






Abstract
The interpretation of serum procalcitonin (PCT) levels in septic patients is facilitated by reviewing the known stimuli that activate the PCT family of genes. Herein we describe 7 pathways that, alone or in combination, can increase serum PCT levels. As a marker of activation of innate immunity, high PCT levels affect clinical diagnosis, can be trended as a measure of “source” control, and can guide duration of antibacterial therapy in septic patients. Low PCT levels reflect little to no activation of an innate immune response, influence the differential diagnosis, and support the discontinuation of empiric antibiotic therapy. Understanding the pathways that result in elevated serum PCT levels is necessary for interpretation and subsequent clinical management.
Procalcitonin (PCT) was first reported as a precursor of calcitonin in 1975 [1]. In 1991, PCT production by medullary carcinoma of the thyroid was described [2]. Elevated PCT levels in burn patients with pneumonia was reported in 1992 [3]. The first article to suggest that PCT levels might discriminate viral from bacterial infection was published in 1993 [4].
PCT elevation after endotoxin injection in human volunteers was reported in 1994 [5], and elevation of PCT due to disseminated candidiasis in a 4-year-old in 1995 [6]. Also in 1995, it was reported that PCT levels varied with the severity of illness in 43 patients with melioidosis [7]. In 1996, there were 4 relevant publications, the first reporting that sequential PCT levels can guide resolution of pneumonia and, hence, treatment duration [8] and the second that immunosuppression did not prevent increases in PCT in septic patients [9]. The third report concerned a prospective study of 337 adults who were admitted with systemic inflammatory response syndrome. In patients with microbiologically documented infection, PCT levels were >0.5 ng/mL, and negative predictive values were very high at lower PCT levels [10]. Finally, in the fourth study, PCT was used successfully as a marker of bacterial infections [11].
Since 1996, the role of PCT levels in patient management has progressively increased. There are now more than 6300 PCT literature citations. The role of PCT in management of intensive care unit patients was summarized in 2017 [12]. The current article describes the recognized pathways to an elevated PCT to facilitate interpretation of both elevated and nonelevated levels.
PCT BIOLOGY
Procalcitonin is the 116 amino acid precursor protein for the 32 amino acid calcium regulating hormone calcitonin. Calcitonin is produced by the thyroid gland C cells in response to elevation in serum calcium concentrations. In contrast, the gene for PCT is found in virtually all human cells and, in the absence of proper stimulus, PCT is constitutively produced at very low levels [13, 14]. In a hamster model of Escherichia coli peritonitis, 2–100-fold increases in PCT messenger RNA occurred rapidly in every major organ, tissue, and cell type [15]. Subsequently, when low doses of lipopolysaccharide (LPS; endotoxin) were infused into human volunteers, there was a 50-800-fold increase in PCT levels within 4–6 hours, and levels peaked after 24 hours [5]. The volunteers also demonstrated concomitant increases in proinflammatory cytokines (tumor necrosis factor [TNF] and interleukin 6 [IL-6]) and the anti-inflammatory interleukin 10 (IL-10) [5].
In distinction to stimulation by bacteria or bacterial LPS, exposure of tissue or organs to only respiratory viruses does not activate PCT genes [13]. In a seminal in vitro experiment, fat cells were shown to produce PCT when incubated with proinflammatory interleukin 1β; interleukin 1 (IL-1)–stimulated PCT production was totally blocked by the presence of interferon (IFN) γ [16, 17]. Hence, PCT blockade by IFN-γ is believed to be the biologic basis of the ability of PCT levels to discriminate viral from bacterial respiratory tract infections.
The ability of PCT levels to distinguish bacterial from viral infection has been clinically confirmed in studies of patients with bacterial versus viral meningitis, virus-only influenza, and other viral respiratory tract infections [18–25]. Of note, bacterial superinfections of viral lower airway infections overcome (trump) IFN- γ suppression of PCT production. Extracellular bacteria can increase PCT levels by either of 2 pathways: (1) direct exposure of patient tissue to bacterial cell wall constituents in the absence of proinflammatory cytokines or (2) host cell exposure and response to the proinflammatory cytokines produced by phagocytic cells [26].
PCT genes are not activated by intracellular cell wall–deficient bacteria (eg, Mycoplasma pneumoniae and Chlamydophila pneumoniae [19]). A summary of microorganisms that do, or do not, increase serum PCT levels is provided in the 2017 review article [12, table 4].
The magnitude of the PCT response to extracelluar bacteria varies. The response to host invasion by gram-negative bacteria is greater than the response to invasion by gram-positive cocci [27, 28]. The mechanism of this difference may be greater concomitant production of IFN- γ in response to gram-positive bacteria [29, 30]. Toxic shock syndrome toxins and clostridial species toxins increase proinflammatory cytokines that, in turn, stimulate production of PCT. Active infections due to rickettsiae, spirochetes, mycobacteria, fungi, or protozoan pathogens vary in their stimulation of PCT [12].
PCT serum levels differ from C-reactive protein (CRP) levels in several ways. CRP levels increase in response to both viral and bacterial infections. CRP levels are suppressed by glucocorticoids and nonsteroidal anti-inflammatory agents, and PCT responses are not suppressed. CRP levels rise in most rheumatologic diseases. In contrast, PCT levels remain low in gout, pseudogout, rheumatoid arthritis, polyarteritis nodosa, and temporal arteritis [31, 32].
IS ELEVATED PCT GOOD OR BAD?
There is no definitive answer. There is no response to giving normal volunteers intravenous PCT. PCT neutralizes LPS in vitro. In vitro, PC increases the production of proinflammatory cytokines by leukocytes [26]. In contrast, in animal models of peritonitis, exogenous PCT versus placebo increased mortality rates in the PCT-dosed animals. In separate experiments, PCT antisera reduced mortality rates in the animals with peritonitis. In sum, the data support the concept that PCT amplifies the acute innate immune inflammatory response to bacterial invasion [26].
INTERPRETING ELEVATED SERUM PCT LEVELS IN PATIENTS IN SHOCK
Invasion of the host by bacteria and other microbes directly activates PCT transcription in virtually all organs and tissue [15]. Furthermore, contact of the invader with phagocytic cells results in release of proinflammatory cytokines that amplify the stimulus for elevating PCT serum levels.
Elevated PCT levels are documented in patients with all varieties of shock syndromes: cardiogenic shock [33], anaphylactic shock [34], distributive shock (eg, bacteremia), mushroom poisoning [35], and posttraumatic crush injury with rhabdomyolysis [36]. To interpret an elevated PCT level, it is helpful to consider the 7 potential reasons for elevation Figure 1. Two can occur in the absence of sepsis or septic shock. The others are pathways to activation of an innate immune response.
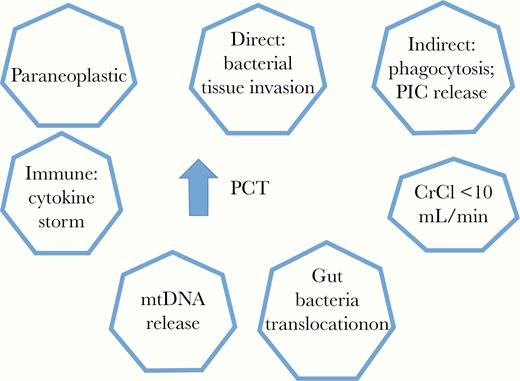
Major variables that can result in elevation of serum levels of procalcitonin (PCT). Abbreviations: CrCl, creatinine clearance; mtDNA, mitochondrial DNA; PIC, proinflammatory cytokine.
Influence of Renal Function on Serum PCT Levels
In volunteers with normal renal function who are injected with endotoxin, PCT levels peak at 24 hours and then decline with a serum half-life that ranges from 24 to 36 hours [5]. Patients with end-stage renal disease and no evidence of infection have elevated serum PCT concentrations. The mechanism may be impaired renal clearance. Alternatively, or in addition, proinflammatory cytokines accumulate in renal failure and may stimulate PCT production [37].
In one well-controlled study, PCT levels were measured before and after hemodialysis 3 times weekly in uninfected patients [38]. The mean level (standard deviation) before dialysis was 0.23 (0.08 ± 0.90) before versus 0.12 (0.03 ± 1.41) after dialysis [38]. Hemodialysis not only removes PCT but also removes some of the proinflammatory cytokines [39].
For clinical cutoffs, it is prudent to obtain PCT levels just before starting a hemodialysis session or any time if patients are critically ill and receiving continuous venous-venous hemodialysis. In septic patients with end-stage renal disease, predialysis serum PCT levels >1 (rarely 1.5) are not due to renal insufficiency and suggest the possibility of active bacterial infection.
Tumors
Neuroendocrine tumors of the C cells of the thyroid (medullary carcinoma), lung, or pancreas can secrete PCT and elevate serum PCT levels into the thousands in the absence of fever or other evidence of an inflammatory response [13, 40].
Bacteremic Shock and PCT
Microbial cell wall constituents (LPS and peptidoglycan) and/or bacterial toxins directly stimulate production of PCT by virtually all tissues, organs, and cell types. In addition, the host inflammatory response generates proinflammatory cytokines (TNF, IL-1, IL-6, and interleukin 2 and 17) that amplify the synthesis of PCT [13]. The intensity of the response varies as follows: (1) PCT levels are higher in response to infection due to gram-negative versus gram-positive bacteria [27, 28]; (2) PCT levels are higher with acute versus subacute or chronic infection (eg, chronic empyema), possibly because of the influence of IL-10 and other concomitant anti-inflammatory cytokines [41, 42]; (3) the peak PCT level varies with the severity of the infection; the PCT peak with nontransmural appendicitis is lower than that in transmural perforated appendicitis with peritonitis [43]; and (4) as described below, comorbid conditions may stimulate additional PCT production by different pathogenic pathways and can reflect multiple concomitant causes of shock (eg, septic shock plus cardiogenic shock).
Translocation of Gut Bacteria
Patients with end-stage hepatic cirrhosis are often admitted for gastrointestinal bleeding and/or encephalopathy. The patients may be afebrile but hypotensive. Assume no evidence of infected ascites and that renal function is adequate and hemoglobin levels are stable. Why is the patient hypotensive? There is mounting evidence that translocation of gut bacteria needs consideration [37, 44–46].
With cirrhosis and portal venous hypertension, gastrointestinal mucosal defense mechanisms break down, allowing enteric flora to penetrate into the submucosa, mesenteric lymph nodes and/or portal venous blood. Systemic venous blood cultures are rarely positive. Laboratory markers of gut translocation include positive tests for serum endotoxin (LPS), LPS-binding protein, soluble CD14 (commercially called presepsin), and serum PCT levels >0.25 ng/mL.
Translocation has been implicated as a cause of elevated PCT levels in patients with mesenteric ischemia [47], hemorrhagic shock [48], cardiogenic shock [33, 49], or anaphylactic shock [34]. Despite the complexities, PCT levels in septic shock patients can help management. Elevated PCT levels do not clarify whether the patient has septic shock rather than other shock syndromes. However, PCT levels <0.5 ng/mL in septic- appearing patients virtually exclude bacteremia, because as the reported negative predictive values are roughly 93% [50–53].
Secondly, sequential PCT levels reflect success, or failure, in improving the process (eg, congestive heart failure) that is driving the translocation [54–56]. Finally, sequential PCT levels can customize the duration of antibacterial therapy. The general guideline is to treat until the PCT level decreases by 80% from its peak (if known) or until the PCT level is <0.5 ng/mL, assuming reasonable renal function [57, 58].
Mitochondrial DNA
Damaged mitochondria induce an acute to subacute inflammatory response believed due to evolutionary history [59, 60]. Mitochondria evolved from saprophytic bacteria that became endosymbionts and eventually mitochondria. The DNA of mitochondria reflect their prokaryotic origin by the presence of CpG DNA repeats. When injured, mitochondrial DNA (mtDNA) is released into the circulation. Owing to similarity to prokaryotes, the host responds with an inflammatory response to include IL-1, TNF, and other proinflammatory cytokines that stimulate the production of PCT [59].
Release of mtDNA in response to myocardial infection, mushroom poisoning with liver necrosis, pulmonary embolus with infarcted lung, trauma with rhadomyolysis, severe burns, and other sterile necrosis syndromes induces an inflammatory response with high PCT levels [61, 62]. Alternatively, invasive bacterial infections damage mitochondria and the released mtDNA induces and/or augments inflammation. Hypoxia of mitochondria can complicate all types of shock, septic and nonseptic, with a subsequently cytokine-mediated inflammatory response [63]. Studies in the critically ill report a correlation of mtDNA levels with increased mortality [63–65].
Neoplasia and Cytokine Storms
The paraneoplastic production of PCT by medullary thyroid, lung, and pancreatic neuroendocrine tumors was mentioned above [2, 13, 40]. In addition, select tumors (eg, some T-cell lymphomas) can produce a macrophage activation syndrome that increases PCT serum levels (also called hemophagocytic lymphohistiocytosis syndrome). This syndrome is typified by phagocytosis of histologic red blood cells by macrophages, highly elevated serum ferritin levels. and overproduction of granulocyte colony-stimulating factor, IL-1, IL-6, and other cytokines that increase PCT serum levels [66–68].
Advances in the immunotherapy of cancer activates T cells, which in turn leads to immune-related adverse events. Both immune checkpoint inhibitor drugs and targeted chimeric antigen receptor-modified T cells can attack nonneoplastic cells with stimulation of proinflammatory cytokines and associated synthesis of PCT [69, 70]. Alemtuzumab (CD20 antibody), interleukin 2, rituximab (CD20 antibody), and other immunomodulator drugs are also associated with high PCT levels [12]. In contrast, some neoplasms known for their association with fever, night sweats, and weight loss (eg, lymphoma, pancreatic cancer, renal cell carcinoma, and sarcoma) are only rarely associated with elevation of serum PCT levels.
Mixed Stimuli
Critically ill patients often have concomitant stimuli that promote transcription of the PCT genes. Consider hypotensive cirrhotic patients with severe portal hypertension, renal insufficiency, evidence of myocardial injury, and new pulmonary infiltrates. There are at least 3 or 4 pathways to an elevation of PCT, and we have limited tools to sort out the contribution of each. In contrast, if, despite comorbid conditions, hypotensive patients have PCT levels <0.05 ng/mL, there is a <5% chance of invasive bacterial infection as a cause of the low blood pressure [12].
USE OF SERUM PCT LEVELS IN SEPTIC PATIENTS
Despite the many variables that can influence serum PCT levels, PCT levels can assist in the management of patients with septic shock. PCT elevation in a patient with reasonable renal function indicates stimulation of the patient’s innate immune response [71]. The specificity for bacterial invasion depends on the clinical setting (pretest probability), the pathogen and its virulence, and host comorbid conditions. Positive predictive values are high in septic intravenous drug users and low in patients with mild lower respiratory tract infection [27, 28, 72].
The magnitude of the peak PCT level is correlates with the intensity of innate immune response (eg, progression from mild nontransmural appendicitis to perforation with peritonitis and bacteremic shock [43]). In contrast, a PCT level ≤0.25 ng/mL in a patient with hemodynamic shock strongly suggests a nonbacterial cause for the patient’s hypotension, because the negative predictive values are >90% [12, 18–25]. A “normal” PCT level is ≤0.05 ng/mL; hence, a value of 0.25 ng/mL may indicate a rising PCT ;eve;. This explains the suggestion to repeat PCT measurement after 4–6 hours before considering discontinuation of empiric antibacterial therapy. Collected case series document the safety of this approach, with the added benefit of fewer antibiotic-associated adverse events [57, 58, 73–77].
The role of PCT levels in patients with hemodynamic shock and severe community-acquired pneumonia is changing. In earlier studies, the microbial etiology of the community-acquired pneumonia was identified in <50% of patients [21–23]. Elevated PCT levels were extensively evaluated as surrogate markers of bacterial invasion. The statistical performance was modest owing to the absence of a clear reference standard and the high frequency of undetected potential pathogens [23].
Potential bacterial and viral pathogens are detected with increasing frequency using rapid molecular polymerase chain reaction diagnosis [24]. The emerging role of PCT is not as a surrogate for detection but as an assist in the adjudication of detected potential viral and bacterial pathogens [23–25].
There are 2 other interpretative roles for PCT in patients with septic shock due to proved, or clinically probable, bacterial infection. Sequential decreases in PCT levels help document ”source control” of clinical syndrome [54–56]. Second, as noted above, the duration of antibacterial therapy can be individualized by repeating measurement of PCT levels every 2–3 days. Three possible end points are suggested for cessation of antibacterial therapy: a decrease of 80% from peak PCT levels or a decrease to ≤0.05 or ≤0.25 ng/mL [54, 55, 57, 58].
AREAS FOR ADDITIONAL STUDY
Several facets of PCT are in need of additional study. First, IFN- γ suppresses PCT gene activation by proinflammatory cytokines; we need similar studies with IL-10 and other anti-inflammatory cytokines. Second, it is unclear whether chronic low level translocation of gut bacteria worsens the prognosis for survival in septic patients. Should placebo-controlled antibiotic trials be conducted in patients with evidence of chronic translocation of gut bacteria? Next, presepsin serum levels can be assessed as a marker of translocation and subsequent elevated PCT in septic patients. Finally, mtDNA levels are needed in order to compare elevated PCT levels in patients with “sterile necrosis” and no evidence of infection versus invasive bacterial infection. It is likely that future studies will combine rapid polymerase chain reaction pathogen detection panels with PCT serum levels and host gene expression dosage [78].
CONCLUSIONS
The following questions should be asked when interpreting serum PCT levels in septic patients. First, is the patient’s clinical syndrome compatible with infection by extracellular bacteria? Second, is the patient’s kidney injury severe enough (creatinine clearance <10 mL/min) to contribute to the patient’s elevated serum PCT level? Third, is the patient a candidate for translocation of gut bacteria owing to concomitant hepatic cirrhosis, stage 4 heart failure, or arterial mesenteric ischemia? If available, what is the serum presepsin level? Fourth, does the patient have enough parenchymal sterile necrosis to release mtDNA and induce an innate immune response? Fifth, if the patient has strikingly elevated PCT levels and no clinical evidence of inflammation, does he or she have a PCT-producing tumor? And, finally, has the patient received immunomodulation therapy for cancer, transplantation, or connective tissue disease that would explain the elevated serum PCT level?
Note
Supplement sponsorship. This supplement is sponsored by bioMérieux, the Gordon and Betty Moore Foundation and Beckman Coulter.
Potential conflicts of interest. Author certifies no potential conflicts of interest. The author has submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}