






Abstract
Streptococcus pneumoniae infection can result in bacteremia with devastating consequences including heart damage. Necroptosis is a proinflammatory form of cell death instigated by pore-forming toxins such as S. pneumoniae pneumolysin. Necroptosis-inhibiting drugs may lessen organ damage during invasive pneumococcal disease (IPD).
In vitro experiments were carried out with human and mouse cardiomyocytes. Long-term cardiac damage was assessed using high-resolution echocardiography in ampicillin-rescued mice 3 months after challenge with S. pneumoniae. Ponatinib, a necroptosis-inhibiting and Food and Drug Administration–approved drug for lymphocytic leukemia treatment, was administered intraperitoneally alongside ampicillin to test its therapeutic efficacy. Histology of heart sections included hematoxylin-eosin staining for overt damage, immunofluorescence for necroptosis, and Sirius red/fast green staining for collagen deposition.
Cardiomyocyte death and heart damage was due to pneumolysin-mediated necroptosis. IPD leads to long-term cardiac damage, as evidenced by de novo collagen deposition in mouse hearts and a decrease in fractional shortening. Adjunct necroptosis inhibition reduced the number of S. pneumoniae foci observed in hearts of acutely infected mice and serum levels of troponin I. Ponatinib reduced collagen deposition and protected heart function in convalescence.
Acute and long-term cardiac damage incurred during IPD is due in part to cardiomyocyte necroptosis. Necroptosis inhibitors may be a viable adjunct therapy.
Streptococcus pneumoniae (the pneumococcus) is a leading cause of community-acquired pneumonia, responsible for 25%–40% of all cases [1]. S. pneumoniae pneumonia has also been directly linked to major adverse cardiac events (MACEs), with Musher et al [2] finding that 20% of individuals hospitalized for S. pneumoniae pneumonia in a Veterans Administration hospital simultaneously experienced cardiac failure, arrhythmia, or infarct, alone or in combination. More recently, Eurich et al [3] showed that patients who bacteremia developed during hospitalization for pneumococcal pneumonia were specifically at risk for MACEs in convalescence (adjusted hazard ratio compared with controls, 2.4; 95% CI, 1.08–5.3). Moreover, the risk of cardiac complications persisted for up to 10 years after the pneumonia episode [3, 4].
In 2014, we showed that pneumococci in the bloodstream were capable of invading the heart. Within the myocardium, S. pneumoniae can replicate to form a focus of infection, disrupt contractility, and kill cardiomyocytes; together contributing to the multifaceted cardiac dysfunction that is known to occur during severe and systemic infections [5–8]. Foci of pneumococci within the myocardium have been termed “cardiac microlesions,” which can vary morphologically. In some instances, cardiac microlesions present as densely packed pneumococci surrounded by the remnants of dead host cells; in others, they contain fewer bacteria in a dispersed state, are associated with dead or dying host cells, and surrounded or infiltrated by immune cells.
We have determined that the morphological properties of these microlesions depend on the pneumococcal strain responsible for the infection, their maturity at the time of sample collection, and the infecting strain’s ability to produce the pore-forming toxin (PFT) pneumolysin [6]. Critically, in mice and nonhuman primates that had been rescued from invasive pneumococcal disease (IPD) with antibiotics, immune cell infiltration and highly disorganized collagen deposition was seen to occur at sites in the heart where microlesions had been present [5, 9]. This cardiac remodeling was reminiscent of what occurs in humans after infarct [5, 9] and was speculated to be an explanation for the increased incidence of MACEs observed in convalescent individuals; however, this remained unproved.
Necroptosis is a programmed mode of cell death that leads to the disruption of cellular membranes and increased inflammation through the release of alarmins [10]. This proinflammatory cell death program is executed on activation of the signaling molecules RIPK1 and RIPK3, followed by the effector protein MLKL (mixed lineage kinase domain like pseudokinase). It is now known that PFTs, such as pneumolysin, induce activation of necroptosis as a result of the sustained ion dysregulation and/or catastrophic energy loss that occur on pore formation [11].
During S. pneumoniae, Staphylococcus aureus, and Serratia marcescens pneumonia, PFT-initiated necroptosis was shown to be involved in the killing of lung macrophages and lung epithelial cells and to contribute directly to pulmonary injury [12, 13]. Further supporting a detrimental role for PFT-mediated necroptosis, S. pneumoniae–infected mice deficient in MLKL had less severe pathological hallmarks of pneumonia and experienced prolonged survival compared with wild-type controls [11]. Thus, pharmacological inhibition of necroptosis may be a therapeutic strategy to protect against the extensive organ injury that occurs during severe S. pneumoniae pneumonia. Importantly, and though pneumolysin-formed pores have been shown to induce calcium dysregulation that impairs cardiomyocyte contractility [10], it was unknown whether cardiomyocytes died as result of pneumolysin-induced necroptosis during severe pneumococcal infections.
The objectives of the current study were (1) to identify the form of cell death experienced by cardiomyocytes exposed to S. pneumoniae, (2) to determine whether S. pneumoniae is responsible for long-term cardiac damage, and (3) to test the potential of Ponatinib, a Food and Drug Administration (FDA)–approved necroptosis inhibitor [14], as a therapeutic adjunct treatment for organ damage during IPD. Our results provide compelling evidence that the severity and long-term negative consequences of pneumococcal disease may be ameliorated through inhibition of necroptosis.
METHODS
Ethics Statement
Animal experiments were approved by the Institutional Animal Care and Use Committees at the University of Texas Health Science Center at San Antonio (no. 13032-34-01C) and the University of Alabama at Birmingham (nos. 20270, 20175, 20479, and 21048).
Bacterial Strains and Media
S. pneumoniae strain TIGR4, its isogenic pneumolysin deficient mutant, and D39 are described elsewhere [15–17]. Pneumococci were grown on blood agar plates or in Todd-Hewitt broth containing 0.5% yeast extract at 37°C and 5% carbon dioxide.
Cell Lines and Assays
Human cardiomyocytes immortalized with SV40 T-antigen (HCSV40) were purchased from ABM. HL-1 atrial cardiomyocytes were a gift from W. Claycomb (Louisiana State University School of Medicine, New Orleans) [18].
Animal Models and Disease Severity Assessments
Male and female 6-week-old BALB/c mice and C57Bl/6 mice were obtained from Jackson Laboratories. MLKL knockout (KO) and RIPK3 KO mice in a C57Bl/6 background were obtained from Warren Alexander (Walter and Eliza Hall Institute of Medical Research Parkville, and colonies maintained at UAB, Australia) and Vishva Dixit (Genentech), respectively [19, 20]. Mice were challenged by intraperitoneal injection with 103 colony forming units (CFUs) of S. pneumoniae suspended in 100 μL of phosphate-buffered saline (PBS) or by forced aspiration with 105 CFUs of S. pneumoniae in 100 μL of PBS.
For rescue experiments, mice received ampicillin (50 mg/kg) intraperitoneally in 100 µL of PBS every 12 hours for 3 days. When indicated, mice also received necrostatin-5 (5 mg/kg) (Sigma) or Ponatinib (1 mg/kg) (Selleckchem), dissolved in dimethyl sulfoxide (DMSO) and diluted with PBS to a total of 100 μL (final <5% DMSO), by intraperitoneal injection every 6 or 12 hours for up to 3 days. Vehicle controls received saline solution with equivalent amounts of DMSO. Cardiac troponin-I in serum samples was measured by means of enzyme-linked immunosorbent assay (G-Biosciences). Cardiac function measurements (ie, ejection fraction, fractional shortening, and myocardium strain) were assessed by echocardiography at the University of Alabama at Birmingham Mouse Cardiovascular Core using the VisualSonics Vevo 3100 imaging system (Fujifilm VisualSonics).
Tissue Staining and Immunofluorescence Microscopy
Methods used for heart and lung processing, hematoxylin-eosin staining, and immunofluorescence staining of sections are detailed in Supplementary Material. Note that we used fluorophore-conjugated monoclonal antibodies against phosphorylated MLKL (p-MLKL), eliminating the need for secondary antibody. Cardiac sections were examined and microlesions enumerated using microscopy. Cardiac sections were stained with hematoxylin-eosin or examined by means of immunofluorescence using antibody against type 4 capsular polysaccharide. Sirius red/fast green staining (Chondrex 9046) was performed to detect collagen. Collagen was measured semiquantitatively in blinded samples with ImageJ software, using threshold analysis of tile-scanned images. Note that thresholds were manually set on the red-green-blue image to visually match the red-stained portions in the color image.
Cell Death Assays
Details on the reagents to block necroptosis and methods used assess cell death are provided in the Supplementary Material.
Statistical Analyses
For multiple-group analyses we used 1-way analysis of variance with Dunn post hoc analysis; Tukey or Sidak posttests were used only when stated, and grouped analyses were performed using 2-way analysis of variance. Summary statistics are presented as means with standard errors of the mean. For nonparametric data sets, we used Mann-Whitney U tests. Statistical analyses were performed using Prism 8 software (GraphPad Software).
RESULTS
Pneumolysin requirement for S. pneumoniae Cardiac Damage
After in vitro challenge, mouse HL-1 cardiomyocytes and HCSV40 were susceptible to killing by 2 unrelated strains of S. pneumoniae: serotype 4 strain TIGR4 and serotype 2 strain D39 (Figure 1A). A requirement for pneumolysin in killing was evidenced by the decreased cytotoxicity observed in HCSV40 exposed to an isogenic mutant of TIGR4 lacking pneumolysin (TIGR4 Δply), as well as in HCSV40 in media containing neutralizing antibody against the toxin (Figure 1B). The contribution of pneumolysin to the pathogenesis of S. pneumoniae–mediated cardiac damage in vivo was also retested and found to be stark.
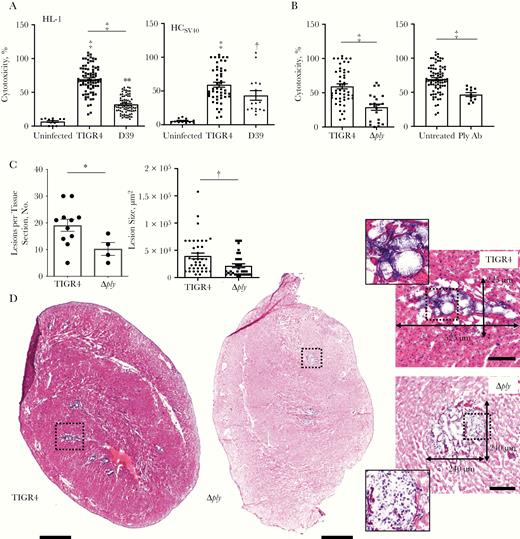
Pneumolysin is required for Streptococcus pneumoniae cardiac damage. A, Cytotoxicity as assessed by the amount of lactate dehydrogenase released into the supernatant by HL-1 mouse cardiomyocytes (black bars) and human cardiomyocytes immortalized with SV40 T-antigen (HCSV40) (white bars) infected with S. pneumoniae strains TIGR4 or D39 or mock-infected (uninfected) after 4 hours at a multiplicity of infection (MOI) of 100. B, Cytotoxicity of HCSV40 infected with TIGR4 or TIGR4 Δply and in the presence of neutralizing monoclonal antibody against pneumolysin (Ply Ab) after 4 hours (MOI, 100). C, Number and size of cardiac microlesions in heart sections from mice infected with TIGR4 or Δply, quantified from hematoxylin-eosin (HE)–stained cardiac sections. D, Representative HE-stained images and number of lesions in heart sections from mice infected intraperitoneally with TIGR4 or Δply. Hearts from infected mice were collected 30 hours after intraperitoneal challenge. Sections were tile scan imaged at ×40 magnification, with the cardiac apex positioned at the bottom of figure; scale bars represent as 500 μm in tile scans and100 μm in lesion panel with 110-μm2 highlighted lesion section. *P ≤ .05; †P ≤ .01; ‡P ≤ .001. (See also Supplementary Figure 1.)
Mice challenged with TIGR4 Δply had fewer and smaller foci of S. pneumoniae in the myocardium (ie, cardiac microlesions) than mice challenged with TIGR4 (Figure 1C–1D). Moreover, and in contrast to TIGR4, cardiac microlesions formed by TIGR4 Δply were characterized by the presence of infiltrated immune cells (Figure 1D), as has been described elsewhere [8]. Notably, TIGR4 infection was associated with detection of the proinflammatory mediators keratinocyte chemoattractant (KC), Interleukin 1α, and interleukin 6, along with a reduction in interferon γ in heart homogenates (Supplementary Figure 1). Interleukin 1α is released during necrotic forms of cell death [21]. Interferon γ is produced by macrophages [22], which have previously been shown to be depleted by heart-invaded S. pneumoniae [23]. Together, these results reconfirmed that S. pneumoniae invades the heart, causing cardiac damage, and that pneumolysin is a key factor in the induction of cardiomyocyte death.
Pneumolysin Killing of Cardiomyocytes Via Necroptosis
PFTs such as pneumolysin trigger necroptosis in pulmonary epithelial cells and macrophages [11–13]. To determine whether cardiomyocytes exposed to S. pneumoniae also underwent necroptosis, we tested whether S. pneumoniae–mediated death of HCSV40 in vitro could be blocked with necroptosis inhibitors. A schematic of the necroptosis pathway and the inhibitors used to block distinct steps in its activation is provided in Figure 2A. Pretreatment of cells with necrostatin-5, an inhibitor of RIPK1 [24], GSK’872, an inhibitor of RIPK3 [25], or necrosulfonamide, an inhibitor of MLKL [26], all reduced cardiomyocyte cytotoxicity after challenge with S. pneumoniae (Figure 2B). Likewise, knockdown of RIPK3 or MLKL with short interfering RNA also reduced S. pneumoniae–mediated cytotoxicity versus control (Figure 2C). Finally, infection of HCSV40 with TIGR4 Δply resulted in reduced levels of detectable p-MLKL versus TIGR4-infected cells (Figure 2D).
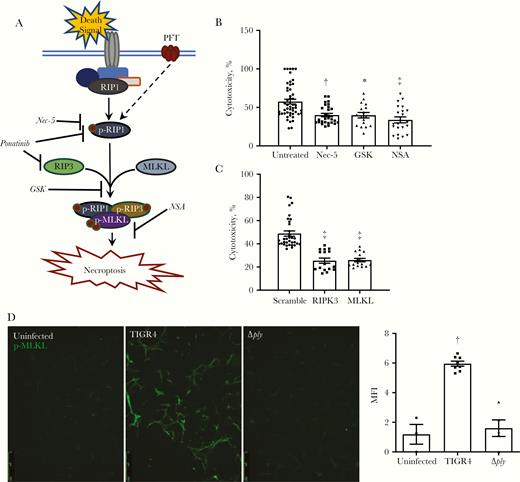
Pneumolysin kills cardiomyocytes via necroptosis in vitro. A, Illustrative model of necroptosis pathway and pharmacological inhibitors used to block activation of the necroptotic mechanism. B, C, Cytotoxicity as quantified by lactate dehydrogenase release from human cardiomyocytes immortalized with SV40 T-antigen (HCSV40) infected with TIGR4 after pretreatment with necrostatin-5 (Nec-5; 10 μmol/L), GSK’872 (GSK; 10 μmol/L), and necrosulfonamide (NSA; 10 μmol/L) (B) or short interfering RNA knockdown of RIPK3 and MLKL (C). Cells were infected for 4 hours at a multiplicity of infection (MOI) of 100. D, Representative immunofluorescence-stained images of HCSV40 infected with TIGR4 or TIGR4 Δply probed for phosphorylated MLKL (p-MLKL) (green). The mean fluorescence intensity (MFI) of the p-MLKL signal was quantified with ImageJ software. Cells were infected for 4 hours at an MOI of 100. Abbreviations: p-RIP1, phosphorylated RIP1; p-RIP3, phosphorylated RIP3. *P ≤ .05; †P ≤ .01; ‡P ≤ .001 (Dunn multiple comparisons posttest).
Within heart tissue of TIGR4-infected mice, p-MLKL, the active form of the necroptosis effector, was detected with immunofluorescence microscopy (Figure 3A and Supplementary Figure 2A). After TIGR4 challenge and compared with wild-type mice, RIPK3 KO and MLKL KO mice had lower levels of serum troponin I (Figure 3B), a marker of cardiac damage that is released by dying cardiomyocytes [27]. Moreover, we detected considerably fewer microlesions in MLKL KO and RIP3 KO mice than in wild-type controls (Figure 3C). It is noteworthy that KO mice had equivalent bacterial burden in the blood and heart compared with wild-type mice (Supplementary Figure 2B). Thus, reduced bacterial entry into the heart is not an explanation for differences in cardiac microlesion frequency. Altogether, these findings demonstrate that S. pneumoniae–derived pneumolysin triggers necroptosis of human and murine cardiomyocytes during acute infection.
![Necroptosis occurs within cardiac microlesions. A, Representative immunofluorescence-stained cardiac microlesion in a cardiac section from a BALBc/J mouse intraperitoneally infected with TIGR4. Nuclei (4’,6-diamidino-2-phenylindole [DAPI]; blue), phosphorylated MLKL (p-MLKL; green), TIGR4 (red), with merged image shown. B, Cardiac troponin I detected in the serum of wild-type C57Bl/6 mice (n = 10; WT), mice deficient in RIPK3 (n = 16; RIPK3 knockout [KO]) or MLKL (n = 16; MLKL KO) of the same background after infection with TIGR4. C, Lesions per tissue section from the same mice as in B. Lesions were counted using immunofluorescence-stained sections. Hearts from infected mice were collected 30 hours after intraperitoneal challenge. For multiple-group comparisons, Dunn or Tukey (C) multiple comparisons tests were used. †P ≤ .01, ‡P ≤ .001. (See also Supplementary Figures 2 and 3.)](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/222/11/10.1093_infdis_jiaa295/5/m_jiaa295_fig3.jpeg?Expires=1749926240&Signature=1XfKuz4VGIZlobZaZ92lZsR~1Lt3DdmyH-8Xx~kBxuNBCJSpTrLZRSaKYQT~yx~NhQRyoZ4wsH1n8Ump11rT0Wp2Rz5oajbdRaNh6sI0OEgIYj06KvL-~BzNQjdzZ~q799sr742JokDbRDkXkbgep7j-klaa05pQrjU9E7csXUADXAeSi5MugvV~XyviAllBfnemHYAo4Q7m2Lx7-KJn4YhIqpzbr6r8GtgzQMdShePEQanG24ELLrFe-KjtREbn6-AUyLQR9lRjnsDthEGFn9k7M8175cqGLK5vDR2hr0YU-ngxHjvwodS33WE9xEOcaQYF~1J9q0HuKw~i2WukoQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Necroptosis occurs within cardiac microlesions. A, Representative immunofluorescence-stained cardiac microlesion in a cardiac section from a BALBc/J mouse intraperitoneally infected with TIGR4. Nuclei (4’,6-diamidino-2-phenylindole [DAPI]; blue), phosphorylated MLKL (p-MLKL; green), TIGR4 (red), with merged image shown. B, Cardiac troponin I detected in the serum of wild-type C57Bl/6 mice (n = 10; WT), mice deficient in RIPK3 (n = 16; RIPK3 knockout [KO]) or MLKL (n = 16; MLKL KO) of the same background after infection with TIGR4. C, Lesions per tissue section from the same mice as in B. Lesions were counted using immunofluorescence-stained sections. Hearts from infected mice were collected 30 hours after intraperitoneal challenge. For multiple-group comparisons, Dunn or Tukey (C) multiple comparisons tests were used. †P ≤ .01, ‡P ≤ .001. (See also Supplementary Figures 2 and 3.)
IPD and Long-term Cardiac Damage
Brown et al [9] previously reported that extensive cardiac remodeling is observed within a S. pneumoniae–injured heart 7–10 days after antimicrobial intervention for IPD. However, it remained unknown whether this collagen persisted or whether the negative effects of S. pneumoniae on heart function were long lasting. Using an antimicrobial intervention model of IPD rescue (schema shown in Figure 4A), we examined heart sections in TIGR4-infected mice that had been rescued by intraperitoneal ampicillin administration beginning 22 hours after S. pneumoniae challenge and continuing for 3 days (mice had clearance of S. pneumoniae confirmed by blood culture). Specifically, hearts from mice 2 weeks, 1 month, and 3 months after infection were examined.
![Invasive pneumococcal disease (IPD) results in long-term cardiac damage. A, Schematic of Streptococcus pneumoniae challenge and antimicrobial intervention used in the current study. B, Percentage of collagen in heart sections from uninfected mice, mice recovered from mild S. pneumoniae infections (<105 colony-forming units [CFUs]/mL blood), and mice that had experienced severe IPD (>105 CFUs/mL blood) 2 weeks, 1 month, and 3 months after antimicrobial treatment. C, Hearts stained with Sirius red/fast green and with median levels of collagen per group and time point are shown as representatives. Areas stained red are collagenous proteins; areas stained green, noncollagenous proteins. Arrows indicate myocardial collagen deposition (MC) and perivascular collagen deposition (PV). D, Ejection fraction and fractional shortening in uninfected mice and in mice that had experienced S. pneumoniae infections, both mild and severe, 3 months after antimicrobial treatment. Shown are representative echocardiographic images demonstrating changes in left ventricular (LV) function between uninfected mice and mice that experienced S. pneumoniae infection and were treated with ampicillin 3 months earlier. Abbreviation: 3D, 3-dimensional. Sidak posttests were used for multiple-group comparisons, and Mann-Whitney U tests for 2-group comparisons. *P ≤ .05; †P ≤ .01; ‡P ≤ .001. (See also Supplementary Figure 3.)](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/222/11/10.1093_infdis_jiaa295/5/m_jiaa295_fig4.jpeg?Expires=1749926240&Signature=rZq~Uyv76ObK9x6w67L~WFn9rwtwShtMFNQTVZScpiMoq2L0DNrTIVZ9pgUku3eR4pg-rQVvnBeno1RQnMuipTt~R0jxqpoCjk~8aA7exBWteHxhw7mxc7IeP9k2CJMO5k7ePmdysl7S7bdD8eOc5Q0veEeUhq2Y5KSUZjtiKmovSYTECUTHC5gqzy3hdY38YAV7U3aomJNeg7kJJi~cLc50evv1O47FE6i8nLm7cSQUM12RYmuwwTHvJ1KEVXKpMDv7Xl2G1G-d7wd2la3qGG4UqpHzKAIkgHdrlGNr5to33j27DiHbt~kzTf48WNwpBOjy~LLxCqFENJ1TAoIF0Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Invasive pneumococcal disease (IPD) results in long-term cardiac damage. A, Schematic of Streptococcus pneumoniae challenge and antimicrobial intervention used in the current study. B, Percentage of collagen in heart sections from uninfected mice, mice recovered from mild S. pneumoniae infections (<105 colony-forming units [CFUs]/mL blood), and mice that had experienced severe IPD (>105 CFUs/mL blood) 2 weeks, 1 month, and 3 months after antimicrobial treatment. C, Hearts stained with Sirius red/fast green and with median levels of collagen per group and time point are shown as representatives. Areas stained red are collagenous proteins; areas stained green, noncollagenous proteins. Arrows indicate myocardial collagen deposition (MC) and perivascular collagen deposition (PV). D, Ejection fraction and fractional shortening in uninfected mice and in mice that had experienced S. pneumoniae infections, both mild and severe, 3 months after antimicrobial treatment. Shown are representative echocardiographic images demonstrating changes in left ventricular (LV) function between uninfected mice and mice that experienced S. pneumoniae infection and were treated with ampicillin 3 months earlier. Abbreviation: 3D, 3-dimensional. Sidak posttests were used for multiple-group comparisons, and Mann-Whitney U tests for 2-group comparisons. *P ≤ .05; †P ≤ .01; ‡P ≤ .001. (See also Supplementary Figure 3.)
Semiquantitation of collagen deposition in cardiac sections with Sirius red/fast green staining (Figure 4B–4C) showed that collagen levels were significantly higher in hearts from mice that had experienced severe disease (>105 CFUs/mL of blood at onset of antibiotic treatment), at all time points, indicating that scar formation happened early and was persistent. Mice with mild bacteremia (<105 CFUs/mL blood at onset of antibiotic treatment) did not have higher levels of collagen in cardiac sections than uninfected mice. In mice with severe disease, collagen deposition was frequently perivascular but also randomly distributed across the myocardium (Figure 4C), as has been reported for the location of S. pneumoniae–induced cardiac lesions in nonhuman primates [9]. In several instances, collagen deposition was seen on the heart periphery, suggesting that pericarditis may have also occurred (Supplementary Figure 3).
Importantly, and regardless of the bacterial burden at the time of antimicrobial intervention, echocardiography of mice revealed that postinfection deficiencies in ejection fraction and fractional shortening persisted as a sign of heart dysfunction in all previously infected mice at 3 months (Figure 4D). Detailed echocardiography demonstrated that contraction in some of the infected mice was now characterized by irregular left ventricle longitudinal and circumferential myocardial strain (Figure 4D). Thus, IPD had physiological consequence, even if long-term remodeling was not evident at staining for collagen.
Therapeutic Inhibition of Necroptosis as Protection Against Organ Damage During IPD
We hypothesized that necroptosis inhibition would be beneficial to the host during IPD. Therapeutic treatment of infected mice with the necrostatin-5 at 5 mg/kg resulted in significantly fewer cardiac microlesions than in mice that received the vehicle control (Figure 5A). Similar to our observation with MLKL and RIPK3 KO mice, necrostatin-5 conferred protection, despite the fact that bacteria numbers in the heart were not diminished (Supplementary Figure 4A). Notably, cardiac microlesions formed in necrostatin-5–treated mice also had infiltrating immune cells (Figure 5B), similar to those caused by TIGR4 Δply (Figure 1D).
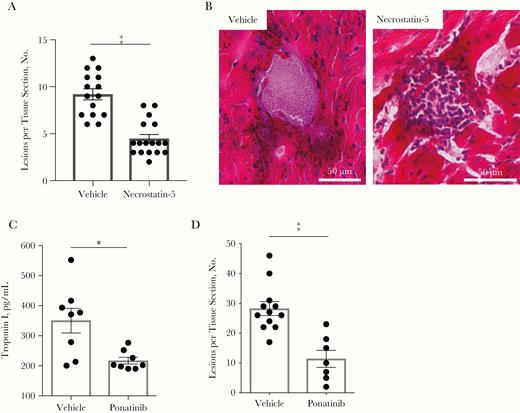
Therapeutic inhibition of necroptosis protect against organ damage during invasive pneumococcal disease. A, Lesions per cardiac tissue section from vehicle-and necrostatin-5–treated mice. B, Representative images of microlesions observed in hematoxylin-eosin–stained cardiac sections from mice treated with a vehicle or necrostatin-5 starting 12 hours before infection and every 12 hours until euthanasia 36 hours after infection. C, Serum cardiac troponin I from C57Bl/6 mice infected intraperitoneally with TIGR4 and treated with 100-µL intraperitoneal injection of vehicle or Ponatinib (1 mg/kg) at 12 and 24 hours after infection and euthanized 30 hours after infection. D, Cardiac microlesions per tissue section from immunofluorescence-stained cardiac sections probed for TIGR4. Mann-Whitney U tests were used for 2-group comparisons. *P ≤ .05; ‡P ≤ .001. (See also Supplementary Figure 4.)
Because necrostatins are not approved for use in humans, we explored suitable alternatives. Ponatinib, a tyrosine kinase inhibitor that is FDA approved for the treatment of chronic myeloid leukemia and Philadelphia-positive acute lymphocytic leukemia, was discovered to also block RIPK1 and RIPK3 activation (Figure 2A) [15]. Ponatinib treatment of mice 12 and 24 hours after S. pneumoniae infection (at 1 mg/kg, without antimicrobials) resulted in reduced levels of troponin I in serum samples (Figure 5C) and formation of fewer cardiac microlesions (Figure 5D). As with necrostatin-5, Ponatinib had no impact on bacterial titers recovered from the blood or heart of infected mice (Supplementary Figure 4B). Finally, Ponatinib was found not to have any direct antimicrobial activity in vitro (Supplementary Figure 4C).
Up to this point, challenge of mice was done by intraperitoneal injection. This challenge route is preferred in the laboratory, because infected mice are synchronized with regard to disease onset and severity and the resultant cardiac damage faithfully recapitulates what is observed to occur in mice in which IPD develops after intratracheal challenge [28]. However, this approach does not provide translatable information on events in the lungs. For this reason, we were compelled to determine whether Ponatinib also protected against airway damage and the subsequent cardiac injury associated with severe pneumonia that results in bacteremia. Therapeutic treatment with Ponatinib after intratracheal challenge of mice with TIGR4 resulted in starkly decreased pulmonary consolidation, albeit considerable immune cell infiltration still occurred (Figure 6A–6C). We also observed a significant reduction in frequency of cardiac microlesions (Figure 6D and Supplementary Figure 5). Although of the mice receiving Ponatinib cleared pneumococci from the airway, there were not significant differences in bacterial burden across the groups (Supplementary Figure 6).
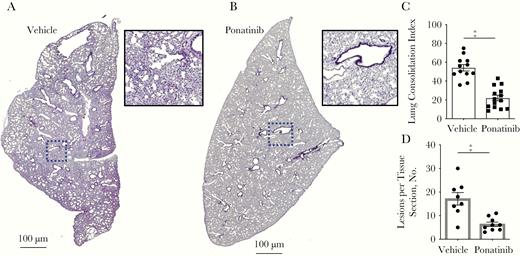
Therapeutic Ponatinib treatment reduces lung and cardiac damage from pneumococcal pneumonia and invasive pneumococcal disease. A, B, Representative hematoxylin-eosin (HE)–stained lung sections from C57Bl/6 mice (n = 12–14) infected intratracheally with TIGR4 and treated intraperitoneally with vehicle (2% dimethyl sulfoxide in phosphate-buffered saline; 0 mg/kg) (A) or ponatinib at 1 mg/kg every 12 hours (B) and euthanized 48 hours after infection. C, HE-stained lung sections were quantified for detectable biomass as an indication of lung consolidation, using ImageJ software. D, Cardiac microlesions per tissue section from these same mice. Mann-Whitney U tests were used for 2-group comparisons. ‡P ≤ .001. (See also Supplementary Figure 5.)
Ponatinib Protection Against Long-term Cardiac Dysfunction
Subsequently, we sought to determine whether Ponatinib treatment conferred protection against cardiac dysfunction in convalescence. As before, mice were challenged intraperitoneally with TIGR4, but in this instance they received Ponatinib along with ampicillin beginning 22 hours after infection and then every 12 hours for 3 days. Only mice in which severe infection had developed (>105 CFUs/mL blood) were used to assess the protective effects of this drug. Mice were assessed 2 weeks, 1 month, and 3 months after infection with high-resolution echocardiography. At these same time points, some mice were euthanized, and their hearts were examined histologically for collagen deposition. Control mice included cohorts that received a vehicle instead of Ponatinib along with ampicillin. Importantly, and at all measured time points, mice that received adjunct Ponatinib had significantly lower collagen levels than those that received vehicle and ampicillin.
Pathological features observed in hearts from infected mice that had not received Ponatinib (eg, remnants of cardiac microlesions, pericarditis, and diffuse collagen deposition) were not present in mice given Ponatinib (Figure 7A and 7B). Further supporting a protective effect for Ponatinib, echocardiography at 3 months after infection showed that fractional shortening values were preserved in mice that had received Ponatinib, compared with the vehicle control group. Similarly, a positive trend was observed for ejection fraction (Figure 7C). Overall, aberrations in left ventricular contraction were diminished in Ponatinib-treated mice versus untreated controls (Figure 7D).
![Ponatinib protects against long-term cardiac dysfunction. Mice were infected with TIGR4 and received Ponatinib treatment (1 mg/kg) or vehicle control in adjunct to antimicrobials. Hearts from mice experiencing severe disease (>105 colony-forming units [CFUs]/mL of blood) at time of intervention were examined. A, Hearts stained with Sirius red/fast green and having median levels of collagen per group and time point are shown as representatives. Collagenous tissue is stained red and noncollagenous proteins, green. B, Collagen levels 2 weeks and 1 month after infection were significantly higher in mice that received vehicle treatment than in those that received Ponatinib. C, Fractional shortening and ejection fraction in Ponatinib- and vehicle-treated convalescent mice 3 months after infection. D, Representative echocardiographic images demonstrating cardiac function preservation in mice receiving Ponatinib 3 months earlier. Abbreviations: 3D, 3-dimensional; LV, left ventricular. Sidak posttests were used for multiple-group comparisons, and Mann-Whitney U tests for 2-group comparisons. *P ≤ .05; †P ≤ .01; ‡P ≤ .001. (See also Supplementary Figure 6.)](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/222/11/10.1093_infdis_jiaa295/5/m_jiaa295_fig7.jpeg?Expires=1749926240&Signature=PD0KqQdh5L-yw6UYNxxoS3uQEN3VoZDk9eCzn3iE5jwfoQkiDB~kiw3Csz2~kEwssAsZEcG7v4DSKTeFUcTeLbk9PIXHtxofK0pUbqZZl69k-YMci1tiUyeBc13n2p77FU3H4PNM68Y5XrTEHpaVzeuse1S6mqHVRSr7EiPbJkj3zK8HUCfbkfKN7PD3luLHjIFayLSgyFKE0k8VAytY-tAPhrSBLl89-a4I0xjzoLYJnaH-0M5qphZCgZpPPKKHRqRetGNVcCrN595TeaBRazqiWzNMkU8NAT1B6OMh-VQuWizU9AA41swKLfapGxp9Fsr2ATCgn0m54FLMLF87ZQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Ponatinib protects against long-term cardiac dysfunction. Mice were infected with TIGR4 and received Ponatinib treatment (1 mg/kg) or vehicle control in adjunct to antimicrobials. Hearts from mice experiencing severe disease (>105 colony-forming units [CFUs]/mL of blood) at time of intervention were examined. A, Hearts stained with Sirius red/fast green and having median levels of collagen per group and time point are shown as representatives. Collagenous tissue is stained red and noncollagenous proteins, green. B, Collagen levels 2 weeks and 1 month after infection were significantly higher in mice that received vehicle treatment than in those that received Ponatinib. C, Fractional shortening and ejection fraction in Ponatinib- and vehicle-treated convalescent mice 3 months after infection. D, Representative echocardiographic images demonstrating cardiac function preservation in mice receiving Ponatinib 3 months earlier. Abbreviations: 3D, 3-dimensional; LV, left ventricular. Sidak posttests were used for multiple-group comparisons, and Mann-Whitney U tests for 2-group comparisons. *P ≤ .05; †P ≤ .01; ‡P ≤ .001. (See also Supplementary Figure 6.)
DISCUSSION
In addition to the physiological stressors imposed by severe systemic infection, considerable clinical, epidemiological, and experimental evidence now supports the notion that S. pneumoniae directly damages the heart and this contributes to MACEs within the hospital setting [2–5, 28, 29]. Specifically, our findings here affirm that S. pneumoniae can cause cardiomyocyte death, as the result of its toxin pneumolysin. Importantly, our results demonstrate that cardiotoxicity is the result of pneumolysin’s ability to trigger a form of programmed cell death known as necroptosis. Although necroptosis had been previously implicated in exacerbating cardiac damage after sterile injury, such as in an ischemia-reperfusion model of infarct [30], and implicated in the depletion of tissue resident macrophages [12, 23], ours is the first report to indicate a role for necroptosis in the death of cardiomyocytes.
The identification of necroptosis as responsible for pneumolysin-instigated cardiomyocyte death raised the possibility that cardiac damage and its associated dysfunction might be preventable. This was demonstrated genetically, using RIPK3 and MLKL KO mice, and pharmacologically, by treatment of infected wild-type mice with necrostatin-5 or Ponatinib. Notably, Ponatinib is approved by the FDA to treat chronic myeloid leukemia and Philadelphia chromosome–positive acute lymphoblastic leukemia [31]. Thus, and as discussed below, it is a potential adjunct therapeutic for IPD. Importantly, MLKL KO mice and wild-type mice treated with necrostatin-5 or Ponatinib had equivalent bacterial burdens in the airway, bloodstream, and heart, compared with control mice after infection. Thus, inhibition of necroptosis does not seem to enhance susceptibility to the infection. The fact that necroptosis inhibition does not affect bacterial burden in the heart suggests that the translocation rate of pneumococci into the myocardium is not affected. The latter has been shown to be mediated by bacterial attachment to laminin receptor and platelet-activating factor receptor on vascular endothelial cells [28].
Importantly, the reduction in cardiac microlesion formation seen in necroptosis-inhibited mice suggests that cardiomyocyte death is an event required for microlesion formation. This aligns well with an earlier observation that S. pneumoniae can be taken up by cardiomyocytes after entering the myocardium and these host cells are killed [9]. We propose that the reason no differences are seen in recoverable CFUs between hearts of necroptosis-inhibited mice and hearts of controls, despite the absence of microlesions, is because pneumococci are continuously invading the heart, and many can be found outside these microlesions [23]. Moreover, well-formed microlesions are composed in part (up to 35%) of dead pneumococci [6]. It is worth noting that cardiac microlesions formed by mice treated with necrostatin-5 mirrored the histological features of cardiac microlesions from mice infected with TIGR4 Δply. That is, these lesions were characterized by immune cell infiltrates, whereas TIGR4 forms microlesions that lack immune cell infiltration [23]. Previously, Gilley et al [23] showed that pneumolysin depletes the heart of cardiac resident macrophages and this inhibits the recruitment of neutrophils. Thus, our results suggest that pneumolysin activity is impaired across multiple cell types, and it may be preserving resident macrophages when necroptosis inhibitors are used.
Our observation that ejection fraction and fractional shortening were diminished in mice that had experienced severe IPD, and not in mice that had received Ponatinib, is strong evidence that pneumolysin-mediated cytotoxicity is indeed a direct contributor to the increased rate of cardiac dysfunction that occurs in convalescent individuals. Furthermore, therapeutic necroptosis inhibition may have long-term beneficial effects for those who experience IPD. Importantly, Ponatinib has a black box warning owing to liver toxicity and its associated risk for thrombosis in patients being treated for leukemia [32]. Its use during IPD, as tested in the current study, would thereby merit considerable caution because of the serious medical complications that are concomitant with IPD as well as the drug’s own negative effects. Critically, we administered the drug within a considerably truncated time frame and at a dose significantly lower than the doses used in murine leukemia models.
Our results open the possibility of repurposing anticancer drugs for infectious disease-related tissue injury in the myocardium, an avenue not explored until this report [33]. Also exciting was our observation that Ponatinib-treated mice were protected against acute pulmonary injury and subsequent cardiac damage after intratracheal challenge. A negative role for necroptosis during pneumonia has already been established, principally using KO mice [12]. Although Ponatinib is certainly a promising potential therapeutic many questions still need to be answered. For example, does Ponatinib, or any other necroptosis inhibitor, have negative consequences during viral coinfection? Does Ponatinib have negative interactions with other classes of antibiotics or treatment modalities? Our positive results suggest that animal studies that carefully test both the efficacy and the toxicity of Ponatinib as adjunct therapy to single and combination antimicrobial therapy during pneumonia are now warranted.
Collectively, our results reaffirm that cardiomyocyte death and myocardium damage occur during IPD. They show for the first time that cardiomyocyte death is the result of pneumolysin-mediated necroptosis. In addition, cardiac damage is long lasting, and its associated dysfunction is a viable explanation for the increased risk of adverse cardiac events reported in convalescent individuals. Our results also show that pulmonary injury and cardiac damage during severe pneumococcal infections are potentially preventable. Pharmacological inhibition of necroptosis reduced the acute injury observed in the lungs and heart during S. pneumoniae pneumonia and IPD and abrogated the cardiac dysfunction seen in convalescent control animals. We conclude that further studies are warranted to study the viability of necroptosis inhibitors in preventing tissue damage during IPD.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. We thank Kelley Bradley, Alexis Herrera, and the University of Alabama at Birmingham Pathology Core Research Lab for their technical assistance, and we thank Luis Reyes for his commentary. We also thank Vishva Dixit (Genentech) and Warren Alexander (Walter and Eliza Hall Institute of Medical Research Parkville, Australia) for the gifts of RIPK3 and MLKL knockout mice, respectively.
Financial support. This work was supported by the National Institutes for Health (grants GM088010 to S. M. B., AI007051 to A. N. R. and N. G. J., HL134640 to K. L. K., and AG055144 and AI114800 to C. J. O.), the American Heart Association (grants 16GRNT30230007 and 18TPA34110195 to C. J. O.), and the Comprehensive Cardiovascular Center and Inflammation, Infection & Immunity joint pilot grant program through The University of Alabama at Birmingham (C. J. O.).
Potential conflicts of interest.All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Presented in part: American Society for Microbiology (ASM) Microbe, Atlanta, Georgia, June 2018, abstract 12; South Eastern Pneumococcal Symposium, Memphis, Tennessee, May 2018; Institutional Research and Academic Career Development Awards (IRACDA) Annual Meeting, Atlanta, Georgia, July 2018.
References
Author notes
S. M. B. and A. N. R. contributed equally to this work.
N. G. J. and C. J. O. contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}