






Abstract
The bacterial pathogen Neisseria gonorrhoeae is able to transmigrate across the mucosal epithelia following the intracellular route and cause disseminated infections. It is currently unknown whether the autophagy pathway is able target intracellular N. gonorrhoeae for destruction in autolysosomes or whether this bacterium is able to escape autophagy-mediated killing. In this study, we demonstrate that during the early stage of epithelial cell invasion, N. gonorrhoeae is targeted by the autophagy pathway and sequestered into double-membrane autophagosomes that subsequently fuse with lysosomes for destruction. However, a subpopulation of the intracellular gonococci is able to escape early autophagy-mediated killing. N. gonorrhoeae is subsequently able to inhibit this pathway, allowing intracellular survival and exocytosis. During this stage, N. gonorrhoeae activates the autophagy repressor mammalian target of rapamycin complex 1 and inhibits autophagosome maturation and lysosome fusion. Thus, our results provide novel insight into the interactions between N. gonorrhoeae and the autophagy pathway during invasion and transcytosis of epithelial cells.
Neisseria gonorrhoeae is the causative agent of the sexually transmitted disease gonorrhea. The World Health Organization estimates that worldwide there are approximately 106 million new cases of gonorrhea annually [1]. N. gonorrhoeae is a human-specific bacterial pathogen that generally colonizes the mucosal epithelial cell layers in the urogenital tract, rectum, and pharynx. Uncomplicated gonococcal infections commonly manifest as urethritis in men and may cause mucopurulent cervicitis in women. However, gonococcal infections of the female reproductive tract are often asymptomatic, but they may lead to serious complications when untreated, including pelvic inflammatory disease, ectopic pregnancy, and infertility [2, 3]. Colonization of the epithelial cell monolayers can lead to the invasion and transmigration of N. gonorrhoeae across the epithelial barrier into the subepithelial spaces [4, 5]. Given that (1) subepithelial N. gonorrhoeae have been discovered in clinical samples from patients with disseminated gonococcal infections [6, 7] and (2) in vitro studies using polarized epithelial cells have shown that N. gonorrhoeae follows the intracellular route to pass epithelial cell layers without breaching the apical tight junctions [8, 9], transcytosis across epithelial cells might be a critical step for N. gonorrhoeae to cause disseminated infections.
For N. gonorrhoeae, multiple surface molecules are able to initiate or mediate colonization and invasion of epithelial cells through direct interactions with cellular receptors or by affecting cellular signaling pathways, including type IV pili [10–13], opacity-associated (Opa) proteins [14, 15], PorB [9, 10, 16], and IgAP [16, 17]. However, the mechanisms involved in transcytosis and intracellular survival are less well understood, although the cellular cytoskeleton and motor proteins appear to be involved in transcytosis [5]. During transcytosis of epithelial cells, N. gonorrhoeae is potentially targeted by the autophagy pathway, which is a cellular maintenance process that targets damaged organelles and macromolecules to the lysosome for degradation. Nonetheless, in addition to cellular maintenance, it is also an important host defense mechanism that is able to target invading bacteria for degradation in the lysosome [18, 19]. However, it is currently unknown whether the autophagy pathway is able to target N. gonorrhoeae during invasion and transcytosis of epithelial cells and limit its ability to survive intracellular. For macrophages only, the addition of phosphoethanolamine to the gonococcal lipooligosaccharide reduces the autophagy flux [20].
In this study, we show that the autophagy pathway is activated during the early stage of epithelial cell invasion and able to target intracellular N. gonorrhoeae and direct N. gonorrhoeae-containing autophagosomes to lysosomes for degradation. However, N. gonorrhoeae is able to inhibit the autophagy pathway during the later stage of invasion, thereby allowing a subpopulation of intracellular bacteria to survive and subsequently show exocytosis. Inhibition of autophagy at this stage is the result of both activation of the autophagy repressor mammalian target of rapamycin complex 1 (mTORC1) and inhibition of autophagosome maturation and fusion with lysosomes.
METHODS
Gentamycin Protection Assays
HeLa human cervix carcinoma cells were seeded in 12-well plates (2 × 105 cells/well) in Roswell Park Memorial Institute (RPMI) 1640 cell culture medium (Biological Industries) supplemented with 10% fetal bovine serum (Bovogen) and grown overnight at 37°C in presence of 5% CO2. HeLa cells were transfected with small interfering ribonucleic acid (siRNA) or plasmid deoxyribonucleic acid (DNA) or incubated with Bafilomycin A1 ([Sigma] 50 nM final concentration, 4 hours), Torin-1 ([Tocris Bioscience] 2 nM final concentration, 2 hours), or 3-methyladenine ([Sigma] 100 mM final concentration, 2 hours) before challenge with N. gonorrhoeae when required. To challenge HeLa cells, N. gonorrhoeae strain American Type Culture Collection 49226 was grown overnight at 37°C in 5% CO2 on GC agar (Oxoid) containing 1% Vitox (Oxoid), suspended in prewarmed cell culture medium, and used at a multiplicity of infection of 100. After 1-hour incubation (0 hours; cell associated), medium was replaced with fresh RPMI 1640 medium containing 200 µg/mL gentamycin (Songon). Samples were taken in a time-series after addition of gentamycin (1–6 hours; intracellular). To quantify the number of viable N. gonorrhoeae, HeLa cells were lysed using 1% Saponin (Sigma), and samples were serial diluted and plated on GC agar containing 1% Vitox. After 48-hour incubation at 37°C and 5% CO2, colony-forming units (CFUs) were enumerated. In a modified gentamycin protection assay, cell culture medium containing gentamycin was replaced after 1-hour incubation using fresh RPMI 1640 medium to allow survival of N. gonorrhoeae that showed exocytosis. The number of viable N. gonorrhoeae inside the HeLa cells and in the culture medium was quantified by serial dilution and plating to determine the CFU counts.
Western Blotting
Samples were boiled for 10 minutes in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis sample buffer and loaded onto 12%–15% SDS polyacrylamide gels. Proteins were transferred to Immobilon-P membranes (Bio-Rad), and membranes were blocked with 3% (w/v) milk solution before incubation with primary antibodies diluted in 5% (w/v) bovine serum albumin: anti-LC3 polyclonal antibodies (Medical and Biological Laboratories), anti-p62 polyclonal antibodies (Proteintec), anti-Phospho-70 S6 Kinase (Thr389) monoclonal antibodies (Abcam), anti-70 S6 Kinase polyclonal antibodies (Abcam), anti-Phospho-4E-BP1 (Thr37/46) monoclonal antibodies (Abcam), anti-4E-BP1 polyclonal antibodies (Abcam), anti-Atg7 monoclonal antibodies (Abcam), or anti-β-tubulin polyclonal antibodies (Proteintec). Membranes were subsequently incubated with secondary antibodies diluted in 1% (w/v) milk solution: horseradish peroxidase (HRP)-conjugated Goat anti-Rabbit IgG (Abbkine) or HRP-conjugated Goat anti-Mouse IgG (Abbkine). Horseradish peroxidase activity was visualized using Enhanced Chemiluminescence (ECL) Western Blotting Substrate (Pierce Biotechnology).
Transfection With Small Interfering Ribonucleic Acids and Plasmids
HeLa cells were transfected with Atg7-specific siRNA (Shanghai GenePharma Corporation) or plasmids expressing GFP-LC3, EGFP-FYVE, or mCherry-GFP-LC3. HeLa cells were transfected with 100 μL OptiMEM medium (Gibco/BRL) containing 1% Lipofectamine2000 (Invitrogen) and 20 pmol siRNA or 1 μg of plasmid DNA.
Confocal Laser Scanning Microscopy
For all confocal laser scanning microscopy experiments, HeLa cells were fixed with 4% paraformaldehyde for 20 minutes. For intracellular LIVE/DEAD staining, permeabilized HeLa cells were incubated with SYTO 9 (1.67 μM) and propidium iodide (10 μM) stains (Thermo) for 15 minutes. For N. gonorrhoeae colocalization and analyses with LC3 or FYVE, HeLa cells were transfected with vectors expressing GFP-LC3, mCherry-GFP-LC3, or EGFP-FYVE and challenged with Dil- or 4’,6-diamidino-2-phenylindole (DAPI)-stained (Beyotime Biotechnology) N. gonorrhoeae. For lysosomal-associated membrane protein (LAMP)-1 colocalization analysis, HeLa cells were challenged with Dil-stained N. gonorrhoeae. After fixing and permeabilization, cells were incubated with rabbit anti-LAMP-1 polyclonal antibodies (Cell Signaling Technology) and Alexa Fluor 488-conjugated goat anti-rabbit IgG (Thermo Fisher). Images were taken on a Zeiss Axiovert confocal microscope (Zeiss). Images were quantified for puncta formation and colocalization with N. gonorrhoeae.
Transmission Electron Microscopy
Samples were fixed with 2.5% glutaraldehyde, incubated with 2% osmium tetroxide in 0.1 M sodium cacodylate buffer, and embedded in epoxy resin (EMBED 812; Electron Microscopy Sciences). Sections were cut at a nominal thickness of 80 nm and stained with uranyl acetate and lead citrate. Sections were examined at 120 kV using a Tecnai G2 Spirit transmission electron microscope (FEI Company), and images were recorded using a Gatan USC1000 2k CCD camera.
RESULTS
A Subpopulation of N.gonorrhoeae Shows Prolonged Intracellular Survival and Exocytosis of HeLa Cells
N. gonorrhoeae invasion, intracellular survival, and exocytosis of HeLa human cervix carcinoma cells were characterized using gentamycin protection assays. Intracellular viable bacterial numbers were highest 1 hour after addition of gentamycin, after which a gradual decline was observed (Figure 1A–C). To investigate whether N. gonorrhoeae is able to show exocytosis, a modified gentamycin protection assay was used that allows survival of bacteria showing exocytosis. Indeed, with the decline of intracellular CFU counts, a gradual increase in extracellular CFU counts in the culture supernatant was observed (Figure 1D). The increase of extracellular CFU counts was not the result of extracellular replication of the bacteria in the cell culture medium (Supplementary Figure 1). Based on the quantification of intracellular and extracellular bacteria, it appears that many invading bacteria are killed, while a subpopulation of the intracellular bacteria is able to survive and subsequently show exocytosis.
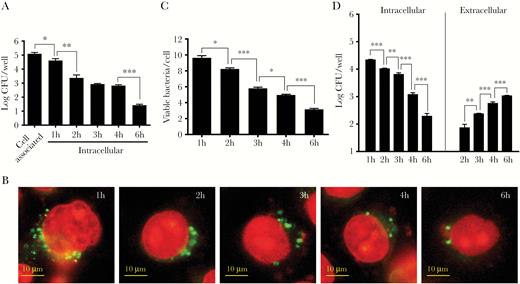
Neisseria gonorrhoeae invasion, intracellular survival, and exocytosis of HeLa cells. (A) Adhesion, invasion, and intracellular survival of N. gonorrhoeae was quantified by gentamycin protection assays. The graph represents the mean and standard deviation (SD) of 3 independent experiments. (B) The viability of intracellular bacteria was visualized by intracellular LIVE/DEAD staining after gentamycin protection assays. Representative images are shown. Viable bacteria are stained with SYTO 9, whereas dead bacteria and the cell nucleus are stained with propidium iodide. (C) The number of viable bacteria after intracellular LIVE/DEAD staining was quantified by image analyses. The graph represents the mean and SD of 3 independent experiments, and at least 100 cells were analyzed for each condition. (D) Exocytosis and intracellular survival of N. gonorrhoeae was quantified in modified gentamycin protection assays in which gentamycin was removed after 1 hour of exposure to allow survival of extracellular bacteria in subsequent time points. The graph represents the mean and SD of 3 independent experiments. Significant differences between time points were identified by Student’s t test. *, P < .05; **, P < .01; ***, P < .005. CFU, colony-forming units.
Invading Gonococci Are Targeted by Autophagy and Captured in Autophagosomes
To investigate the activity of the autophagy pathway during N. gonorrhoeae invasion of epithelial cells, 2 of the major autophagy activity marker proteins, p62 and microtubule-associated protein 1 light chain 3 (LC3), were studied by Western analysis. Both P62 and LC3-II appeared to accumulate in time during the invasion process (Figure 2A and Supplementary Figure 2). To investigate whether accumulation was the result of increased flux or inhibition of recycling, the autophagosome-lysosome fusion step was interrupted by inhibition of the V-ATPase with BafA1. Further accumulation of p62 and LC3-II levels was observed after BafA1 treatment compared with the vehicle control (Figure 2B and Supplementary Figure 3), indicating that the autophagy flux was induced during cell invasion but simultaneously recycling appeared to be blocked. In addition, given that LC3-I also accumulated, it appeared that the process of LC3 conjugation with phosphatidylethanolamine (PE), which is essential for autophagosome formation, might also be affected. To investigate autophagosome formation and direct targeting of N. gonorrhoeae by the autophagy pathway during invasion of epithelial cells, LC3 activity and colocalization with N. gonorrhoeae were investigated by confocal microscopy. During the first hour of invasion, an increase in total LC3 puncta and colocalized puncta with N. gonorrhoeae were observed, whereas in subsequent time points the number of LC3 puncta and colocalized puncta dropped significantly (Figure 2C and D). In a control experiment, HeLa cells were incubated with 3-MA, which inhibits the class III phosphatidylinositol-3-OH kinase (PI3KC3) complex and prevents LC3-PE conjugation and formation of autophagosomes. Indeed, Western analysis confirmed that LC3 processing was inhibited, whereas p62 accumulation to similar levels as the vehicle control was still apparent (Supplementary Figure 4). Furthermore, a major reduction in LC3 puncta and colocalization with N. gonorrhoeae was observed by confocal microscopy (Figure 2D and Supplementary Figure 4). Subsequent transmission electron microscopy (TEM) analysis showed that N. gonorrhoeae was frequently present inside double-membrane compartments or associated with autophagic structures (Figure 2E), which confirmed that N. gonorrhoeae indeed ended up in autophagosomes during cell invasion. Overall, these data showed that during the early stage of N. gonorrhoeae invasion of epithelial cells, the autophagy pathway is activated and able to target N. gonorrhoeae, whereas during the later stage of the invasion process this pathway appears to be inhibited.
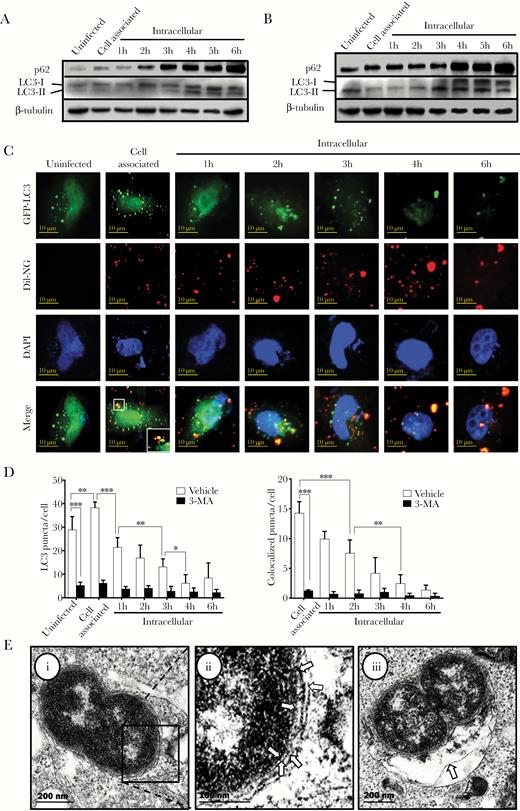
The autophagy pathway is activated and targets Neisseria gonorrhoeae during cell invasion. (A) Western analysis of the autophagy marker proteins p62 and LC3 during N. gonorrhoeae invasion of HeLa cells in gentamycin protection assays. Representative images are shown of at least 3 independent biological repeats. β-tubulin was included as loading control. (B) Western analysis of p62 and LC3 in gentamycin protection assays after pretreatment with the V-ATPase inhibitor bafilomycin A1 (BafA1). Representative images are shown of at least 3 independent biological repeats. (C) Confocal microscopy analysis of GFP-LC3 puncta and colocalization with Dil-stained N. gonorrhoeae in gentamycin protection assays. The HeLa cell nucleus was stained with 4’,6-diamidino-2-phenylindole (DAPI). (D) Quantification of total LC3 puncta and colocalization of LC3 puncta with N. gonorrhoeae from panel C and 3-MA pretreated control cells. The graphs represent the mean and standard deviation of 3 independent experiments, and at least 100 cells were analyzed for each repeat. Significant differences between time points and between treatment and control were identified by Student’s t test. *, P < .05; **, P < .01; ***, P < .005. (E) Transmission electron microscopy analysis of intracellular N. gonorrhoeae after 1 hour of invasion in HeLa cells. (i) N. gonorrhoeae-containing autophagosome. Arrows in image (ii) indicate the vacuolar and double autophagic membrane surrounding N. gonorrhoeae, which is the hallmark of autophagosomes. The arrow in image (iii) indicates additional autophagic structures that appear to fuse with the N. gonorrhoeae-containing autophagosome.
The Autophagy Pathway Limits Early Intracellular Gonococcal Survival
To investigate whether early activation of the autophagy pathway has an impact on N. gonorrhoeae invasion, intracellular survival, and exocytosis, gentamycin protection assays were performed using HeLa cells transfected with Atg7-specific siRNA, which is an essential component of the canonical autophagy pathway. Transfection with Atg7-specific siRNA resulted in a major reduction of Atg7 (Figure 3A). Further Western analysis of p62 and LC3-II levels in Atg7 siRNA-treated cells showed that p62 still accumulated during the course of the infection, whereas LC3 conversion to LC3-II was almost completely abolished (Figure 3B and Supplementary Figure 5). Subsequent quantification of viable bacteria showed that both intracellular survival (Figure 3C) and exocytosis (Figure 3D) were 2- to 3-fold enhanced in Atg7 siRNA-transfected cells compared with control cells. To corroborate these results, gentamycin protection assays were performed after pretreatment of the HeLa cells with BafA1 and 3-MA. Similarly to what was observed for the Atg7-siRNA transfected cells, intracellular survival and exocytosis was enhanced in HeLa cells pretreated with either BafA1 or 3-MA (Supplementary Figure 6). Therefore, these results show that the autophagy pathway impacts intracellular survival of N. gonorrhoeae in the early stage of invasion, but based on the similar trend in temporal decline of intracellular viable bacteria with or without inhibition of the autophagy pathway and the subsequent exocytosis of N. gonorrhoeae, it again appears that N. gonorrhoeae is able to inhibit the autophagy pathway during the later stage of cell invasion.
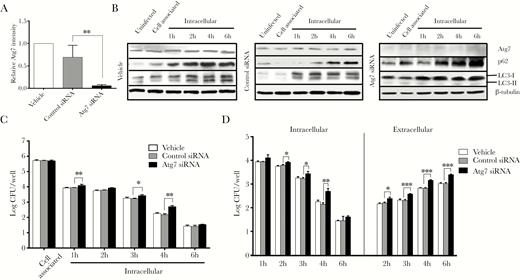
Neisseria gonorrhoeae shows enhanced intracellular survival and exocytosis after Atg7 knockdown. HeLa cells were transfected with Atg7-specific small interfering ribonucleic acid (siRNA) or control siRNA and used in gentamycin protection assays with N. gonorrhoeae. (A) Quantification of Atg7 levels in uninfected HeLa cells after transfection with Atg7 or control siRNA, as determined by Western blot analysis. The Atg7 levels were normalized according to β-tubulin levels. The graph represents the mean and standard deviation (SD) of 3 independent experiments. (B) Western blot analysis of Atg7, p62, and LC3 levels during N. gonorrhoeae invasion of HeLa cells in gentamycin protection assays. Representative images are shown of at least 3 independent experiments. β-tubulin was included as loading control. (C) Quantification of adhesion, invasion, and intracellular survival of N. gonorrhoeae in transfected HeLa cells. The graph expresses the mean and SD of 3 independent biological repeats. (D) Quantification of exocytosis and intracellular survival of N. gonorrhoeae in modified gentamycin protection assays using transfected HeLa cells. The graph represents the mean and SD of 3 independent experiments. Significant differences between Atg7 siRNA transfected cells and control cells were identified by Student’s t test. *, P < .05; **, P < .01; ***, P < .005. CFU, colony-forming units.
Autophagosome Maturation Is Inhibited During the Late Stage of Invasion
To investigate maturation of the N. gonorrhoeae-containing autophagosomes into autolysosomes, HeLa cells were transfected with a vector expressing a mCherry-GFP-LC3 fusion protein and subsequently used in gentamycin protection assays. GFP is acid sensitive, and its fluorescent activity is therefore lost in the low-pH environment of the autolysosome, whereas mCherry is more acid stable and therefore still visible in this environment. Accumulation of both GFP+-mCherry+ and GFP−-mCherry+ LC3 puncta was apparent after 1 hour of invasion (Figure 4A and B); in particular, the number of GFP−-mCherry+ puncta was highly elevated compared with uninfected cells (12-fold). In subsequent time points, the total number of GFP−-mCherry+ puncta dropped quickly, whereas for GFP+-mCherry+ puncta a more gradual decline was observed. Further analyses of colocalization of GFP−-mCherry+ and GFP+-mCherry+ puncta with DAPI-stained N. gonorrhoeae showed that during the first hour of cell invasion, most of the bacteria colocalized with GFP−-mCherry+ puncta. In subsequent time points, colocalization of N. gonorrhoeae with GFP−-mCherry+ LC3 puncta disappeared quickly and became undetectable. In contrast, colocalization of N gonorrhoeae with GFP+-mCherry+ LC3 puncta was highest after 1 hour of gentamycin treatment and showed a gradual decline in later time points. As control, a major reduction in GFP+-mCherry+, GFP−-mCherry+, and colocalized LC3 puncta was observed in 3-MA pretreated cells (Supplementary Figure 7). Subsequent TEM analyses confirmed that during the early stage of epithelial cell invasion, N. gonorrhoeae-containing autophagosomes were able to fuse with endosomes or lysosomes, which resulted in the degradation of N. gonorrhoeae in these autolysosomes (Figure 4C). To further confirm fusion with lysosomes was inhibited during the later stage of cell invasion, colocalization of N. gonorrhoeae and the lysosomal marker protein LAMP-1 was investigated by confocal microscopy after endogenous immunostaining of LAMP-1. The total number of LAMP-1 puncta increased during the first hour of cell invasion, indicating that lysosome biogenesis might initially be induced, whereas afterwards a gradual decline was observed to levels below that of uninfected cells (Figure 5A and B). Likewise, colocalization of LAMP-1 puncta with Dil-stained N. gonorrhoeae was highest after the first hour and a gradual decline was again observed. As control, cells were treated with 3-MA, which indeed resulted in a marked reduction in total and colocalized puncta (Figure 5B and Supplementary Figure 8). From these results it is apparent that during the first hours of N. gonorrhoeae invasion, autophagosomes containing N. gonorrhoeae are able to mature into autolysosomes, thereby killing the bacterium, whereas during the later stage of invasion autophagosome maturation and fusion with lysosomes is inhibited.
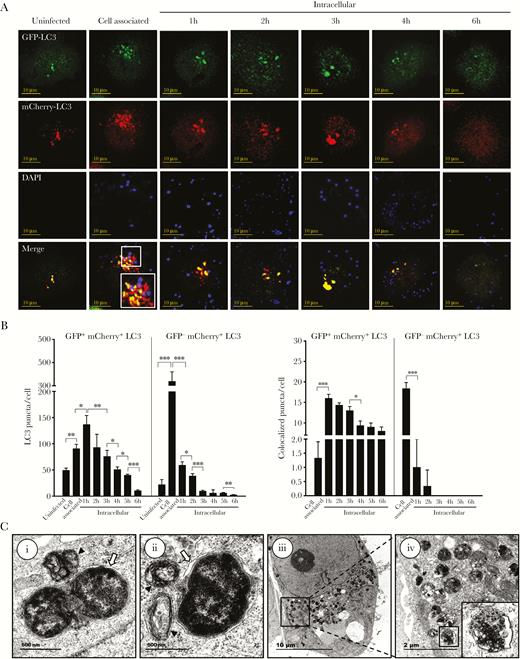
Neisseria gonorrhoeae inhibits autophagosome maturation. (A) Confocal microscopy analysis of gentamycin protection assays using HeLa cells transfected with a vector expressing mCherry-GFP-LC3. Cytosolic LC3 puncta are shown as GFP+-mCherry+, whereas lysosomal LC3 puncta are shown as GFP−-mCherry+. N. gonorrhoeae was stained with 4’,6-diamidino-2-phenylindole (DAPI) before the assays. Representative images are shown of assays performed on 3 separate occasions. (B) Quantification of the total GFP+-mCherry+ and GFP−-mCherry+ LC3 puncta and colocalization with N. gonorrhoeae in panel A. The graph represents the mean and standard deviation (SD) of 3 independent repeats, and at least 100 cells were analyzed for each repeat. Significant differences between time points were identified by Student’s t test. *, P < .05; **, P < .01; ***, P < .005. (C) Transmission electron microscopy analysis of intracellular N. gonorrhoeae in gentamycin protection assays after 1 hour of gentamycin treatment. (i) N. gonorrhoeae-containing autophagosome appears to fuse with a lysosome. (ii) N. gonorrhoeae looks already partly degraded inside an autolysosome, whereas more endosomes/lysosomes appear to fuse. (iii and iv) Numerous autolysosomes are visible at this stage containing N. gonorrhoeae undergoing degradation. White arrows indicate double-membrane autophagic structures surrounding N. gonorrhoeae, and black triangles indicate lysosomes and endosomes that seem to fuse with the N. gonorrhoeae-containing autophagosomes.
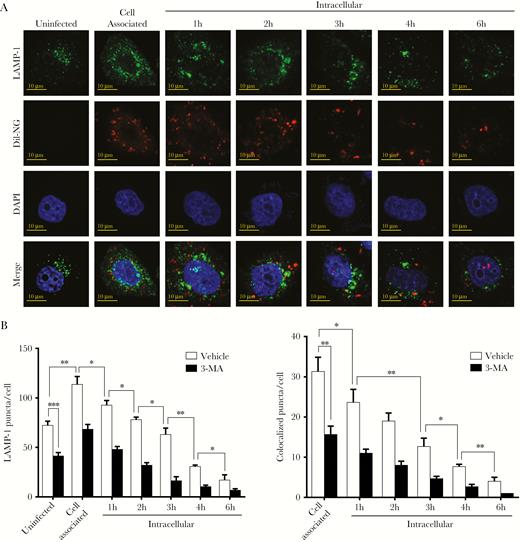
Neisseria gonorrhoeae inhibits colocalization and fusion with lysosomes. (A) Confocal microscopy analysis of lysosomal-associated membrane protein (LAMP)-1 puncta and colocalization with Dil-stained N. gonorrhoeae in gentamycin protection assays. The HeLa cell nucleus was stained with 4’,6-diamidino-2-phenylindole (DAPI). Representative images are shown. (B) Quantification of total LAMP-1 puncta and colocalization with N. gonorrhoeae in panel A and 3-MA pretreated control cells. The graph represents the mean and standard deviation of at least 100 cells per time point. Significant differences were identified by Student’s t test. *, P < .05; **, P < .01; ***, P < .005.
Phosphatidylinositol-3-Phosphate Levels and Colocalization With Neisseria gonorrhoeae Are Reduced During the Late Stage of Invasion
Western analyses showed that LC3-I also accumulated during N. gonorrhoeae cell invasion (Figure 2A), which indicates that LC3 processing might be affected. LC3 conjugation with PE at the phagophore and autophagosome is dependent on the Atg16L1 complex, which is recruited to this site through the formation of phosphatidylinositol-3-phosphate (PI3P). The formation of PI3P by the PI3KC3 complex recruits downstream proteins containing a FYVE zinc-finger domain, which therefore can serve as a marker for the generation of PI3P. To visualize generation and colocalization of PI3P during N. gonorrhoeae cell invasion, HeLa cells were transfected with a vector expressing GFP-FYVE and used in gentamycin protection assays. Indeed, colocalization of N. gonorrhoeae with GFP-FYVE puncta was observed on confocal microscopy images (Figure 6A). The total number of GFP-FYVE puncta was significantly elevated after 1 hour of cell invasion compared with uninfected cells, and afterwards the number of puncta dropped quickly to levels below observed for uninfected cells (Figure 6A and B). Further analyses of colocalization between N. gonorrhoeae and GFP-FYVE puncta showed that the number of colocalized puncta was highest after 1 hour of gentamycin treatment and afterwards colocalization declined. As control, cells were treated with 3-MA, which indeed resulted in a marked reduction in total and colocalized puncta (Figure 6B and Supplementary Figure 9). From these results, it appears that besides autophagosome maturation, generation of PI3P is also inhibited during the later stage of cell invasion.
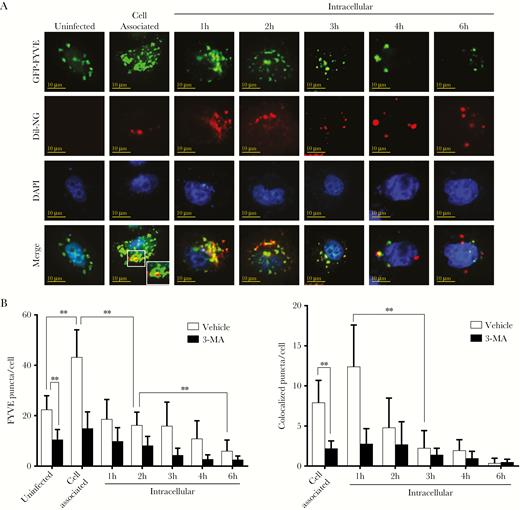
Neisseria gonorrhoeae inhibits phosphatidylinositol-3-phosphate generation and colocalization. HeLa cells were transfected with a vector expressing GFP-FYVE and used in gentamycin protection assays with N. gonorrhoeae. (A) Confocal microscopy analysis of GFP-FYVE puncta and colocalization with Dil-stained N. gonorrhoeae in gentamycin protection assays. The HeLa cell nucleus was stained with 4’,6-diamidino-2-phenylindole (DAPI). Representative images are shown of 3 independent repeats. (B) Quantification of total GFP-FYVE puncta and colocalization with N. gonorrhoeae in panel A and 3-MA pretreated control cells. The graph represents the mean and standard deviation of 3 independent repeats, and at least 100 cells were analyzed for each repeat. Significant differences between time points were identified by Student’s t test. **, P < .01.
Neisseria gonorrhoeae Inhibits Canonical Autophagy Through Mammalian Target of Rapamycin Complex 1 Activation
The canonical autophagy pathway is regulated by mTORC1, which inhibits the ULK1 complex by phosphorylation, thereby preventing its recruitment to the phagophore assembly sites. However, mTORC1 also phosphorylates 2 other downstream proteins that are not directly feeding into the canonical autophagy pathway, namely, 4EBP1 and p70S6K. Therefore, the phosphorylation state of these 2 proteins is frequently used as an indicator for mTORC1 activity. Western analysis indeed showed that the levels of phosphorylated 4EBP1 and p70S6K were both elevated during the invasion assays, whereas no changes in the nonphosphorylated controls were observed (Figure 7A and Supplementary Figure 10). To further study the impact of mTORC1 on N. gonorrhoeae cell invasion and intracellular survival, gentamycin protection assays were performed after pretreatment of the HeLa cells with Torin1, which is a well defined chemical inhibitor of mTORC1. Western analysis on Torin1-pretreated cells indeed confirmed activation of the autophagy pathway, since p62 accumulation was largely prevented and LC3-II levels were significantly reduced (Supplementary Figure 11). However, no significant differences in invasion, intracellular survival, and exocytosis were observed between the Torin1-pretreated and vehicle control cells (Figure 7B and Supplementary Figure 12). To investigate autophagosome maturation during N. gonorrhoeae invasion in Torin1-pretreated cells, HeLa cells were again transfected with the mCherry-GFP-LC3-expression vector. Although Torin1-pretreated cells initially showed significantly higher numbers of GFP+-mCherry+ and GFP−-mCherry+ puncta and colocalized puncta, after 2 hours no differences were observed anymore compared with the vehicle control (Figure 7C and D and Supplementary Figure 13). Therefore, these results show that N. gonorrhoeae inhibits the canonical autophagy pathway during cell invasion through activation of mTORC1. However, in addition, N. gonorrhoeae also inhibits autophagosome maturation and fusion with lysosomes independent of mTORC1 activation.
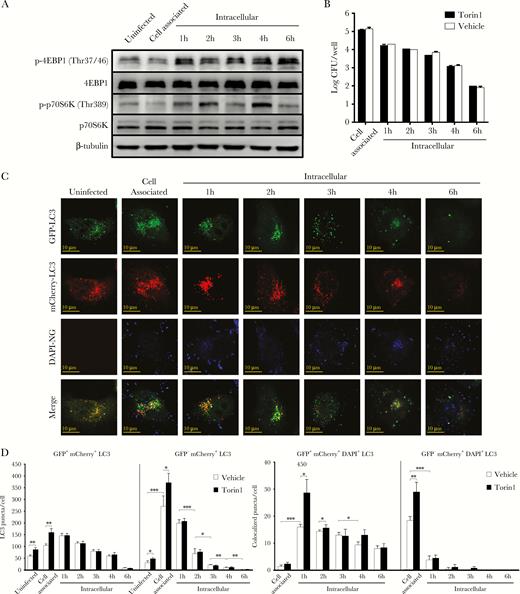
Neisseria gonorrhoeae activates the autophagy repressor mammalian target of rapamycin complex 1 (mTORC1) during cell invasion. (A) Western analysis of the phosphorylated and nonphosphorylated mTORC1 substrate proteins 4EBP1 and p70S6K during N. gonorrhoeae invasion of HeLa cells in gentamycin protection assays. β-tubulin was included as loading control. (B) Quantification of adhesion, invasion, and intracellular survival of N. gonorrhoeae in Torin1 pretreated HeLa cells. The graph expresses the mean and standard deviation (SD) of 3 independent experiments. (C) Confocal microscopy analysis of gentamycin protection assays using HeLa cells transfected with a vector expressing mCherry-GFP-LC3 and pre-treated with Torin1. Cytosolic LC3 puncta are shown as GFP+-mCherry+, whereas lysosomal LC3 puncta are shown as GFP−-mCherry+. N. gonorrhoeae was stained with DAPI before the assays. Representative images are shown of assays performed on 3 separate occasions. (D) Quantification of the total GFP+-mCherry+ and GFP−-mCherry+ LC3 puncta and colocalization with N. gonorrhoeae using Torin1-treated cells in panel C and vehicle control cells in Supplementary Figure S13. The graph represents the mean and SD of 3 independent repeats, and at least 100 cells were analyzed for each repeat. Significant differences between time points were identified by Student’s t test. *, P < .05; **, P < .01; ***, P < .005.
DISCUSSION
During an infection, N. gonorrhoeae colonizes the epithelial mucosa that lines the urogenital tract. In addition, N. gonorrhoeae is able to cause disseminated infections, which requires transmigration across the epithelial cell monolayer, likely following the intracellular route. However, during transcytosis of epithelial cells, intracellular gonococci are exposed to the autophagy pathway, which is able to clear invading bacterial pathogens. For a number of bacterial pathogens, it has already been shown that autophagy is able to restrict their intracellular growth or eliminate the infection (reviewed in [21, 22]). However, for N. gonorrhoeae, little was known about its interaction with the autophagy pathway and the impact of autophagy on intracellular survival. In this study, we showed that autophagy indeed had an impact on intracellular survival of N. gonorrhoeae. Upon invasion, the autophagy pathway was activated and able to capture invading gonococci in double-membrane autophagosomes that subsequently were able to mature and fuse with lysosomes for their degradation. Based on our TEM analyses, N. gonorrhoeae appeared to reside in a vacuole while being targeted by autophagy, similar to what has been observed for a number of other bacteria, including Salmonella typhimurium [23] and Mycobacterium tuberculosis [24]. However, in contrast to these bacteria, the primary vacuole directly surrounding N. gonorrhoeae was very tight, which might explain why there are some conflicting studies about whether intracellular N. gonorrhoeae resides in vacuoles [25].
Although most of the invading gonococci initially ended up in autolysosomes for their degradation, a subpopulation of intracellular bacteria was able to escape autophagy-mediated killing. N. gonorrhoeae was subsequently able to repress the autophagy pathway, which allowed for prolonged intracellular survival and exocytosis. During this later intracellular stage, the autophagy repressor mTORC1 was activated, which resulted in suppression of autophagy signaling. Interference of canonical autophagy signaling is a common strategy for intracellular pathogens. Similar to our observation for N. gonorrhoeae, S. typhimurium is also able to inhibit autophagy at the later stage of invasion through activation of mTORC1 [26, 27]. For N. gonorrhoeae, we furthermore showed that autophagosome maturation was inhibited and that LC3, PI3P, and LAMP-1 activity and colocalization with N. gonorrhoeae were reduced. These observations might be explained by mTORC1 activation and subsequent autophagy inhibition. However, given that Torin1-mediated mTORC1 repression did not negatively impact intracellular survival and only resulted in induced autophagy activity at the early stage of cell invasion, N. gonorrhoeae likely has additional means to inhibit autophagosome maturation and lysosome fusion. For several other bacterial pathogens, mechanisms involved in avoiding or stalling of autophagosomal maturation processes have already been identified. For instance, Legionella pneumophila is able to both deconjugate LC3 and PE on autophagosomes [28] and deplete PI3P [29, 30], thereby inhibiting the autophagosome maturation processes, whereas M. tuberculosis hydrolyses PI3P through secretion of a lipid phosphatase [31]. For other bacteria, stalling of autophagosome maturation has been shown, whereas their exact mechanisms are currently still unknown [32–34]. These bacteria reside in nondegradative autophagososmes and block fusion with late endosomes or lysosomes. For invasive strains of Escherichia coli that reside in immature autophagosomes, it has been shown that activation of autophagy through repression of mTORC1 restricted intracellular growth [35], which is in contrast to our observation that forced activation of autophagy through repression of mTORC1 did not affect intracellular survival.
CONCLUSIONS
In conclusion, our study showed that N. gonorrhoeae is targeted by autophagy upon epithelial cell invasion and sequestered in autophagosomes that subsequently matured into autolysosomes for elimination. However, a subpopulation of invading gonococci is able to survive this initial stage of cell invasion and autophagy-mediated killing. This subpopulation is able to show prolonged intracellular survival and exocytosis due to inhibition of the autophagy pathway at this stage as a result of activation of the canonical autophagy pathway repressor mTORC1 and inhibition of autophagosome maturation and lysosome fusion. Overall, our results provide novel insight into intracellular N. gonorrhoeae-autophagy interactions, which has significant meaning given that transcytosis of epithelial cells is expected to be a prerequisite for N. gonorrhoeae to cause disseminated infections.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Disclaimer. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. None of the authors have a commercial or other association that might pose a conflict of interest.
Acknowledgments. We thank Professor Hongguang Xia for providing the mCherry-GFP-LC3 expression vector. We thank the staff at the Imaging Center of the Zhejiang University School of Medicine for assistance with confocal and electron microscopy.
Financial support. This work was funded by Zhejiang University (start-up funding; to S. v. d. V.); the National Natural Science Foundation (31771525 and 91754113; to Q. S.); the Ministry of Science and Technology of the People’s Republic of China (2017YFA0503402; to Q. S.); and the Zhejiang Provincial Funds for Distinguished Young Scientists (LR15C070001; to Q. S.).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}