






Abstract
Cronobacter sakazakii causes meningitis and necrotizing enterocolitis in premature infants. However, its virulence determinants, especially those specific for strains associated with neonate infections, remain largely unknown.
In this study, we performed a comparative genomic analysis of 209 C. sakazakii genomes, and 8 clonal groups (CGs) were revealed.
CG1 and CG2 were found to be significantly associated with neonate infections, and significantly prevalent genes in these 2 CGs were identified. Of these, a gene encoding the LysR-type regulator, CklR, was shown to contribute to bacterial pathogenicity based on animal experiments. We found that CklR directly binds and activates the suf Fe-S cluster biosynthesis operon, and high expression of the suf operon increases bacterial resistance to oxidative stress, which increases survival within the host. This leads to a high degree of bacteremia, which contributes to the development of meningitis.
Our work revealed a novel virulence factor specific to predominant pathogenic C. sakazakii strains.
Cronobacter sakazakii (formerly Enterobacter sakazakii) is responsible for up to 80% of infant deaths associated with infection [1], and it is associated with outbreaks of life-threatening necrotizing enterocolitis, meningitis, and sepsis in neonates and infants [2]. It is a major foodborne pathogen isolated from powdered infant formula. The C. sakazakii fatality rates range from 40% to 80%, and 20% of survivors develop chronic and serious neurological and developmental disorders [1, 3]. The International Commission on Microbiological Specification for Foods has classified C. sakazakii as a “severe hazard for restricted populations, life-threatening or with substantial chronic sequelae over long duration” [4]. One of the most severe outbreaks of C. sakazakii was in a neonatal intensive care unit in France in 1994, and it lasted more than 3 months, infecting 17 neonates and claiming the lives of 3 [5].
As an orally ingested pathogen that causes systemic infections, C. sakazakii first adheres to and invades the intestine [6]. It can then cross the intestinal epithelial barrier, enter the bloodstream, transcytose the blood-brain barrier, and survive in cerebrospinal fluid (CSF) [4]. This organism can also survive within macrophages and probably use macrophages as a vehicle to invade other body organs [7]. Although some virulence factors involved in C. sakazakii pathogenicity have been reported, the mechanisms of pathogenicity remain largely unknown. Major reported candidate virulence factors of C. sakazakii include outer membrane proteins OmpA and OmpX, which contribute to cell invasion [8, 9], ZpX, a metalloprotease that can cause cell deformation [10], Cpa, which can confer serum resistance [11], the iuc iron acquisition system, which plays a role in iron transport and regulation [11], type III hemolysin Hly, which has hemolytic activity [12], and a heat-stable enterotoxin [13].
A multilocus sequence type (MLST) scheme has been established for Cronobacter [14]. Among defined sequence types (STs), C. sakazakii ST4 was reported to be predominantly associated with neonatal meningitis [15], and ST12 strains were frequently associated with necrotizing enterocolitis [16]. However, the genetic basis for their association with virulence is still unclear, mainly due to the limited information on virulence characteristics within the whole genome. Because most known C. sakazakii virulence genes such as ompA are widely distributed in clinical and environment strains, they cannot be used to differentiate pathogenic and non-pathogenic strains.
In this study, we performed comparative genomics analysis of 209 available C. sakazakii genomes including 59 sequenced herein. Phylogenetic analysis divided these C. sakazakii genomes into 8 distinct clonal groups (CGs), and most clinical strains were within CG1 and CG2. Animal experiments showed that one gene (cklR), which encodes a LysR-type regulator and was prevalent in both CG1 and CG2, plays an important role in virulence by promoting survival within the host. Furthermore, we showed that CklR directly activates the expression of the suf Fe-S cluster biosynthesis operon to increase bacterial resistance to oxidative stress, thus contributing to bacterial survival. This work provides important insight into the pathogenicity of C. sakazakii and forms the basis for the further analysis of key factors that contribute to neonatal infections.
MATERIALS AND METHODS
Ethics Statement
The animal research methods and experimental procedures were approved by the Institutional Animal Care Committee at Nankai University, Tianjin, China. Sick animals were monitored more frequently and euthanized when necessary to prevent suffering.
Bacterial Strains, Plasmid, and Growth Conditions
The bacterial strains, plasmids, and primers used in this study are listed in Supplementary Tables S1 and S2. The mutant strains were generated using the λ Red recombinase system. To generate ΔcklR and ΔsufD mutant strains, the clkR and sufD gene fragments were replaced by kanamycin resistance cassette and chloramphenicol acetyltransferase cassette, respectively.
Genome Sequencing and Assembly
Whole-genome sequencing was performed using Solexa paired-end sequencing technology. The Illumina data were de novo assembled using Velvet. Annotation was performed using Glimmer 3.0. The complete genome of C. sakazakii G4023 was sequenced by PacBio RS II and assembled using HGAP.
Phylogeny and Population Genetic Analyses
Ortherfinder [17] was performed to identify conserved genomic single core genes. Single-nucleotide polymorphisms (SNPs) due to recombination were removed by ClonalFrameML [18], and SNPs due to mutation were used to construct the phylogenetic tree [19]. Population analysis was performed using nonspatial BAPS 6.0 [20].
Comparative Genomics Analyses
The large-scale BLAST score ratio (LS-BSR) pipeline [21] was used to create pangenomes from genome assemblies. The included post-matrix Python script compare_BSR.py from LS-BSR was used to identify unique coding sequences. The presence or absence of virulence genes in these genomes was analyzed using Python script ls_bsr.py.
Ribonucleic Acid Sequencing and Data Analysis
The ribonucleic acid (RNA) samples were prepared, the quality of raw reads was assessed, and low-quality reads were discarded. The treated reads were then mapped to the genome. The RPKM (reads per kilobase million) method was used to quantify gene expression levels. Differential gene expression was analyzed using the edgeR package.
Chromatin Immunoprecipitation Sequencing and Data Analysis
Chromatin immunoprecipitation was performed as previously described with minor changes [22]. Chromatin immunoprecipitation sequencing (ChIP-Seq) reads were mapped to the C. sakazakii wild-type genomes, using TopHat with 2 mismatches allowed. The enriched peaks were identified using MACS software.
Newborn Rat Model
Rat infections were performed as previously described with minor changes [8]. For survival, at defined time intervals, the survival of infected animals was recorded. For colony-forming unit enumeration experiments, postinfection, the amount of bacteria in blood, CSF, liver, and spleen were determined by plating on Luria-Bertani (LB) agar plates.
Cell Invasion Assays
The bacterial invasion was performed as described previously, with some modifications [23]. For Caco-2 invasion assays, bacterial cells were added to the cell monolayers at a multiplicity of infection (MOI) of 10. For human brain microvascular endothelial cell (HBMEC) invasion assays, bacteria were added to the cell monolayers at an MOI of 100. Results were expressed as percentage relative invasion.
Macrophage Survival Assay
Macrophage survival was assessed as described previously with minor changes [23]. Cronobacter sakazakii strains added to monolayers of RAW264.7 at an MOI of 100. The number of bacteria inside macrophages was determined by plating serial dilutions on LB agar plates.
Quantitative Reverse-Transcription Polymerase Chain Reaction
Quantitative reverse-transcription polymerase chain reaction (RT-PCR) was performed using the 7500 Real-Time PCR system. Three replicates were performed.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay were performed as described previously with some modifications [24]. Gel shift assays were performed by incubating purified deoxyribonucleic acid (DNA) fragment with various concentrations of purified His6 protein in a solution containing the band-shift buffer.
Statistical Analysis
Statistical analysis of all data was performed using GraphPad Prism 7. Statistical significance was analyzed by performing an unpaired Student’s t test, χ2 test, or log-rank test in correlative situations: * or #, P < .05; ** or ##, P < .01; *** or ###, P < .001; ns, not significant.
Accession Numbers
The sequence data have been deposited in GenBank under the accession numbers SAMN10531409–SAMN10531466 and SAMN10518520. Ribonucleic acid sequencing (RNA-Seq) and ChIP-Seq data have been deposited in the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) database under accession codes SRR8275733 and SRR8275734 for RNA-Seq and SRR8276977 and SRR8276978 for ChIP-Seq.
RESULTS
Population Structure of Cronobacter sakazakii
To investigate the population structure of C. sakazakii species at the whole genome level and the relationship between pathogenicity and specific groups or lineages, we sequenced 59 isolates from different sources by Illumina sequencing. In addition, 150 available C. sakazakii genomes were downloaded from GenBank and included (Supplementary Table S3). Genome comparison revealed 887 core genes shared by the 209 genomes. Single-nucleotide polymorphisms within these 887 genes were further screened, and SNPs due to recombination were removed by ClonalFrameML [18], whereas SNPs due to mutation were used to construct a phylogenetic tree using PhyML [19]. Phylogenetic analysis using the maximum-likelihood algorithm divided 209 C. sakazakii genomes into 8 distinct CGs (Figure 1). This was confirmed using the population genetic statistical framework, Bayesian Analysis of Population Structure (BAPS) [20]. As shown in Figure 1, the number of strains within different CGs ranged from 86 (CG1, 41.1%) to 6 (CG6, 2.9%). Analyzing the distribution of different CGs showed that clinical strains were significantly more prevalent in CG1 and CG2 (χ2 test, P < .001) (Figure 1 and Supplementary Table S3), because 39 strains (45.3%) within CG1 and 8 (100%) within CG2 were isolated from clinical samples (75.7% [28 of 39] clinical strains in CG1 and 75.0% [6 of 8] clinical strains in CG2 were isolated from neonatal patients), but only 13.1% strains (14 of 107) within the other 6 CGs were isolated from clinical samples (Supplementary Table S3).
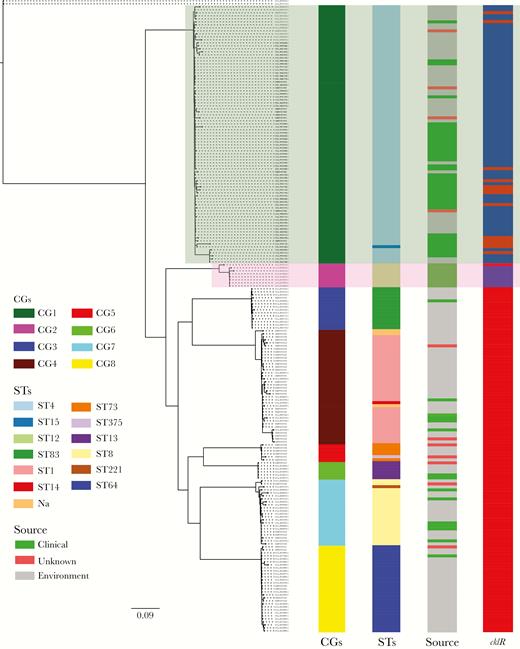
Whole-genome phylogeny of Cronobacter sakazakii. The maximum-likelihood phylogeny tree was constructed based on single-nucleotide polymorphisms across the whole genome excluding those resulting from recombinations (2 Cronobacter malonaticus strains were used as the outgroup). Clonal groups (CGs), sequence types (STs), and sources of strains are shown. The presence (blue) or absence (red) of cklR among these 209 C. sakazakii genomes is also shown. Light green background and pink background represent CG1 and CG2, respectively.
We also performed MLST analysis of these genomes using Cronobacter PubMLST database (https://pubmlst.org/cronobacter/). Of 209 genomes, 206 were divided into 12 STs (Figure 1 and Supplementary Table S3), and 3 strains were not classified into known STs.
These data showed highly consistent results between molecular typing based on MLST and whole genomes; specifically, CG1, CG2, CG3, CG4, CG5, CG6, CG7, and CG8 mainly corresponded to ST4, ST12, ST83, ST1, ST73, ST13, ST8, and ST64, respectively (Figure 1). It is worth noting that ST4 and ST12 strains have been frequently associated with neonatal meningitis and necrotizing enterocolitis, respectively [19, 25, 26], which is consistent with our results indicating that clinical strains are closely related to CG1 and CG2. However, there were also some discrepancies between the results of these 2 typing methods (Figure 1). For example, the clinical strain C. sakazakii 4 (GenBank accession number GCA_001972225.1) belonging to ST15 was classified as CG1, which is closely related to pathogenic C. sakazakii strains (Figure 1). This indicates that whole-genome analyses can provide unprecedented phylogenetic resolution to identify strains with potential to cause neonate infections.
We analyzed the distribution of 29 known virulence genes of C. sakazakii [8–11, 23, 25, 26] among the 8 CGs (Supplementary Figure S1). However, no obvious differences were observed among CGs. In addition, CG1 and CG2 strains did not contain more virulence genes than those of other CGs, and none of these factors were closely associated with CG1 and CG2. Thus, it is highly likely that there are other unidentified genes that contribute to virulence of CG1 and CG2.
To identify genes more frequently associated with CG1 and CG2, we used LS-BSR analysis to analyze the whole genomes of these 209 strains [21]. Forty-four and 119 genes were found to be significantly prevalent in CG1 and CG2, respectively, and 13 genes were significantly co-prevalent in both CG1 and CG2 (Supplementary Figure S2 and Supplementary Table S4). Although the function of most of these 13 genes can not be predicted, we found that one of these 13 genes (GenBank accession number WP_015386764.1) encodes a product of the LysR-type transcriptional regulator family. It contains a bacterial regulatory HTH domain and a LysR substrate-binding domain. We named this cklR, and we further analyzed its role in the C. sakazakii virulence.
cklR was found in 81.6% of CG1 strains and 87.5% of CG2 strains but not in the other 6 CGs. cklR was also not found in other Enterobacteriaceae such as Escherichia coli, Shgella, salmonella. In addition, CklR shared no sequence identity with a reported LysR-type regulator of C. sakazakii, which plays a role in bacterial invasion [25], indicating that these 2 regulators have different functions in C. sakazakii.
CklR Contributes to Cronobacter sakazakii Virulence by Promoting Survival Within the Host
A cklR-mutant (ΔcklR) was generated in C. sakazakii G4023, which belongs to CG1. ΔcklR and wild-type strain exhibited similar growth rates in LB medium (Supplementary Figure S3A). Three groups of 3-day-old Sprague-Dawley rats were orally infected with wild-type, ΔcklR, and complement ΔcklR+ strains, and survival was monitored for >108 hours. Animals infected with wild-type and ΔcklR+ strains started to die by 12 hours, and all died within 72 hours (Figure 2A). In contrast, animals infected with ΔcklR started to die at 24 hours, and all died within 108 hours postinfection. Survival curves identified a significant difference between groups infected with wild-type/ΔcklR+ strains and the group infected with ΔcklR (log-rank test, P < .01) (Figure 2A), indicating that cklR deletion severely attenuates C. sakazakii virulence in rat pups after oral infection.

CklR contributes to the virulence of Cronobacter sakazakii. (A) Survival curves comprising 108-hour postoral infection of corresponding strains. Data presented are the combination of 3 independent experiments (log-rank test; **, P < .01). (B–D) Bacterial counts recovered from the blood, spleen, and liver of orally infected rat pups. At 48 hours postinfection, blood collected from the facial vein, as well as spleen and liver, which were harvested and homogenized, were used for colony enumeration. Data are presented as means ± standard deviation; n = 3 (Student’s t test; **, P < .01 and ***, P < .001). For B, the development of meningitis in rat pups, defined as positive cerebrospinal fluid cultures, is shown by solid circles. Differences between the frequencies of the development of meningitis were determined (χ2 test; #, P < .05). CFU, colony-forming units; WT, wild type.
Then, we investigated whether CklR affects C. sakazakii colonization within rat pups. Animals were orally infected with ΔcklR or wild-type strains, and bacterial loads in the blood, liver, and spleen were analyzed. Results showed that colonization of ΔcklR in these organs was decreased by 19- to 30-fold, compared with that with the wild-type strain (Figure 2B–D).
To cause systemic infection, C. sakazakii must infect cells of the inner gut, cross the gut barrier to reach the bloodstream, and penetrate the blood-brain barrier. To investigate whether cklR regulates virulence by affecting cell invasion, the ability of wild-type and ΔcklR strains to invade Caco-2 cells and HBMECs was investigated. Cell invasion assays showed that invasion was similar between ΔcklR and wild-type strains (Figure 3A and B). These data suggested that cklR does not play a role in cell invasion, and thus it was thought that cklR might contribute to colonization ability to promote survival within animals.

Invasion of Caco-2 (A) and human brain microvascular endothelial cells (HBMECs) (B) by wild-type (WT) and ΔcklR Cronobacter sakazakii strains. Data are presented as means ± standard deviation; n = 3 (Student’s t test; ns, no statistical difference).
For meningitis-causing bacteria, some bacteremia is required for the onset of meningitis [8]. Our data showed that a high level of bacteremia is significantly associated with the development of meningitis defined as positive CSF cultures [27] in animals (Figure 2B). In contrast, the ability of ΔcklR to cause meningitis was significantly impaired. Thus, bacteremia caused by the ΔcklR strain might not have reached the level (Figure 2B) required for meningitis, mainly due to decreased survival within the host.
Ribonucleic Acid Sequencing and Chromatin Immunoprecipitation Sequencing Analysis Reveals That cklR Activates Expression of the suf Operon
Transcriptome analyses were performed to reveal differences in global gene expression profiles between ΔcklR and wild-type strains grown in LB medium. We identified 768 genes that were differentially expressed (±1.5-fold) between ΔcklR and wild-type strains, among which 47.0% (361 of 768) and 53.0% (407 of 768) of genes exhibited higher or lower transcription levels, respectively, in ΔcklR (details in Supplementary Table S5). The RNA-Seq results were confirmed by RT-PCR analysis (targeting 6 genes) under the same conditions (Supplementary Figure S4). Among the differentially expressed genes, we found that 6 genes (sufA, sufB, sufC, sufD, sufS, and sufE) within an operon (suf operon) were all down-regulated in ΔcklR.
Next, the C. sakazakii genome was screened by ChIP-Seq to reveal possible CklR-binding sites. After removing nonspecific binding regions for which read counts from mock-IP samples were greater than those of IP samples, 153 potential binding sites were detected. Among these, we found a binding site within sufD of the suf operon. The Suf system was postulated to have adapted to synthesize Fe-S clusters under stress conditions [28], which is important for bacterial virulence and survival [29, 30]. The proteins encoded by 6 genes of the C. sakazakii suf operon shared 73.6%–93.2% sequence identity to corresponding homologs of E coli K12 (GenBank accession number GCA_000005845.2).
Some LysR-type regulators such as CatR and MetR can bind to coding sequences downstream of the transcriptional start site of the operon to regulate operon expression [31, 32]. It was suggested that these regulators act as antirepressors that can relieve the binding of other negative regulators to the operon [31, 32]. These regulators might also affect DNA-loop formation by changing the architecture of nucleoprotein complexes to influence operon expression [31–33]. Thus, it was hypothesized that CklR binds sufD to regulate the suf operon.
CklR Directly Activates suf Operon Expression to Increase Bacterial Resistance to Oxidative Stress and Contribute to Survival in the Host
Electrophoretic mobility shift assays were performed with purified CklR-His6 protein to test whether CklR directly binds sufD. As shown in Figure 4A, with increasing concentrations of CklR, slow migrating bands were observed for the CklR-sufD complex, whereas no retarded bands were observed for the negative control, indicating that CklR directly binds sufD in vitro. Quantitative RT-PCR assays showed that sufD, sufC, and sufB expression was decreased by 2.2-, 1.8-, and 2.6-fold, respectively, in the ΔcklR strain (Figure 4B). These data suggested that CklR directly up-regulates expression of the suf operon.
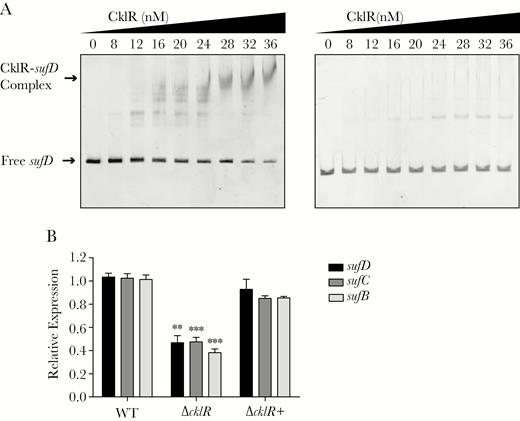
Cronobacter sakazakii CklR directly regulates expression of the suf operon. (A) Electrophoretic mobility shift assays of sufD fragment with purified CklR-His6 protein. 16s ribosomal deoxyribonucleic acid (A, right) was used as a negative control. (B) The expression of sufB, sufC, and sufD in wild-type (WT), ΔcklR, and ΔcklR+ strains, as revealed by quantitative reverse-transcription polymerase chain reaction. Data are presented as means ± standard deviation; n = 3 (Student’s t test; **, P < .01 and ***, P < .001).
We also performed animal experiments to analyze the role of the suf operon in C. sakazakii virulence. A sufD-mutant (ΔsufD) was generated, which exhibited similar growth rates with wild type (Supplementary Figure S3B). Rat pups were orally infected with of ΔsufD, wild-type, and ΔsufD+ complement strains, and survival was monitored for >108 hours (Figure 5A). Survival curves identified a significant difference between groups infected with wild-type/ΔsufD+ strains and the group infected with ΔsufD.
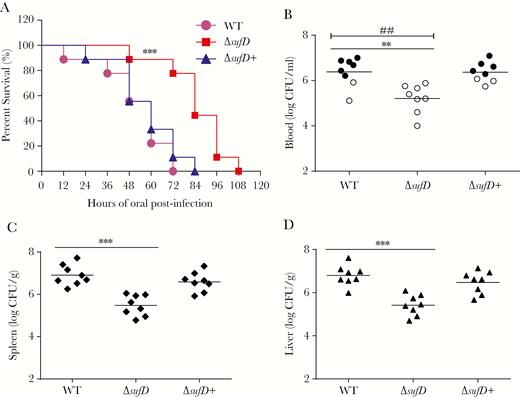
The suf operon contributes to the virulence of Cronobacter sakazakii. (A) Survival curves comprising 108 hours after oral infection with corresponding strains. Data presented are a combination of 3 independent experiments (log-rank test; ***, P < .001). (B–D) Bacterial counts from blood, spleen, and liver of orally infected rat pups. At 48 hours postinfection, blood collected from the facial vein, and spleen and liver, which were harvested and homogenized, was used for colony enumeration. Data are presented as means ± standard deviation; n = 3 (Student’s t test; **, P < .01 and ***, P < .001). For B, the development of meningitis in rat pups, defined as positive cerebrospinal fluid cultures, is shown by solid circles. Differences between the frequencies of the development of meningitis were determined (χ2 test; ##, P < .01). CFU, colony-forming units; WT, wild type.
In addition, bacterial colonization experiments also showed that the colonization of ΔsufD in blood, liver, and spleen was decreased by 14- to 29-fold compared with that of the wild-type strain (Figure 5B–D). Complementation of ΔsufD with the wild-type gene restored the colonization defect of ΔsufD. These results showed that sufD deletion attenuates the virulence of C. sakazakii toward rat pups.
The suf operon has been reported to contribute to bacterial oxidative-stress survival [29, 30]. Thus, we tested the role of cklR and the suf operon in bacterial resistance to oxidative stress by exposing wild-type, ΔcklR, ΔcklR+, ΔsufD, and ΔsufD+ to H2O2. In the presence of 25 mM H2O2 for 20 minutes, mutants exhibited significantly lower survival (ΔcklR, 21.3-fold; ΔsufD, 10.6-fold) than wild-type strains (Figure 6). These data suggest that cklR and sufD increase C. sakazakii survival in response to oxidative stress.
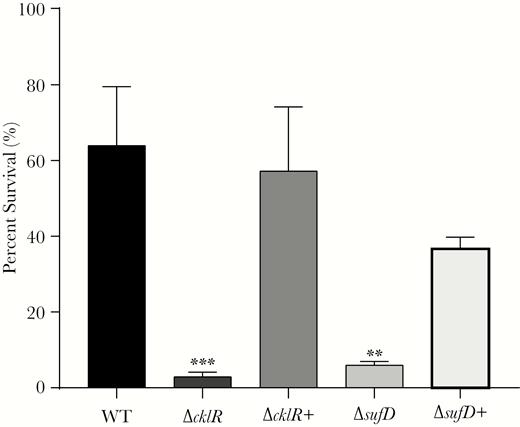
Sensitivities to hydrogen peroxide of Cronobacter sakazakii strains. Survival of C. sakazakii wild-type (WT), ΔcklR, ΔcklR+, ΔsufD, and ΔsufD+ strains in the presence of hydrogen peroxide. Data are presented as means ± standard deviation; n = 3 (Student’s t test; **, P < .01 and ***, P < .001).
It has been reported that C. sakazakii can tolerate the intracellular environment of macrophages and uses these cells as a vehicle to invade other organs [7, 23]. As bacteria encounter oxidative stress within macrophages and cklR and the suf operon contribute to survival in response to oxidative stress, we analyzed survival in RAW 264.7 cells. Results showed that survival of ΔcklR and ΔsufD strains was obviously decreased compared with that of the wild-type strain (Figure 7A and B). Complementing mutant strains with wild-type genes restored this defect. Quantitative RT-PCR also showed that when the wild-type strain was cultured in macrophages, the expression of cklR, sufB, sufC, and sufD was increased by 10.0-, 2.5-, 2.4-, 3.3-fold, respectively, compared with that when grown in LB medium (Figure 7C). Furthermore, in macrophages, the expression of sufB, sufC, and sufD was decreased by 2.1-, 1.9-, and 2.8-fold in the ΔcklR stain, compared to that in the wild-type strain (Figure 7C). Complementation of ΔcklR restored expression of the suf operon to wild-type levels within macrophages. Thus, it is highly possible that CklR contributes to C. sakazakii virulence by activating the expression of the suf operon, which increases bacterial survival within macrophages by protecting bacteria from oxidative stress.
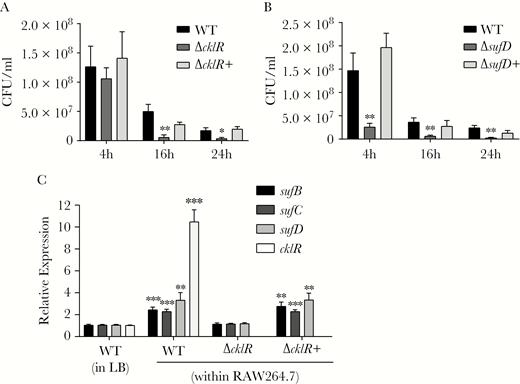
cklR and the suf operon are required for the survival of Cronobacter sakazakii within macrophages. (A and B) The survival of C. sakazakii was attenuated in RAW264.7 macrophage cells due to the absence of cklR and sufD. (C) Expression of sufB, sufC, sufD, and cklR in C. sakazakii wild-type (WT), ΔcklR, and ΔcklR+ strains within RAW264.7 macrophage cells or in Luria-Bertani medium, as revealed by quantitative reverse-transcription polymerase chain reaction. Data are presented as means ± standard deviation; n = 3 (Student’s t test: *, P < .05; **, P < .01; ***, P < .001).
DISCUSSION
In this study, we characterized a specific LysR-type regulator (CklR) that is significantly associated with prevalent C. sakazakii strains that cause neonate infections. In addition to the suf operon, our data showed that the expression of several other genes including sodC, sodA, btuE, katE, and msrAB, which are involved in the antioxidant defense system, was also decreased in ΔcklR (Supplementary Table S5), indicating that CklR can positively affect the expression of these genes. However, our ChIP-Seq analysis did not detect the binding of CklR within DNA region associated with these antioxidant defense genes, indicating that CklR probably indirectly regulates the expression of these genes, possibly contributing to bacterial resistance to oxidative stress; however, the underlying molecular mechanism needs to be further elucidated.
Fe-S cluster metalloproteins perform essential functions in all domains. The Suf Fe-S cluster biosynthesis operon is required for the fitness and survival of many bacterial pathogens such as Staphylococcus aureus [29], Shigella flexneri [30], and Erwinia chrysanthemi [34]. Host phagocytic cells including neutrophils and macrophages produce reactive oxygen species (ROS) to restrict bacterial pathogen infection [35]. Thus, resistance to ROS is essential for the survival and virulence of bacterial pathogens within the host [36]. Protein-associated and solvent-exposed Fe-S clusters are a primary target of ROS damage [37]. For example, mutations in the suf operon of S aureus obviously increase bacterial sensitivity to ROS and decrease the ability to survive challenge by human polymorphonuclear neutrophils [29]. In this study, we showed that a mutation in the suf operon and down-regulation of suf expression (by deleting cklR) both increase the sensitivity of C. sakazakii to H2O2 and inhibit survival within macrophages, indicating that cklR and the suf operon contribute to the virulence of CG1 and CG2 C. sakazakii by improving resistance to oxidative stress encountered within the host.
CONCLUSIONS
Our analysis revealed that CG1 and CG2, which mainly corresponds to ST4 and ST12, respectively, are significantly associated with neonate infections. However, previously, no specific traits or factors had been identified in these 2 important C. sakazakii pathogenic groups [16]. As described, all known virulence factors were equally distributed among all C. sakazakii CGs, and thus these might not determine the high virulence of CG1 and CG2 compared to that of other C. sakazakii strains (Supplementary Figure S1). In this study, we showed that cklR is possibly closely related to the predominance of these 2 CGs in neonate infection. In addition to cklR, several other genes were also found to be prevalent in either CG1, CG2, or both (Supplementary Table S4), and their functions require further investigation. It should be noted that there were also clinical strains in other CGs. For example, C. sakazakii ATCC BAA-894, belonging to ST1, was found to be within CG4, whereas C. sakazakii ATCC 29544, belonging to ST8, was within CG7, and both were isolated from clinical samples [38, 39]. However, cklR were not found in these genomes. It is likely that these strains probably have other regulators for suf operon or utilize other mechanisms to increase resistance to oxidative stress.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. We thank Dr. Yamin Sun and Wenchao Lin for technical support of transcriptome and genome analysis.
Financial support. This work was funded by the National Science and Technology Major Project of China(No. 2017ZX10104002-001-006; to B. L.) and the National Natural Science Foundation of China (Nos. 81471904, 81772148, and 81611530714; to B. L.).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
References
Author notes
M. W., L. W., and P. W. contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}