






Abstract
The recent increase in babies born with brain and eye malformations in Brazil is associated with Zika virus (ZIKV) infection in utero. ZIKV alters host DNA methylation in vitro. Using genome-wide DNA methylation profiling we compared 18 babies born with congenital ZIKV microcephaly with 20 controls. We found ZIKV-associated alteration of host methylation patterns, notably at RABGAP1L which is important in brain development, at viral host immunity genes MX1 and ISG15, and in an epigenetic module containing the causal microcephaly gene MCPH1. Our data support the hypothesis that clinical signs of congenital ZIKV are associated with changes in DNA methylation.
The recent increase in babies born with brain and eye malformations in Brazil has been shown to be associated with the Zika virus (ZIKV) outbreak [1]. All clinically affected babies have neurological effects associated with congenital ZIKV infection, with microcephaly, anomalies of skull shape, and redundancy of scalp consistent with the fetal brain disruption sequence present in 70% of infants [2].
Many parallels can be drawn between clinical signs of congenital ZIKV with those associated with congenital cytomegalovirus and congenital toxoplasmosis [3]. We previously showed that clinical signs in congenital toxoplasmosis are associated with epigenetically regulated genes involved in severe inherited forms of ocular disease [4]. Furthermore, we have shown that Toxoplasma gondii infection in cell culture alters global patterns of host DNA methylation, including gene pathways that influence neural and retinal development [5]. Similarly, studies in an organoid model of the developing human brain demonstrated that ZIKV alters DNA methylation of neural genes [6]. These studies support the hypothesis that clinical signs associated with congenital ZIKV may have an underlying epigenetic basis. This could include perturbation of the normal expression pattern of genes involved in the virus/host cells interactions, in genes regulating the immune response, or in genes directly regulating developmental pathways in the brain.
To determine whether ZIKV modulates host cell epigenetic profiles in vivo, we here examine genome-wide DNA methylation profiles for babies born with congenital ZIKV microcephaly and compare these with babies exposed to ZIKV in utero but born without clinical signs, and with control unaffected unexposed babies. We find no evidence for differential methylation between exposed and unexposed unaffected babies. However, when comparing affected babies with unaffected unexposed babies we find clear evidence for ZIKV-associated alteration in host gene methylation patterns.
METHODS
Study Population
Subjects were recruited through the Pediatric Hospital of the Federal University of Rio Grande do Norte or through visits to households that had cases suspected of microcephaly in Natal and other cities where cases were reported during the 2014–2016 ZIKV outbreak, Rio Grande do Norte, Brazil, including a group of children born in São Luis, Maranhão, during 2018. A subgroup of the Maranhão children had evidence of ZIKV exposure in utero.
Ethics
This study was undertaken with ethical approval from the institutional review board of the Universidade Federal do Rio Grande do Nortem, Comissão Nacional de Ética em Pesquisa CONEP (CAAE 53111416.7.0000.5537) and from the Federal University of Maranhão, CONEP (CAAE 56480316.4.0000.5086). Written consent was obtained from the parents of babies who ranged in age from 5 months to 40 months at the time of sample collection. The individual consent included an option to accept or refuse continued use of their genetic or clinical data in further studies. The parents or legal guardian of all subjects included in the study had given consent for storage and future use of deidentified DNA samples and data for their children.
Description of Clinical Data Collection and Definition of Microcephaly Phenotype
Samples were taken by professional clinical staff either at the clinics or through household visits. Three groups of babies were studied: (1) babies diagnosed with ZIKV-associated microcephaly at birth (n = 18; 16 born in 2015, 1 born in 2016, 1 born in 2018); (2) unaffected babies known to be exposed to ZIKV in utero (ie, mothers were infected with ZIKV during pregnancy; n = 7; all born in 2018 in São Luis, Maranhão); and (3) unaffected unexposed control babies (n = 20; 17 born 2015–2017 in Rio Grande do Norte; 3 born in 2018 in Maranhão). All 7 babies exposed in utero but without clinical signs were positive by polymerase chain reaction (PCR) for ZIKV in the placenta [7]. For the 18 babies diagnosed with ZIKV-associated microcephaly, the mean head circumference at birth was 28.7 cm (SD, 2.5 cm), with a range of 23 to 32 cm, consistent with the December 2015 Brazilian Ministry of Health protocol (head circumference ≤ 33 cm) for case ascertainment. Further descriptions of clinical data and processing of methylation arrays are provided in Supplementary Methods.
Data Analysis
We used R version 3.6.1 for all analyses and a detailed description can be found in Supplementary Methods.
RESULTS
Study Cohort
There were more females in the unexposed control group (11/20; 55%) when compared to the microcephaly cases (7/18, 39%) and the exposed unaffected babies (2/7; 29%). The mean age of cases was 25.6 months (SD, 8.7 months) and for controls was 24.1 months (SD, 11.7 months). The exposed babies were younger with a mean age of 6.7 months (SD, 0.5 months).
Differential Methylation Analysis of CpG Sites
We found significantly differentially methylated cytosine-phosphate-guanine dinucleotides (CpGs) when comparing microcephaly cases to controls, with the strongest evidence at regions on chromosomes 1 and 21 (Figure 1A and Supplementary Table 1). There was a single significantly differentially methylated CpG when comparing microcephaly cases to exposed babies (Supplementary Figure 1), but no significant results when comparing exposed to controls (Supplementary Figure 2). Given these findings, results henceforth focus on the comparison of microcephaly cases to controls.
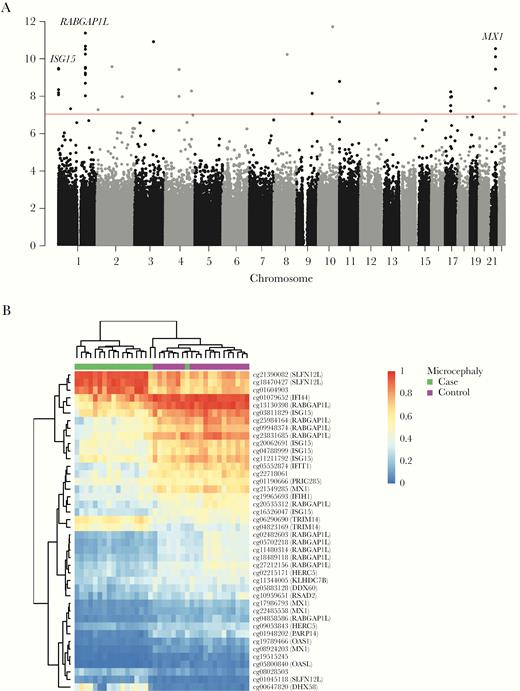
A, Manhattan plot showing results of differential methylation tests of 747 178 CpG sites for microcephaly cases versus controls. The red line indicates the experiment-wide significance threshold of 9 × 10−8. Genes associated with multiple CpGs in a region that were also detected as differentially methylated regions with DMRcate are highlighted. B, Heatmap showing beta values of differentially methylated CpGs (P value < 9 × 10−8). Probes that target genes from the UCSC database have the gene symbol listed in brackets. See online version for color figures.
The strongest region of association included CpGs associated with RAB GTPase activating protein 1 like (RABGAP1L), which were less methylated in the cases compared to the controls (Figure 1A and 1B and Supplementary Table 1). We also found MX dynamin like GTPase 1 (MX1) and ubiquitin like modifier (ISG15) to be strongly associated with promoter methylation differences between cases and controls (Figure 1A) and these genes have previously been implicated in ZIKV response. CpGs for both MX1 and ISG15 were less methylated in cases compared to controls (Figure 1B and Supplementary Table 1).
Analysis of CpG site P values from the differential methylation analysis using methylGSA revealed gene sets primarily involved in viral and immune response processes (Supplementary Figure 3). When considering Gene Ontology terms, there was enrichment for type I interferon response (GO:0034340; P value = 3.8 × 10–19), production (GO:0032606; P value = 1.6 × 10–15), and signaling (GO:0060337; P value = 4.9 × 10–19). Terms related to the viral response included regulation of viral process (GO:0050792; P value = 3.2 × 10–15), genome replication (GO:0019079; P value = 3.1 × 10–13), and life cycle (GO:1903900; P value = 1.8 × 10–13). For Reactome terms, enrichment was for interferon α/β signaling (HSA-909733; P value = 1.4 × 10–17) and antiviral mechanism by IFN-stimulated genes (HSA-1169410; P value = 1.8 × 10–16).
Differential Methylation Analysis of Regions
De novo identification of differentially methylated regions (DMRs) confirmed association with RABGAP1L, ISG15, and MX1 (Supplementary Table 2). The top-ranked DMR is associated with RABGAP1L (chr1:174 842 938–174 844 560) and involved 12 CpGs with a mean difference in methylation of −0.17, and the second-ranked DMR is associated with ISG15 (chr1:948 814–949 893) and involved 9 CpGs with a mean difference of −0.11. The DMR for MX1 (chr21:42 797 468–42 799 141) involved 13 CpGs with a smaller mean difference in methylation (−0.05) to that seen for RABGAP1L and ISG15.
Differential Methylation Interactome Hotspots
We used a functional supervised algorithm to identify differentially regulated modules of genes that may be contributing to the microcephaly phenotype. We identified 7 epigenetic modules (EpiMods) ranging in size from 21 to 68 genes (Supplementary Table 3). One of the EpiMods contains 37 genes centered around transcription factor Dp-1 (TFDP1) and this module also contains a known causal microcephaly gene, microcephalin 1 (MCPH1), which was differentially methylated (Figure 2). The module also contains a number of E2F family member genes that TFDP1 is known to regulate during cell cycle progression and DNA replication [8].
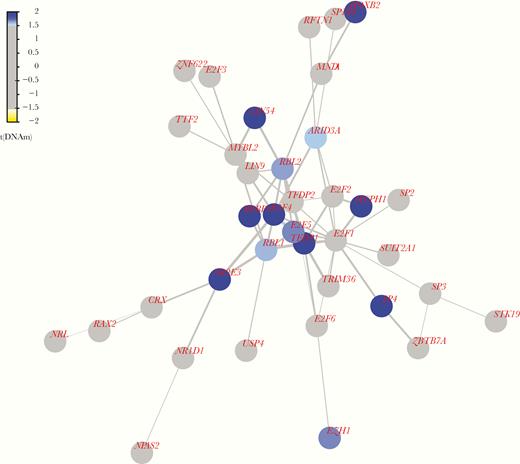
Epigenetic module centered around TFDP1. Color coding of nodes represents the test statistic from differential methylation analysis at the promoter of the gene, and edge widths show the average of the test statistics for the genes at the nodes of the edge. Blue indicates that the promoter is hypermethylated in the microcephaly cases compared to the controls and yellow indicates that the promoter is hypomethylated. See online version for color figures.
The EpiMod centered around EPH receptor A3 (EPHA3) contains 21 genes, including a number of ephrins and ephrin-related receptors that are important during nervous system development [9] (Supplementary Figure 4). All of the differentially methylated nodes in the aforementioned EpiMods were hypermethylated in cases compared to controls, suggesting downregulation of gene expression.
DISCUSSION
Here we present results of epigenetic profiling studies that demonstrate differential methylation patterns in babies with clinical signs associated with congenital ZIKV. Of interest, while we found no difference between babies known to be exposed to ZIKV in utero (as evidenced by positive PCR in the placenta) and unaffected control babies, we did find compelling evidence for differential methylation of genes, pathways, and networks that relate to host-virus interaction in babies born with clinical signs associated with ZIKV infection in utero.
In comparing methylation profiles, we found CpGs in the region of RABGAP1L to be most significantly different between affected cases and unexposed controls. Evidence for its involvement in the ZIKV immune response is lacking and we did not find any publications reporting an association between RABGAP1L and ZIKV or microcephaly. RABGAP1L is a GTPase activating protein (GAP) involved in regulation of intracellular membrane trafficking [10]. This gene has 23 transcripts, with the principal isoform coding for 815 amino acids, and all other protein-coding transcripts being smaller (based on Ensembl GRCh37 release 98). Our region of association is within intron 19 of the principal isoform and within the promoter region of a shorter transcript (ENST00000478442.1), which codes for a truncated (72 amino acids) version of the protein. This truncated protein does not contain the Rab-GAP Tre2-Bub2-Cdc16 (TBC) functional domain so presumably does not have the same function as the full-length protein. Hence, we are unable to assign functional significance to this association. However, we did investigate FANTOM5 promoter expression for the principal transcript versus the truncated transcript (Supplementary Figure 5) and found that expression of the principal transcript is higher for most (937/988) samples. FANTOM5 samples where expression is higher for the truncated transcript include embryonic stem cells, embryoid body cells, induced pluripotent stem cells (differentiation to neuron), and monocytes. This suggests that ZIKV may affect methylation of a transcript that is important in stem cells of the developing embryo, including stem cells involved in brain development.
CpGs in the region of MX1 and ISG15 were also found to be significantly differentially methylated in cases compared to controls. MX1 has been shown to be an important interferon-stimulated gene (ISG) in the antiviral response to ZIKV at the fetomaternal interface, where increased expression inhibits replication of the virus [11]. ISG15 is also an ISG and was found to inhibit ZIKV replication in human corneal cells, by restricting infectivity and host cell binding [12]. Given the reduced methylation, expression of MX1 and ISG15 should be higher in the cases, supporting the role of these genes in the antiviral response to ZIKV.
We detected a downregulated EpiMod centered around TFDP1 that is characterized by genes involved in the DNA damage response, including known microcephaly gene MCPH1. Mutations in MCPH1 cause primary autosomal recessive microcephaly and this was the first gene to be implicated in the disease. How MCPH1 is related to the microcephaly phenotype is not well understood; however, a study in mouse brain development found that deficiency of Mcph1 affects neuroprogenitor production leading to reductions in the number of neurons and hence brain size [13]. These effects were due to the role Mcph1 plays in DNA damage response, where its deficiency resulted in disruption of cell cycle regulation, resulting in neuroprogenitors prematurely exiting the cell cycle. Therefore, downregulation of this DNA damage response EpiMod could be contributing to the microcephaly phenotype.
An EpiMod centered around EPHA3 was also found to be associated with the microcephaly phenotype and it contains a number of genes involved in nervous system development. A previous study of cerebral organoids found that ZIKV disrupts TLR3-mediated networks of neurogenesis by downregulating positive regulators of nervous system development, including EPHA3 [14]. Another study, using a human glioblastoma cell line, found that ZIKV dysregulates axonal guidance signaling proteins including EPHA3, which showed significantly reduced expression [15]. Hence, downregulation of the EpiMod centered around EPHA3 may similarly be due to the effects of ZIKV infection on brain development, which consequently leads to microcephaly.
Overall, our data are consistent with epigenetic changes associated with ZIKV infection in utero playing an important role in determining congenital developmental anomalies. Retention of this epigenetic signature could continue to influence development into childhood.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Author contributions. D. A. analyzed and interpreted the methylation array data and drafted the manuscript. J. I. C. F. N. conducted field work, subject recruitment, and clinical ascertainment. C. R. M. S. collected and processed blood, and performed serological assays. J. G. V. performed serological assays for ZIKV and DENV. J. M. G. D. performed qRT-PCR of RNA isolated from blood and placenta. M. D. S. B. N. and R. C. C. B. aided recruitment of ZIKV children exposed intrautero and their mothers. N. M. R. A. aided patient recruitment and ascertainment of phenotypes. T. L. interpreted the methylation array analysis. J. M. B. co-led the study, reviewed the clinical database, and assisted in drafting the manuscript. S. M. B. J. was lead investigator of the study in Brazil, assisted with clinical evaluation of participants, sample collection, preparation of samples for methylation arrays, data cleaning for clinical database, and assisted with preparation of the manuscript. All authors reviewed and approved the final manuscript.
Financial support. This work was supported in part by Wesfarmers (Seed Grant to J. B. at the Telethon Kids Institute); Coordination for the Improvement of Higher Education Personnel, Ministry of Education, Brazil (grant number 440893/2016-0); and National Council for Scientific and Technological Development (CNPq), Brazil (grant number 88881.130729/2016-01), PPSUS – FAPEMA/MSDECIT/ CNPq/SES/Nº 008/2016.
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
Author notes
T. L., J. M. B., and S. M. B. J. are joint senior authors.


{kind=link}
{kind=link}