






Abstract
Understanding hepatitis C virus (HCV) transmission among people who inject drugs (PWID) is essential for HCV elimination. We aimed to differentiate reinfections from treatment failures and to identify transmission linkages and associated factors in a cohort of PWID receiving opioid agonist therapy (OAT).
We analyzed baseline and follow-up specimens from 150 PWID from 3 OAT clinics in the Bronx, New York. Next-generation sequencing data from the hypervariable region 1 of HCV were analyzed using Global Hepatitis Outbreak and Surveillance Technology.
There were 3 transmission linkages between study participants. Sustained virologic response (SVR) was not achieved in 9 participants: 7 had follow-up specimens with similar sequences to baseline, and 2 died. In 4 additional participants, SVR was achieved but the participants were viremic at later follow-up: 2 were reinfected with different strains, 1 had a late treatment failure, and 1 was transiently viremic 17 months after treatment. All transmission linkages were from the same OAT clinic and involved spousal or common-law partnerships.
This study highlights the use of next-generation sequencing as an important tool for identifying viral transmission and to help distinguish relapse and reinfection among PWID. Results reinforce the need for harm reduction interventions among couples and those who report ongoing risk factors after SVR.
Hepatitis C virus (HCV) infection is the leading cause of end-stage liver disease, hepatocellular carcinoma, and liver transplantation in the United States [1–3]. HCV-related deaths have surpassed those from HIV, and the disease burden is predicted to increase up to 3-fold over the course of the next 10-20 years [4, 5]. People who inject drugs (PWID) are at the heart of the HCV epidemic, comprising 60% of all HCV-infected persons in the United States and >80% of new HCV infections [6, 7].
Direct-acting antiviral therapies have revolutionized HCV treatment. The current all oral regimens have cure rates of >90% for all HCV genotypes with few adverse effects [8]. To achieve the World Health Organization’s goal for HCV elimination by 2030 [9], prevention of HCV transmission among PWID is critical [10]. For PWID who undergo HCV treatment, prevention efforts need to avert (1) forward transmission among those who fail treatment and (2) reinfection in those who are treated and in whom HCV cure is achieved. Differentiating reinfection from treatment failure has important clinical implications. Most individuals who are reinfected after HCV treatment exhibit high-risk behaviors and may be at higher theoretical risk for forward transmission to others [11]. Conversely, individuals who fail treatment are at high risk of having resistance-associated substitutions (RASs) after HCV treatment [12]. RASs can complicate retreatment efforts and can be transmitted to others with ongoing high-risk behaviors [13].
Next-generation sequencing (NGS) can be used to assess the degree of similarity among HCV viral strains within and between individuals. As such, one application of NGS is for differentiating reinfection from treatment failure. If an individual has a genetically different viral strain after treatment, this implies a reinfection. Recurrence of a similar viral strain at follow-up suggests treatment failure. Additional uses of NGS include identification of superinfections (ie, infections with multiple viral strains) associated with multiple exposures [14–17], as well as the degree of similarity of HCV viral strains between individuals. This technology has been used to demonstrate transmission linkages in networks of PWID [18–21] and in closed prison settings [22]. NGS generates multiple viral sequences from infected individuals, which can be used to construct phylogenetic networks allowing the historical relationships between infected individuals in a population to be modeled [23]. Distinct transmission clusters can be identified and associated with individual level characteristics that account for their transmission [24, 25]. Intrahost viral heterogeneity can also provide an estimate on the duration of time since transmission [26].
To our knowledge, no studies have addressed transmission networks among PWID engaged in opioid agonist therapy (OAT). Nationwide, >375 000 patients receive OAT (methadone or buprenorphine) from approximately 1500 opioid treatment programs (OTPs) [27], and conservative estimates suggest that >60% of PWID in OTPs are infected with HCV [6]. OAT has been shown to effectively reduce the incidence of HCV infection [28]. However, a subset of PWID engaged in OAT continues to inject leading to ongoing risk exposure.
The current study had 2 aims: (1) to evaluate the use of NGS in differentiating reinfections from treatment failures, and (2) to identify transmission linkages and associated factors among a cohort of 150 PWID being treated for HCV infection at 3 OTPs in Bronx, New York. We propose that these findings will have important implications to inform targeted interventions among PWID on OAT.
METHODS
Study Participants, Setting, and Specimen Handling
Participants in this study were part of the PREVAIL study, a randomized controlled trial examining the effectiveness of 3 models of HCV care (directly observed therapy, group treatment, and self-administered individual treatment) among 150 PWID receiving OAT with genotype 1 HCV infections [29]. This study took place in 3 OTPs in Bronx, New York. The 3 models of HCV care were distributed evenly within OTPs because randomization occurred in each OTP. The study was approved by the institutional review board of Albert Einstein College of Medicine.
As part of the trial, we developed a biorepository that contained samples from the following PREVAIL study visits: baseline, treatment week 4, end of treatment (EOT), and posttreatment weeks 4 (PT4), 12 (PT12), and 24 (PT24) [30]. In the parent study we assessed for sustained virologic response (SVR), or HCV cure, denoted by undetectable HCV RNA at PT12 using the COBAS Ampliprep/Taqman assay (version 2.0; Roche Diagnostics). The PT24 study visit was used to assess short-term reinfection rates. We retained 114 participants in whom SVR was achieved for reenrollment in an extension study to assess for reinfection and ongoing risk behaviors [31]. PREVAIL extension study visits were targeted every 6 months after PT24 for up to 2 years. We collected blood samples at extension study visits, which were added to our biorepository. All samples were sent to Monogram Biosciences for assessment of RASs. If HCV RNA was found to be detectable, deidentified blood samples were sent from our biorepository to the Centers for Disease Control, Division of Viral Hepatitis Laboratory, for NGS.
Global Hepatitis Outbreak and Surveillance Technology and Phylogenetic Analysis
Global Hepatitis Outbreak and Surveillance Technology (GHOST) is a cloud-based technology that enables users, regardless of computational expertise, to use NGS amplicon data of viral hepatitis target genes to analyze and visualize transmission clusters in an independent, accurate, and reproducible way [32]. To perform genetic analysis, we applied NGS to the hypervariable region 1 (HVR1) of the HCV genome, as described elsewhere [33]. HVR1 is the most variable; hence, it is commonly used in molecular epidemiologic studies to detect clusters of persons infected via transmission events [34]. The NGS raw data were subject to quality control in the GHOST pipeline, generating individual haplotypes with associated frequencies.
All haplotypes were then genotyped by matching to the HVR1 database in GHOST. To detect and visualize linkages, all haplotypes with frequency ≥10 were analyzed by measuring the genetic distances between sequences and creating linkages where the distance was below the experimentally established cutoff of 0.037 to generate networks [32]. For phylogenetic analysis, all haplotypes with frequency ≥2 were used to create alignments and build neighbor joining trees using Kimura 80 distance parameters. The trees were presented as radial phylograms in CLC Genomics Workbench software (version 11) (Qiagen Aarhus). All sequences from this study were submitted to GenBank.
Differentiating Reinfection From Treatment Failure
For participants who were viremic after their baseline study visit, we created phylogenetic trees using sequences from all available time points and compared on-treatment and posttreatment viral strains to assess whether the participant became viremic with a similar viral strain to his or her baseline (suggesting treatment failure) or a different viral strain (suggesting reinfection) using the same genetic distance cutoff of 0.037. We also report the presence of RASs using Sanger-based consensus sequencing relative to H77 (genotype 1a; GenBank accession no. AF009606) and Con1 (genotype 1b; GenBank accession no. AJ238799) reference sequences to explain the characterized treatment failures and help differentiate late treatment failure from reinfection.
Study Measures, Outcomes, and Data Analysis
We used GHOST to examine whether there were any transmission linkages among PREVAIL participants [32, 35]. Phylogenetic analysis was used to differentiate treatment failure from reinfection. Data on injection drug use (IDU) and HCV treatment adherence, described elsewhere [30], were used to support the characterization of reinfection versus treatment failure in certain cases.
We then performed an integrated analysis of molecular, demographic (age, sex, race/ethnicity), social network (Norbeck social support questionnaire) [36], and geographic data (OTP facility). Our primary outcome was the presence of transmission pairs or clusters among PREVAIL participants at baseline, follow-up, or extension study time points. By restricting our cohort to a single borough in New York City, we anticipated that transmission networks would be evident because such connections have been found among small samples of blood donors in large geographic areas [37]. We hypothesized that patients attending the same OTP would be more likely to be linked than patients attending different OTPs. A post hoc analysis was conducted to assess social network and treatment setting characteristics associated with transmission linkages.
RESULTS
Of the 150 PREVAIL study participants, the majority (75%) reported a history of IDU. IDU was reported in the 30 days before HCV treatment, PT12, and PT24 by 59 (52%), 55 (49%), and 53 (47%), respectively. In the extension study cohort, 22 (19%) reported ongoing IDU. Full demographic and risk behaviors for the parent and extension studies are reported elsewhere [29–31]. Among all PREVAIL participants, 128 (85%) were infected with HCV genotype 1a and 22 (15%) with genotype 1b (Figure 1). Using NGS, 1 participant with genotype 1a (participant 2041) was found to have a baseline genotype 3a coinfection (data not shown).
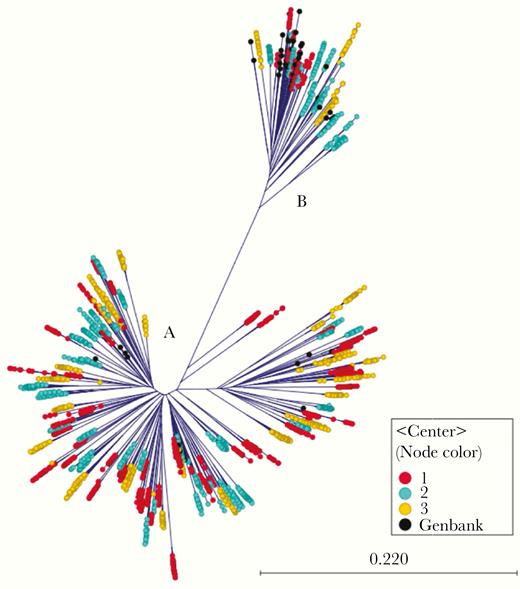
Radial phylogram showing all hepatitis C virus (HCV) genotype 1a (n = 128; n = 3622 haplotypes) and genotype 1b (n = 22; n = 602 haplotypes) isolates at baseline, with color coding to indicate opioid treatment program center.
Transmission Linkages
At baseline, there were 2 pairs of participants who were linked by transmission (Figure 2A). Both pairs of participants attended the same OTPs and were involved in spousal or common-law partnerships. Intermixing of intrahost HCV variants and a greater genetic diversity of HCV population in one of the partnerships demonstrates that the female partner (participant 1026) was infected more recently than the male partner (participant 1014) suggesting that the male partner may have transmitted HCV to the female partner, though additional individuals in this transmission chain cannot be ruled out (Figure 2B). Separation of intrahost HCV populations from participants 2022 and 2033 suggests that transmission occurred in a more distant past or that both participants were exposed to HCV from an unidentified source (Figure 2C). An additional transmission linkage was identified after treatment between participants 1075 (male) and 1046 (female), who both also attended the same OTP but who were not in a spousal or common-law relationship and will be discussed later.
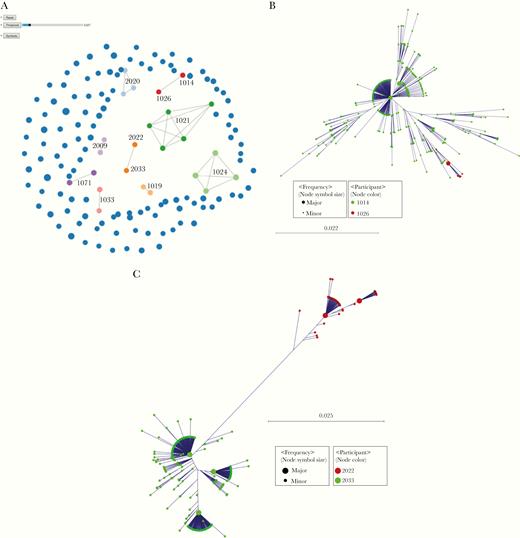
Transmission clusters identified using Global Hepatitis Outbreak and Surveillance Technology (GHOST). A, Blue circles indicate participants not related to anyone else in the network. The size of the circle is proportional to the number of different haplotypes found in the participants. Connected circles of the same color represent follow-up specimens from the same participants or transmission if they link different participants.. Two pairs were linked by transmission at baseline (pairs 1014-1026 and 2022-2033). B, Baseline transmission cluster of participants 1014 and 1026 (n = 449 haplotypes; frequency, >2). C, Baseline transmission cluster of participants 2022 and 2033 (n = 449 haplotypes; frequency, >5).
Treatment Failures
SVR was not achieved in 9 participants. Two died during HCV treatment and therefore did not have follow-up specimens, leaving 7 participants who had treatment failure at the EOT or PT12 time points with pretreatment and posttreatment sequences. These participants (participants 1021, 1024, 1035, 1046, 1080, 2009, and 2020) had similar sequences at baseline and after treatment, consistent with treatment failure (Figure 3A and 3B).


Phylogenetic trees of hepatitis C virus (HCV) hypervariable region 1 variants sampled at baseline and follow-up time-points from participants in the PREVAIL in whom treatment failed. Abbreviations: BL, baseline; EOT, end of treatment; PT4, posttreatment week 4; PT12, posttreatment week 12; PT24, posttreatment week 24; TW4, treatment week 4. A, HCV genotype 1a (participants 1024, 1035, 1046, 1080, 2009, and 2020) (n = 795 haplotypes; frequency, >5). B, HCV genotype 1b (participant 1021) (n = 117 haplotypes; frequency, >10). C, HCV genotype 1a (participant 2020 (n = 409 haplotypes; frequency, >2). Arrow indicates a haplotype present at baseline at low frequency. D, Participant 2009. The major haplotypes at EOT and PT4 are identical (n = 730 haplotypes; frequency, >2).
Of the 7 participants with treatment failures, with baseline and on-treatment or posttreatment detectable sequences, 3 were treated with sofosbuvir-ribavirin and 4 with sofosbuvir-ledipasvir. Among the 3 with sofosbuvir-ribavirin treatment failure, 1 had HCV NS5b (sofosbuvir)–specific RASs emerging in their on-treatment or posttreatment specimens. Among the 4 with sofosbuvir-ledipasvir treatment failures 3 had NS5a (ledipasvir)–specific RASs emerging in their on-treatment or posttreatment specimens.
Two participants, 2009 and 2020, had follow-up haplotypes that seemed to be genetically distant from their baseline (Figures 3C and 3D). In these participants, data on RASs, IDU, and/or HCV treatment adherence were used as complementary information to establish treatment failure. Participant 2020 (Figure 3C) was treated with sofosbuvir-ledipasvir. This participant had HCV genotype 1a infection with 2 distinct viral strains at baseline. Sofosbuvir-ledipasvir cleared the dominant strain, and phylogenetic analysis confirmed that the infection observed during follow-up resulted from an overgrowth of the minority strain that was present at baseline. At baseline, there were no NS5a mutations detected by consensus sequencing. At the PT12 and PT24 study visits, we observed Q30H, Y93H RASs conferring ledipasvir resistance.
Participant 2009 (Figure 3D) was treated with sofosbuvir-ribavirin. This participant’s HCV RNA levels were never fully suppressed. The participant’s baseline specimen was phylogenetically distinct from EOT, but the major haplotypes at EOT and PT4 were identical. Reinfection with a similar strain was possible, particularly since the blood sample that was sent for phylogenetic analysis was obtained 15 days after the participant completed treatment; however, this participant had a Q80K mutation at all available time points from baseline to PT4 and denied IDU 30 days before the baseline and EOT visits. This, and the participant’s average daily adherence of 45%, suggests treatment failure.
Late Treatment Failure Versus Reinfections
Four participants were viremic after SVR. Participant 1019 had a very heterogeneous genotype 1a viral population at baseline, suggesting infection with >1 HCV strain. This participant had SVR achieved with sofosbuvir-ribavirin but had a late treatment failure with viremia at the PT24 time point. Phylogenetic analysis suggests that the strain observed during follow-up likely resulted from an evolution of minority variants (Figure 4). It is possible that participant 1019 was reinfected by a partner with a similar strain; however, this is unlikely owing to the presence of similar NS5b RASs at the PT24 time point compared with treatment week 4, as well as a highly heterogeneous HCV population at extended 12-month follow-up (EXT12). Moreover, this participant’s average daily adherence was 53%, which provides further support to probable treatment failure. In participant 2019, the baseline genotype 1a infection cleared; however, a new genotype 1a HCV population emerged at PT24. The new population expanded at EXT12, suggesting a possible reinfection with a different HCV strain (Figure 5A).
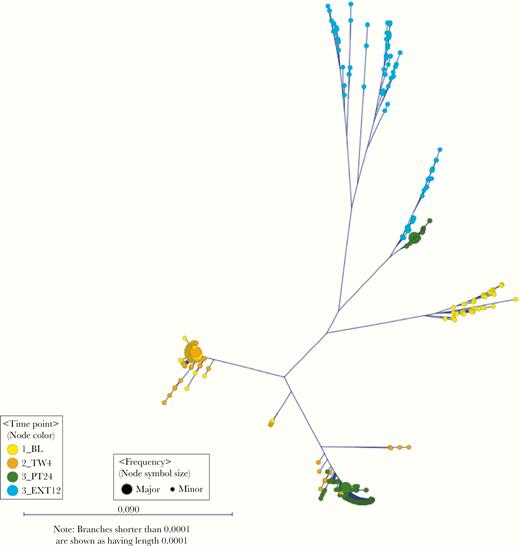
Phylogenetic tree of hepatitis C virus hypervariable region 1 variants sampled from participant 1019 at different time points (n = 730 haplotypes; frequency, >5), in whom sofosbuvir-ribavirin treatment failed.


Phylogenetic trees of hepatitis C virus (HCV) hypervariable region 1 (HVR1) variants sampled at baseline and follow-up time points from participants in the PREVAIL with probable reinfections. A, Phylogenetic tree of HCV HVR1 variants sampled from participant 2019 at different time points (n = 1018 haplotypes; frequency, >2). B, Phylogenetic tree of HCV HVR1 variants sampled from participants 1075 and 1046 at different time points, excluding the genotype 3a strain unrelated to any PREVAIL study members (n = 397 haplotypes; frequency, >2). Abbreviations: BL, baseline; EXT6, extended 6-month follow-up; EXT12, extended 12-month follow-up; PT4, posttreatment week 4; PT12, posttreatment week 12; PT24, posttreatment week 24.
Participant 1075 was reinfected with phylogenetically distinct strains. The baseline original 1a infection had cleared, and this participant was subsequently reinfected with a distinct 1a strain shared by another PREVAIL study participant (participant 1046), and a genotype 3a strain unrelated to any PREVAIL study members (Figure 5B). Participant 1020 was only transiently viremic 17 months after treatment, with no specimen sent for NGS. Participants 2019, 1075, and 1020 reported ongoing high-risk behaviors, which is supportive of reinfections in these participants [38].
DISCUSSION
This is the first study to use NGS to characterize HCV phylogenetic networks in a system of urban OTPs. Although OAT provides relative stability from active drug use, understanding patterns of transmission in this setting is important because a subset of OAT recipients continues to inject, leading to ongoing risk exposure. Using NGS, we were able to detect baseline transmission linkages, all of which occurred among spousal or common-law partners. We also identified one transmission linkage after reinfection. All transmission linkages occurred within the same OTPs. Use of NGS in conjunction with epidemiologic and clinical data was instrumental in differentiating HCV treatment failures from reinfections. Although NGS has been used in several studies to help differentiate treatment failure from reinfection [39, 40], this is among the first studies to present phylogenetic data.
The first study to describe a phylogenetic analysis among a large network of PWID was conducted in Melbourne, Australia. In this study, 18 clusters comprised 66 distinct pairs were identified among 138 PWID. In total 51 of 138 participants (37%) had related infections and 12 of 66 pairs (18%) were injection partners [21]. The number of transmission linkages observed in our study is low by comparison. This may be explained by the differences in study settings, recruitment methods and genetic methods used to identify transmission linkages. Aitken et al [21] used respondent-driven sampling as a recruitment strategy in an inner municipality in Melbourne known to have a large illicit drug market. More recent studies reporting higher numbers of transmission linkages took similar approaches to recruitment using respondent-driven or chain-referral sampling [18].
Our findings are consistent with those of studies conducted in more controlled settings. For example, Bretaña et al [22] identified 3 transmission clusters among a small population (n = 79) of PWID living with HCV in an Australian prison. In addition, the phylogenetic proximity of consensus sequences from infected persons in those studies is less accurate [in] identifying transmission than the NGS-based GHOST approach that we used in the current study [32]. Thus, differences in molecular technologies may also contribute to variation in the detection of actual transmission clusters.
Although we did not detect large transmission clusters among PREVAIL study participants, we detected 3 distinct pairs of interconnected HCV infections. We hypothesized that transmission linkages would more likely be found among members of the same OTP facility, which was indeed the case for all 3 linkages we identified. Transmission linkages were further characterized by the Norbeck social support questionnaire, revealing the 2 baseline transmission pairs were involved in spousal or common-law partnerships. Moreover, both individuals in these pairs referenced each other as primary sources of social support in these questionnaires. Sexual transmission of HCV among heterosexual monogamous couples is unlikely [41]. However, PWID who cohabitate and have a sexual relationship with their injection partners are more likely to engage in high-risk injection practices [42]. This finding has important implications for counseling PWID regarding the risk of transmission to their partners, providing sterile injecting equipment to these individuals, and, perhaps, prioritizing treatment of such pairs to prevent HCV reinfection after treatment of one infected partner.
Phylogenetic analysis has been used to help distinguish treatment failure from reinfection [39, 40]. However, few studies thus far have presented phylogenetic data [43, 44]. Distinguishing reinfection from treatment failure has implications for selecting optimal retreatment strategies. For those who experience treatment failure, RASs are common, particularly with NS5a inhibitor–containing regimens, such as the ledipasvir-containing regimens used in the current study [12]. Those with treatment failure need to be considered for retreatment with second-line direct-acting antiviral regimens. In our study, we demonstrated the utility of phylogenetic analysis to help differentiate late treatment failure from reinfection. Late treatment failure is rare [43]; however, if it is not detected and reinfection is suspected, treating an infection as a primary infection could lead to the use of a first-line regimen, which may not be as effective.
However, distinguishing late treatment failures from reinfections may not be as straightforward as application of NGS to genetic characterization of HCV strains. In our study, participants 1019 and 2019 present a serious challenge to this simple characterization. Their HCV genetic histories are similar. Both were infected with a heterogeneous HCV population at baseline, which cleared on treatment. Both could have experienced HCV reinfection with a genetically distant HCV strain at PT24, which somewhat expanded at EXT12. Most important, a new highly heterogeneous and genetically distant HCV population emerged at EXT12 in both participants. Emergence of such heterogeneous population is inconsistent with recent reinfection. Recent infection is generally associated with a dense and simple, starlike intrahost HCV population [14] and frequently observed during outbreak investigations [24, 25]. The high genetic diversity of these late populations suggests a long history of HCV infection in participants 1019 and 2019.
Emergence of such highly diverse and genetically distant HCV population after treatment is unusual. It may indicate that these subpopulations originated from minority HCV variants surviving on treatment. This scenario is supported by the fact that heterogeneous subpopulations identified at late stages of HCV infection frequently remain cryptic during early stages of infection [14]. A significant genetic distance between the baseline and EXT12 HCV populations and among members of this late population indicates infection of participants 1019 and 2019 with >1 HCV strain, which is not unusual among PWID [45]. This observation warrants further investigation because it has significant implications for clinical management of PWID with late treatment failures. With the emergence of an HCV population genetically distant from baseline, and seemingly resistant to treatment, late treatment failures may be misclassified as reinfection, potentially leading to inappropriate retreatment.
For those who are reinfected, the implications are different but also important because these individuals may exhibit ongoing high-risk drug use. Here we present NGS data for 2 of 3 individuals who appeared to be reinfected in the PREVAIL study. Phylogenetic data suggest new infections with HCV genotype 1a and 3a strains in participant 1075. This participant shared HCV genotype 1a strain with participant 1046 and had HCV genotype 3a strain; both were different from the HCV strain identified at baseline. It is important to note that, in contrast to participants 1019 and 2019, the new HCV populations in participant 1075 were tight, which is consistent with recent infections. Given that individuals who are reinfected may have ongoing high-risk behaviors, they should be considered for targeted intervention and harm reduction [31].In the current study, phylogenetic analysis also distinguished a genotype 3 infection at baseline, which has important implications for settings in which pangenotypic regimens are not yet being, because since these individuals may be prone to treatment failure with regimens directed at other genotypes (eg, 1 or 4 [43]).
The current study has some limitations. First, by performing phylogenetic analysis of 150 PWID enrolled in a randomized controlled trial, we did not use respondent-driven sampling, which is commonly used for network analyses. Therefore, we did not necessarily include other individuals from our OTP programs in Bronx, New York, who are injection partners with study participants. Second, given the small number of transmission linkages, we were not able to identify definitive factors associated with HCV transmission. Third, we cannot exclude the possibility that individuals who have a viral relapse with a similar strain were reinfected by the same injection partner they were injecting with before HCV treatment. Fourth, owing to specimen volume limitation, we were unable to perform deep sequencing of HCV populations to detect minority variants suggested to be ancestors of intrahost HCV subpopulations found after treatment.
In conclusion, we identified 3 HCV phylogenetically related pairs, 2 at baseline and 1 at reinfection. Two baseline transmission linkages occurred in spousal or common-law partnerships. All transmission linkages were detected among participants from the same OTPs. NGS is an important tool to differentiate relapse from reinfection. Given efforts to expand OAT in response to the opioid epidemic, it is essential to ascertain the corresponding effects on HCV epidemiology among those at ongoing risk. The application of phylogenetic analysis to local/regional HCV epidemics may assist with the design of targeted prevention strategies. If these high-risk networks can be identified, harm reduction and HCV treatment services can be delivered to these groups, which will be critical for HCV elimination efforts.
Notes
Disclaimer. The findings and conclusions in this report are those of the authors and do not represent the official position of the Centers for Disease Control and Prevention. Use of trade names and commercial sources is for identification only and does not constitute endorsement by the US Department of Health and Human Services or the Centers for Disease Control and Prevention.
Financial support. This study was funded by the National Institute of Drug Abuse (grants R01 DA034086 and K99DA043011), Gilead Sciences, and the Albert Einstein College of Medicine Liver Center (pilot grant NIDDK P30DK41296).
Potential conflicts of interest. M. J. A. has participated in educational activities supported by independent educational grants from AbbVie and Gilead Sciences. A. H. L. has served on advisory board for Merck Pharmaceuticals, AbbVie, and Gilead Sciences and has received research grants from Merck Pharmaceuticals and Gilead Sciences. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}