinvasion, immunopathogenesis, and development of severe VZV CNS infection. Although the cohort of patients is of limited size, we believe the results are important may be generalizable for larger populations of patients with VZV encephalitis.
Among the main findings was the identification of several variants in genes related to VZV sensing and triggering of innate antiviral responses. First, cytosolic DNA sensors were represented by both cGAS, POL III, and TLR9, in which we found potentially pathogenic variants. Whole exome sequencing analysis of the present VZV cohort revealed 2 variants, different from the ones in VZV disease [17, 19, 20], including a PORL3A variant in P5 and a POLR3E variant in P10, of whom only P5 showed impaired antiviral IFN responses. It is interesting to note that Ramanathan et al [41] recently described the identification of RNA polymerase III deficiency due to a POLR3E variant in an infant with severe recurrent viral infections, and they suggested that POL III deficiency deficiency may lead to a broader susceptibly to multiple viral infections rather than being specific to VZV. In the present study, we only included patients with a narrow phenotype related to severe VZV encephalitis but no previously described history of viral infections, thus precluding specific conclusions as to the specificity of virus susceptibility in POL III deficiency.
The identification of variants in the POL III pathway has raised the possibility of the existence of RIG-I variants in VZV susceptibility, since RIG-I is downstream in POL III signaling, recognizing the 5’ triphosphorylated RNA species generated by the enzymatic activity of POL III [21]. Furthermore, it was reported that overexpression of RIG-I causes suppression of VZV replication in human dermal fibroblast [42]. In the present study, we identified variants in several genes that are important in RIG-I signaling, including TRIM38, DHX36, TANK, and DDX60 in P6, P8, P9, and P11, respectively. Among these, the TRIM38 variant in P6 may represent a particularly good candidate, given that antiviral responses to VZV in cells from P6 showed significantly decreased IFN and CXCL10 responses. These variants may be pathogenic and thus suggest a role for RIG-I downstream of POL III in VZV sensing and support a nonredundant role of the POL III-RIG-I pathway in raising innate antiviral responses towards VZV. Further support for a role of type I IFN responses were indicated by the presence of variants in genes encoding signaling molecules regulating IRF1 and IRF7 activities.
The WES analysis also yielded several variants in autophagy-related genes. Autophagy has emerged as a part of the immune system and acts by protecting against invading pathogens [43]. The autophagy pathway consists of more than 30 different autophagy-related proteins, which all participate in the degradation of organelles and foreign material to maintain cellular homeostasis [34]. It is interesting to note that several studies point towards a particularly important antiviral role of autophagy in the CNS, to protect non-renewable neurons against virus and prevent excessive IFN production [44]. This is supported by 2 recent studies demonstrating autophagy defects in patients with herpes simplex virus-2 recurrent lymphocytic Mollaret’s meningitis and poliomyelitis [45, 46]. These studies from our group were the first to describe defective autophagy in patients with viral CNS infection and suggest that autophagy defects may represent IEIs. However, it remains unexplored whether autophagy defects might also play a role in CNS infections caused by other viruses, such as VZV. However, the evidence for the role of autophagy during VZV infection is conflicting [47]. Although one study found that VZV-induced autophagy is important for viral replication [48], another study demonstrated that VZV blocks autophagic flux resulting in increased VZV replication [49]. The autophagy variants identified in the present study were located in different genes, including ATG2A, ATG2B, ATG9A, ULK1, ULK2, TGM2, and TSC1, all participating in important steps in the autophagy pathway. Perhaps the most likely disease-causing of these might be the single or combined effect of ATG2A and TGM2 in P2, as well as the TSC1 variant in P11, because these patients also exhibited significantly increased VZV replication. Moreover, it is notable that ATG2A was affected in P2, and ATG2B in both P1 and P7, suggesting a particularly important role of these molecules for autophagy to proceed. Altogether, the identification of genetic variants in several autophagy genes supports the hypothesis that autophagy might play an important antiviral part in the CNS during VZV infection in humans, although further functional studies are needed to elaborate the specific mechanistic role of these autophagy-gene-variants in VZV CNS disease.
In 4 of the 17 patients (P14–17), no obvious potential disease-causing variants were identified. Several issues may explain this. First, because we only investigated the coding parts of the genome, important variants in introns and regulatory sequences may be responsible for disease in these patients. It also remains entirely possible that a digenic or polygenic etiology, which was not revealed by the current approach, may underlie some cases of VZV CNS infection in the present cohort. Finally, other factors are undoubtedly important for viral control during primary infection or for maintenance of VZV latency during reactivation. It is unfortunate that the present cohort does not have a sufficient size to make a distinction between encephalitis with and without vasculitis based on specific gene variants. Therefore, it remains a possibility that patients with CNS vasculitis as part of VZV neuroinfection might share certain common pathophysiological characteristics with defined immunological and genetic disturbances, for example leading to interferonopathy or other conditions associated with development of vascular inflammation [50]. In this context, it might be of relevance that 1 patient, P8, with CNS vasculitis displayed increased levels of CXCL10 and IFNλ.
CONCLUSIONS
In the present study involving 17 patients with severe CNS encephalitis, 4 of whom also suffered from CNS vasculitis, we identified genetic variants of potential pathogenic role in the majority. However, functional data did not uniformly confirm the genetic findings in these patients, leaving the precise role of these variants in disease pathogenesis partially unresolved. With the available patient material and limited immunological functional assays, these proposed variants are therefore not to be considered fully functionally validated. The variants and data presented here should serve as a platform for generating hypotheses for future studies addressing molecular genetic and immunological factors associated with susceptibility to severe complicated VZV CNS infection. Overall, we focused on innate immune molecules and signaling pathways and identified several variants in innate cytosolic DNA (and RNA) sensors as well as another major category involving autophagy pathways. Thus, our study confirms and extends previous literature and provides further evidence for a role of innate cytosolic immune responses in antiviral VZV immunity. Moreover, the study suggests a contribution of autophagy to antiviral responses to VZV in the brain and indicates that disturbances in autophagy pathways may represent IEIs predisposing to VZV infection in the CNS.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. We thank the patients participating in this study. We also thank the Danish Study Group for helpful discussions, patient inclusion, and data management (Members: Lykke Larsen, Lothar Wiese, Jannik Helweg-Larsen, Christian Brandt, Birgitte R. Hansen, Merete Storgaard, Anne Mette Lebech, Jacob Bodilsen, Henrik Nielsen, and Hans Rudolf Lüttichau). We would also like to thank Maria Johansson at the laboratory of Clinical microbiology, Sahlgrenska University Hospital for skilful technical assistance.
Author contributions. T. H. M. and A. G. conceived the idea; T. T., K. S., M. S., M. H., P. N., M. C.-T., M. R. F., and A. G. cared for patients and/or collected and isolated patient material; H. N. and J. B. cared for patients and provided information from the DASGIB database to identify patients with previous varicella zoster virus central nervous system infection; M. M. T. performed experiments and analyzed data together with T. H. M.; M. M. T. and T. H. M. wrote the first draft of the manuscript; all authors contributed, read, commented, and approved the final version of the manuscript.
Financial support. T. M. was funded by The Independent Research Fund Denmark (4004-00047B), Aarhus University Research Fund (AUFF-E-2015-FLS-66), and The Lundbeck Foundation (R268-2016–3927).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.


{kind=link}
{kind=link}
{kind=link}
{kind=link}