






Abstract
Human rhinoviruses (HRVs) are the commonest cause of the common cold. While HRV is less pathogenic than other respiratory viruses, it is frequently associated with exacerbation of chronic respiratory diseases such as rhinosinusitis and asthma. Nasal epithelial cells are the first sites of viral contact, immune initiation, and airway interconnectivity, but there are limited studies on HRV infection of nasal epithelial cells. Hence, we established a model of HRV infection of in vitro–differentiated human nasal epithelial cells (hNECs) derived from multiple individuals. Through HRV infection of hNECs, we found that HRV mainly targeted ciliated cells and preferentially induced type I and III interferon antiviral pathways. Quantitative polymerase chain reaction analysis of inflammatory genes suggested predominant type 1 immunity signaling and recruitment, with secreted CXCL9, IP-10, CXCL11, and RANTES as likely initiators of airway inflammatory responses. Additionally, we further explored HRV bidirectional release from the hNECs and identified 11 associated genes. Other HRV interactions were also identified through a systematic comparison with influenza A virus infection of hNECs. Overall, this in vitro hNEC HRV infection model provides a platform for repeatable and controlled studies of different individuals, thus providing novel insights into the roles of human nasal epithelium in HRV interaction and immune initiation.
The human airway is constantly in direct contact with the external environment [1]. Hence, the airway mucosa plays significant roles in clearing foreign insults and maintaining homeostasis through physical and immune responses connecting downstream responses involving the subepithelia and lower airways [2]. Therefore, the biological networks between external stimuli from the environment, the airway epithelium, and the immune system are crucial in the pathogenesis of airway diseases [3].
Acute respiratory tract infections are frequently caused by viral infections and result in significant productivity loss and financial burden on the healthcare system worldwide [4]. Among them, human rhinovirus (HRV) infections are the major cause of upper respiratory tract infections, accounting for >50% of all cases. While most HRV infections are self-limiting, they trigger inflammatory responses via release of chemokines and cytokines into the airways, recruiting immune cells (ie, neutrophils, eosinophils, and T cells) that can increase the risk of developing allergic airway inflammation or aggravate preexisting airway inflammation. Such interactions contribute to the association of HRV with more-severe clinical complications, such as asthma and chronic obstructive pulmonary disease, leading to life-threatening conditions [5, 6].
HRV-induced immune responses have been an active area for research using in vitro cell models [7–11] and mouse models [12–15]. Previously, most studies on HRV pathogenesis and immune responses elicited by HRV-infected host systems were based on the lower respiratory airway, owing to the greater severity of exacerbations of respiratory diseases and limited focus on upper airway models. Thus, much less is known about HRV replication dynamics and immune responses in the upper respiratory mucosa, the primary site for HRV replication. Recently, some studies used nasal epithelial cells to investigate HRV infection, although they were limited to evaluation of specific immune responses or viral replication [16–18]. Therefore, considering that HRV interactions with the healthy nasal epithelium may contribute to immune cascades predisposing individuals to developing allergic reactions [19], there has yet to be a holistic study of HRV responses and pathogenesis initiated from the primary site of viral replication in its native state.
To accomplish this, we isolated and characterized adult human nasal epithelial stem/progenitor cells (hNESPCs) from nasal biopsy specimens from different individuals. These in vitro–passaged hNESPCs retain the potential to fully differentiate into a stratified mucociliary airway epithelium composed of both ciliated columnar cells and goblet cells via air-liquid interface culture, constituting an in vitro system of pure differentiated human nasal epithelial cells (hNECs) in its native state for investigating epithelial interactions with HRV [20, 21]. This in vitro hNEC model provides a reproducible platform for comprehensive elucidation of primary interactions between HRV and susceptible nasal epithelia from different individuals and of their roles in airway inflammation [22, 23]. The aim of this study is therefore to characterize HRV infection and its interactions in the nasal epithelium and to identify key interaction factors contributed by the nasal epithelium.
MATERIALS AND METHODS
Derivation of hNESPCs and In Vitro Differentiation of hNECs
Approval to conduct this study was obtained from the National Healthcare Group Domain-Specific Board of Singapore (protocol D/11/228) and institutional review board of the National University of Singapore (protocol 13–509). The hNECs were differentiated from hNESPCs according to a previously standardized protocol [23]. hNESPCs were derived from 12 healthy donors aged 22–60 years (including 1 smoker and 4 atopic subjects) from different ethnic groups who underwent septal plastic surgery in National University Hospital, Singapore. At the time of collection, all subjects were healthy, free of symptoms of upper respiratory tract infection, and had not used any glucocorticoids or antibiotics within 3 months before surgery. The medical backgrounds of the donors and samples are summarized in Supplementary Table 1. The detailed procedure of hNEC cultures are found in the Supplementary Materials.
Inoculation of HRV in Fully Differentiated hNECs and Viral Quantification
HRV-A16 strain 11757 (ATCC VR-283, Manassas, VA) was propagated in a HeLa cell line (HeLa Ohio, ECACC 84121901, Porton Down, United Kingdom).
HRV was diluted using B-ALI growth medium and inoculated at multiplicity of infection of 2.5 into the apical chamber of 12-well Transwell plates, followed by incubation for 24 hours at 33°C. B-ALI growth medium without virus was added as an uninfected control. Viral inoculum was removed after 24 h. The HRV-infected and uninfected control hNECs were then incubated at 33°C for up to 96 hours after infection. Apical secretion, basolateral medium, and hNEC lysate were collected to perform relevant assays 48, 72, and 96 hours after infection. A detailed procedure is found in the Supplementary Materials.
Cytospin Preparation and Immunofluorescence Staining
At each time point, single-cell suspensions (1 × 105–2 × 105 cells) were dissociated from the Transwell by use of a 0.5× trypsin/ethylenediaminetetraacetic acid solution (Gibco) at 37ºC. Dissociated cells were fixed in 4% formaldehyde at room temperature for 10 minutes, washed twice with 1× Dulbecco’s phosphate-buffered saline, and centrifuged. Cytospin slides (2 × 104 cells/slide) were prepared from the washed cell suspension using Thermo Shandon Cytospin 3 centrifuge (Thermo Fisher Scientific, Waltham, MA) at 500 rpm for 5 minutes and were stored at −20°C until staining. To identify the main HRV-infected cell type, costaining was performed using an antibody to detect the HRV structural protein VP2 and its precursor, VP0, with antibodies targeting cellular markers of ciliated cells (α-tubulin), goblet cells (MUC5AC), or basal cells (p63) of the nasal epithelium. The detailed immunofluorescence staining procedure is available in the Supplementary Materials.
Total RNA and Viral RNA Extraction, Reverse Transcription, and Real-Time Quantitative PCR (qPCR)
Total RNA and viral RNA were extracted using the mirVana miRNA isolation kit (Life Technologies, Grand Island, NY) and the QIAamp viral RNA mini kit (Qiagen, Hilden, Germany), respectively. Total RNA and viral RNA (1000 ng each) were subjected to complementary DNA (cDNA) synthesis, using the Maxima First-Strand cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA). The messenger RNA (mRNA) expression profiles at different stages of HRV infection were analyzed and normalized against the housekeeping gene RPL13A. The mRNA expression profiles 48, 72, and 96 hours after infection were presented as fold changes from values for the uninfected control. The detailed real-time qPCR protocol is specified in Supplementary Materials.
Luminex and FITC Annexin V Apoptotic Assays
Detailed protocols are found in the Supplementary Materials.
Statistical Analyses
All data were analyzed using GraphPad Prism 6 software (San Diego, CA). For real-time qPCR, since the distribution of the gene expression level (2-ΔΔCt) was not normal (or Gaussian), owing to variability in different individual-derived hNECs, median values with interquartile ranges were used in statistical analyses. The significance level was calculated using 1-way analysis of variance (for nonparametric, grouped data) with the Dunn multiple comparison test. Data are presented as fold changes. P values of <.05 are considered significant.
RESULTS
In Vitro–Differentiated hNECs Sustain HRV Infection via Infection of Ciliated Cells
As shown in Figure S1A, a standard curve (with a correlation coefficient of 0.99) was established via amplification of HRV transcripts in serially diluted stock. Initially, we studied host innate mRNA responses between 0 and 96 hours after infection and observed relatively mild responses during the early stage of viral infection (0, 12, and 24 hours after infection; Figure S2). Therefore, shifting our focus to later time points, we observed that viral loads in cell lysate, apical secretion, and basal medium peaked 48 hours after infection and were sustained throughout 72 and 96 hours after infection, with interindividual differences signifying differential susceptibility of nasal epithelium toward HRV (shown by large median range of approximately 1–2 logs difference; Figure 1A). Furthermore, viral progeny and mRNAs were detected in both the apical secretion and basal medium, suggesting a bidirectional viral release (Figure 1A). This bidirectional release occurred despite the epithelium remaining intact, with transepithelial electrical resistance reading remaining >1000 Ω/cm2 up to 6 days after infection [21, 24] (Figure S1B). Coimmunofluorescence staining of HRV VP proteins with 3 major types of hNEC markers revealed that ciliated cells but not goblet and basal cells were infected by HRV (Figure 1B). Interestingly, when further probed for FOXJ1, a key ciliogenesis marker, no significant reduction was observed despite infection of ciliated cells (Figure 1C). This may be attributed to the minimal apoptotic or necrotic cell death in HRV infection (Figure S3). On the other hand, the MUC5AC goblet cell marker was significantly upregulated, which is typically observed during HRV infection, thus confirming that hNECs elicit proper HRV responses in vitro [25–27] (Figure 1C).
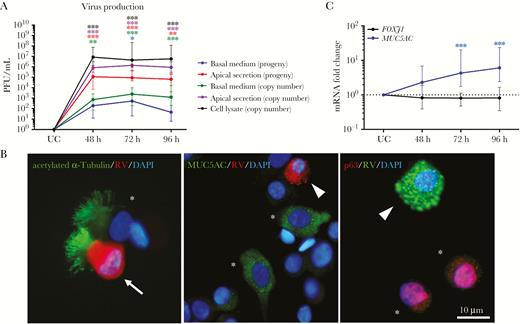
Human rhinovirus (HRV) replication in human nasal epithelial cells (hNECs). A, HRV titers were calculated by plotting a standard curve, using cycle threshold values of viral complementary DNA. Virus progeny were detected using a plaque assay. B, Representative images of costained infected α-tubulin–positive ciliated cells (white arrow) with the structural protein (VP) of HRV, showing that ciliated cells were the major cell type infected by HRV, and uninfected ciliated cells (white asterisk) 48 hours after infection (left panel); representative images of uninfected MUC5AC-positive goblet cells (white asterisk) and p63-positive basal cells (white asterisk; middle); and infected cells that were not of goblet or basal cell origin (white triangle) 48 hours after infection (right). No infected goblet and basal cells costained with HRV VP were detected. Scale bar, 10 µm. C, Intracellular messenger RNA (mRNA) expression levels for the markers FOXJ1 (ciliated cells) and MUC5AC (goblet cells). The y-axis shows the normalized fold-change in levels in infected cells as compared to those in uninfected control (UC) cells. Median and interquartile ranges are shown. The significance level was calculated using 1-way analysis of variance (for nonparametric, grouped data) and the Dunn multiple comparisons test. PFU, plaque-forming units. *P < .05, **P < .01, and ***P < .001, compared with UC cells (n = 12).
Initial HRV Detection Involves RIG-I and TLR7 Induction of Type I and III IFN Pathways
We further evaluated innate immune responses within HRV-infected epithelial cells via qPCR, the sequential and spatiotemporal reactions of viral sensors, and antiviral effectors reported in experimental models of infection with respiratory viruses [23]. We followed the sequence of HRV receptor, pathogen sensors, early effectors, cytokines, and chemokines (Figures 2–4, S4-S6). We observed an increase in ICAM-1 (Figure S4A), indicating the retention of nasal epithelial properties following in vitro differentiation [23, 24]. On the other hand, the most prominent changes in mRNA levels of pattern-recognition receptors (Figure 2A and 2B and Supplementary Figure 4B and 4C) were RIG-I and TLR7 (Figure 2A and 2B) and effector genes IFN-β, IL29/IFN-λ1, IRF7, IFN-α2, and ISG15 (Figure 2C and 2D and Supplementary Figure 4E, 4H, and 4I). Interestingly, enhanced expression of IFN mRNAs only correlated slightly with their respective secreted proteins, which were not statistically significant, suggesting low-level secretion of these proteins during HRV infection (Figure 2C and 2D and Supplementary Figure 4G and 4H). These findings indicate that the initial events of a clinical HRV infection contributed by the nasal epithelium were type I and III IFN responses.
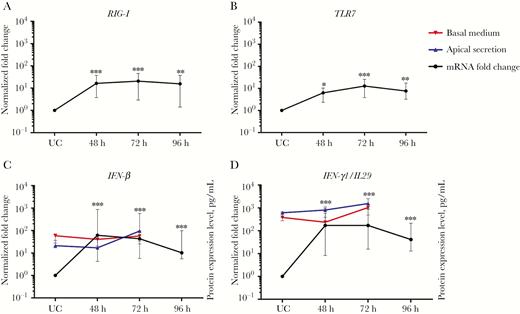
Main pathogen sensing and initiation of antiviral signaling in human nasal epithelial cells (hNECs), as measured by intracellular messenger RNA (mRNA) levels. Key pathogen sensors were analyzed to determine the main pathogen sensors that were important in human rhinovirus (HRV) infection. The main pathogen sensors and effectors were RIG-I (A), TLR7 (B), IFN-β (C), and IL29/IFN-λ1 (D; n = 12). Each plot shows the intracellular mRNA expression levels as median fold changes with interquartile ranges at each time-point, compared with uninfected control (UC) cells. The left y-axes show the normalized fold change in values for infected cells, compared with UC cells. The right y-axes of panels C and D show protein expression levels for apical (blue) and basal (red) secretion (n = 5). HRV infection preferentially induced the type I and III interferon pathways. The significance level was calculated using 1-way analysis of variance (for nonparametric, grouped data) and the Dunn multiple comparisons test. *P < .05, **P < .01, and ***P < .001, compared with UC cells.
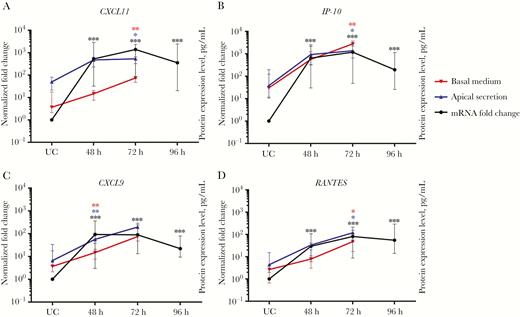
Main induction of chemokines in human nasal epithelial cells (hNECs) for the recruitment of immune cells, as measured by intracellular messenger RNA (mRNA) levels. The main chemokines observed in response to human rhinovirus (HRV) infection were CXCL11 (A), IP-10 (B), CXCL9 (C), and RANTES (D; n = 12). Each plot shows the intracellular mRNA expression levels as median fold changes with interquartile ranges at each time point as compared to values for uninfected control (UC) cells. The left y-axes show the normalized fold change in values for infected cells, compared with UC cells. The right y-axes show protein expression levels for apical (blue) and basal (red) secretion for each chemokine (n = 5). Following HRV infection, the key chemokines induced in hNECs were CXCL9, IP-10, CXCL11, and RANTES. The significance level was calculated using 1-way analysis of variance (for nonparametric, grouped data) and the Dunn multiple comparisons test. *P < .05, **P < .01, and ***P < .001, compared with UC cells.
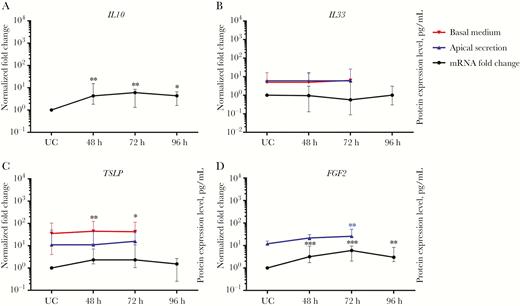
Activation of genes related to inflammatory response and airway remodeling in human nasal epithelial cells (hNECs), as measured by intracellular messenger RNA (mRNA) levels. The main cytokines and other airway remodeling genes screened during HRV infection were IL10 (A), IL33 (B), TSLP (C), and FGF2 (D; n = 12). Each plot shows the intracellular mRNA expression levels as median fold changes with interquartile range of each time point, compared with UC cells. The left y-axes show the normalized fold change in values for infected cells, compared with UC cells. The right y-axes show protein expression levels for apical (blue) and basal (red) secretion for each cytokine (n = 5). IL10 and FGF2 were strongly induced in hNECs following HRV infection. The significance level was calculated using 1-way analysis of variance (for nonparametric, grouped data) and the Dunn multiple comparisons test. *P < .05, **P < .01, and ***P < .001, compared with UC cells.
hNECs Predominantly Increase Expression of Type 1 Immunity Cytokines and Chemokines and of the Airway Remodeling Gene FGF2 in Response to HRV
Following intracellular responses, hNEC-initiated inflammatory responses were predominantly type 1 responses (T-helper type 1 biased) through relatively higher (>50-fold) expression of CXCL11, IP-10, CXCL9, and RANTES mRNAs (Figure 3A–D) and moderate (>2-fold) increase of CCL20, CX3CL1, IL27, IL6, TNFα, and IL1α mRNAs (Figure S5A-B, S6A, S6C-E). Type 2 responses (T-helper type 2 biased) thought to be expressed by airway epithelium were subdued. Only IL10 and TSLP levels were moderately increased (Figure 4A and 4C), while IL25 levels remained constant (Figure S6B) with transient increase in early infection (data not shown). IL33 expression remained constant (Figure 4B), unlike previous studies using bronchial cell cultures, a difference that should be further explored [28, 29]. Other cytokines and chemokines CCL7, CCL11, CCL22, CXCL6, IL1β, IL5, and IL8 were only increased at 1 specific time point or not at all after infection, implying their limited role in the HRV-infected nasal epithelium (Figure S5C-G; S6F-G). Interestingly, moderate FGF2 expression was noted following HRV infection, indicating possible nasal epithelium-initiated airway remodeling (Figure 4D). Secretory protein analysis of selected genes via Luminex further revealed significantly enhanced secretion of RANTES, CXCL9, IP-10, CXCL11, and FGF2 (Figure 3A–D and Figure 4D). These 5 proteins secreted by hNECs were likely crucial initial response factors of HRV-induced pathogenesis, inflammation, and remodeling, culminating in HRV-associated complications.
Analysis of Correlation of Immune Response Genes with HRV Bidirectional Release
Upon elucidating HRV kinetics and hNEC-initiated responses, a distinct feature of HRV infection was the bidirectional release of viral progeny, which may account for the HRV-specific immune responses possibly due to early viral exposure to immune cells in the subepithelial mucosa. Using linear mixed-effect regression analysis, we analyzed the immune response genes that correlated with HRV release across the epithelium into the basal compartment. Of the 38 genes tested, 17 differentially regulated genes were significantly correlated with the apical and total HRV titers; whereas 12 differentially regulated genes were significantly correlated with HRV in basal medium (ie, CX3CL1, CXCL9, CCL22, RIG-I, TLR7, IFN-β, CXCL11, ISG15, IL10, ICAM-1, FGF2, and IRF7; Supplementary Table 2). Among them, FGF2 and IRF7 were solely correlated with the basal HRV titer. The augmented expression of these 12 genes may contribute to the eventual bidirectional release during HRV infection across the multilayered nasal epithelium.
Comparison of HRV and Influenza A Virus (IAV) Infection in hNECs for Identifying Factors Influencing HRV Pathogenesis
To further elucidate the specific features of HRV pathogenesis in hNECs, we compared HRV infection kinetics and responses with our previous IAV infection model of hNECs similarly differentiated from hNESPCs [23] (Table 1). Notable similarities were that both infections caused viral-receptor upregulation, with the hNECs relying mainly on RIG-I and TLR7 pathogen sensors for respiratory viral detection. In terms of differences, the HRV load increased later but was sustained (after peaking 72 hours after infection) for up to 14 days after infection, with minimal cell death (data not shown), whereas the IAV load increased faster and decreased as early as 72 hours after infection, owing to extensive cell death. While innate immune responses were generally similar, HRV induced a more moderate response at both the mRNA and secreted-protein levels. HRV specifically infected ciliated cells of hNECs and induced MUC5AC expression. A noteworthy feature of HRV infection (not seen in the more virulent IAV infection) is that HRV progeny were detected in the basal medium, despite both viruses not infecting basal cells. These different hNEC interactions between HRV and IAV may contribute to a better understanding of the frequent associations of HRV with airway complications such as rhinosinusitis and asthma.
Characteristics of Human Rhinovirus (HRV) and Influenza Virus Infection in In Vitro–Differentiated Human Nasal Epithelial Cells (hNECs)
| Characteristic | HRV | Influenza Virusa |
|---|---|---|
| MOI needed | 2.5 | 0.1 |
| Viral kinetics | Late increase (48 h after infection), sustained up to 96 h | Increases as early as 16 h after infection and decreases by 72 h, with almost all cells dead by 96 h |
| Viral presence in basal medium | Yes | No |
| Virus receptor upregulation | Yes, ICAM-1 | Yes, α-2,6 sialic acid receptor |
| Magnitude of immune response expression | Moderate (range, 10–1000-fold) | Strong (range, 10–10000-fold) |
| Key pathogen sensors | RIG-I, TLR7 | RIG-I, TLR7 |
| Key immune response, fold upregulationb | ||
| CXCL11 | 1397 | 14735c |
| IP-10 | 1180 | 11000c |
| IL29 | 170d | 119 |
| RANTES | 80.6 | 140e |
| IFN-b1 | 61.4 | 188e |
| ISG15 | 59.5 | 715c |
| RIG-I | 20.7 | 38.5e |
| IRF7 | 14.1 | 86.8e |
| TLR7 | 12.7 | …f |
| CXCL9 | 88.8 | …f |
| IL6 | …f | 35.2 |
| IL1a | …f | 28.1 |
| Apoptosis and cell death | Minimal | Mostly present, strong |
| Main infected cell type(s) | Ciliated cells | Ciliated and goblet cells |
| MUC5AC expression | Increased MUC5AC transcripts | No induction of MUC5AC transcripts |
| Characteristic | HRV | Influenza Virusa |
|---|---|---|
| MOI needed | 2.5 | 0.1 |
| Viral kinetics | Late increase (48 h after infection), sustained up to 96 h | Increases as early as 16 h after infection and decreases by 72 h, with almost all cells dead by 96 h |
| Viral presence in basal medium | Yes | No |
| Virus receptor upregulation | Yes, ICAM-1 | Yes, α-2,6 sialic acid receptor |
| Magnitude of immune response expression | Moderate (range, 10–1000-fold) | Strong (range, 10–10000-fold) |
| Key pathogen sensors | RIG-I, TLR7 | RIG-I, TLR7 |
| Key immune response, fold upregulationb | ||
| CXCL11 | 1397 | 14735c |
| IP-10 | 1180 | 11000c |
| IL29 | 170d | 119 |
| RANTES | 80.6 | 140e |
| IFN-b1 | 61.4 | 188e |
| ISG15 | 59.5 | 715c |
| RIG-I | 20.7 | 38.5e |
| IRF7 | 14.1 | 86.8e |
| TLR7 | 12.7 | …f |
| CXCL9 | 88.8 | …f |
| IL6 | …f | 35.2 |
| IL1a | …f | 28.1 |
| Apoptosis and cell death | Minimal | Mostly present, strong |
| Main infected cell type(s) | Ciliated cells | Ciliated and goblet cells |
| MUC5AC expression | Increased MUC5AC transcripts | No induction of MUC5AC transcripts |
Abbreviation: MOI, multiplicity of infection.
aData are from [23].
bData are for the 10 genes with the greatest upregulation.
cMore than 10 times the increase of HRV-infected hNECs.
dLess than 10 times the increase of influenza virus–infected hNECs.
eLess than 10 times the increase of HRV-infected hNECs.
fData not included (not part of 10 genes with greatest upregulation).
Characteristics of Human Rhinovirus (HRV) and Influenza Virus Infection in In Vitro–Differentiated Human Nasal Epithelial Cells (hNECs)
| Characteristic | HRV | Influenza Virusa |
|---|---|---|
| MOI needed | 2.5 | 0.1 |
| Viral kinetics | Late increase (48 h after infection), sustained up to 96 h | Increases as early as 16 h after infection and decreases by 72 h, with almost all cells dead by 96 h |
| Viral presence in basal medium | Yes | No |
| Virus receptor upregulation | Yes, ICAM-1 | Yes, α-2,6 sialic acid receptor |
| Magnitude of immune response expression | Moderate (range, 10–1000-fold) | Strong (range, 10–10000-fold) |
| Key pathogen sensors | RIG-I, TLR7 | RIG-I, TLR7 |
| Key immune response, fold upregulationb | ||
| CXCL11 | 1397 | 14735c |
| IP-10 | 1180 | 11000c |
| IL29 | 170d | 119 |
| RANTES | 80.6 | 140e |
| IFN-b1 | 61.4 | 188e |
| ISG15 | 59.5 | 715c |
| RIG-I | 20.7 | 38.5e |
| IRF7 | 14.1 | 86.8e |
| TLR7 | 12.7 | …f |
| CXCL9 | 88.8 | …f |
| IL6 | …f | 35.2 |
| IL1a | …f | 28.1 |
| Apoptosis and cell death | Minimal | Mostly present, strong |
| Main infected cell type(s) | Ciliated cells | Ciliated and goblet cells |
| MUC5AC expression | Increased MUC5AC transcripts | No induction of MUC5AC transcripts |
| Characteristic | HRV | Influenza Virusa |
|---|---|---|
| MOI needed | 2.5 | 0.1 |
| Viral kinetics | Late increase (48 h after infection), sustained up to 96 h | Increases as early as 16 h after infection and decreases by 72 h, with almost all cells dead by 96 h |
| Viral presence in basal medium | Yes | No |
| Virus receptor upregulation | Yes, ICAM-1 | Yes, α-2,6 sialic acid receptor |
| Magnitude of immune response expression | Moderate (range, 10–1000-fold) | Strong (range, 10–10000-fold) |
| Key pathogen sensors | RIG-I, TLR7 | RIG-I, TLR7 |
| Key immune response, fold upregulationb | ||
| CXCL11 | 1397 | 14735c |
| IP-10 | 1180 | 11000c |
| IL29 | 170d | 119 |
| RANTES | 80.6 | 140e |
| IFN-b1 | 61.4 | 188e |
| ISG15 | 59.5 | 715c |
| RIG-I | 20.7 | 38.5e |
| IRF7 | 14.1 | 86.8e |
| TLR7 | 12.7 | …f |
| CXCL9 | 88.8 | …f |
| IL6 | …f | 35.2 |
| IL1a | …f | 28.1 |
| Apoptosis and cell death | Minimal | Mostly present, strong |
| Main infected cell type(s) | Ciliated cells | Ciliated and goblet cells |
| MUC5AC expression | Increased MUC5AC transcripts | No induction of MUC5AC transcripts |
Abbreviation: MOI, multiplicity of infection.
aData are from [23].
bData are for the 10 genes with the greatest upregulation.
cMore than 10 times the increase of HRV-infected hNECs.
dLess than 10 times the increase of influenza virus–infected hNECs.
eLess than 10 times the increase of HRV-infected hNECs.
fData not included (not part of 10 genes with greatest upregulation).
DISCUSSION
Using an in vitro–differentiated hNEC model, our study is the first to highlight key features of HRV infection in healthy, authentic nasal epithelium by air-liquid interface culture and their responses via holistic analyses of infection in their immunologically native state. The model expanded on recent studies [16–18] to establish pure nasal epithelium for HRV infection, where we focused on immune responses triggered by interactions between the nasal epithelial barrier and HRV. The study was intended to serve as a normalized baseline for HRV infection in the native nasal epithelial barrier, to be compared with other disease states with respect to the alterations in gene expression in the nasal epithelium. We used a common HRV strain (HRV16) belonging to the HRV-A serotype commonly used in human studies and bearing similarities to the HRV-C serotype in immune responses, to exemplify representative responses against common HRV infections [30–33]. Additionally, this model also offers a platform to reiterate variations of pure epithelial innate responses against HRV across different individuals, given that certain innate immune deficiency states can contribute to differential susceptibility to HRV [34]. This model also facilitates experimental simulations in a controlled and reproducible manner via fresh differentiation from frozen hNESPCs, thus obviating repeated collection of the primary tissues. While many studies concentrated on infections of the lower airway, owing to more-severe disease manifestations, studying the primary infection site at the nasal epithelium is equally important because the initial responses against invading pathogens can subsequently influence predisposition to lower airway complications, owing to their interconnectivity.
We hypothesize that the nasal epithelium alone provides an ideal environment for highly efficient HRV replication and for inducing significant innate responses even before complications manifest in the lower airway. This is attributed to the nasal epithelium expressing greater levels of HRV receptor (ICAM-1) as compared to the other airway compartments, rendering it the primary HRV infection site [35]. Following infection, we noted an increased HRV titer as early as 12 hours after infection, while host responses exhibited clear increase starting 24 hours after infection and persisted up to 96 hours after infection. We therefore focused on the viral kinetics, tropism, and hNEC responses during the intermediate-to-late stages of HRV infection (48–96 hours after infection). We found HRV titers in both apical secretion and basal medium, implying that infectious virus progeny release is bidirectional, with a greater proportion being released outward toward the nasal airway lumen, rather than downward toward the subepithelial mucosa. This finding, made possible by our model, documents the bidirectional release of HRV progeny through an intact epithelium similar to other viruses, such as hantavirus [36]. This is unlike the apical-only release of other respiratory viruses, such as respiratory syncytial virus (RSV) [37] and IAV [23, 36], in differentiated multilayer airway epithelial cell cultures. This finding further confirmed that HRV bidirectional release at the nasal epithelium involved infectious progeny, instead of only HRV RNA presence reported in a previous study [38]. The bidirectional release of HRV into the subepithelial mucosa may contribute to its distinct pathogenesis, in which inflammatory responses occur without epithelial barrier destruction, unlike other respiratory viruses, such as IAV and RSV [35, 39]. The bidirectional release suggests that HRV may interact directly with the immune cells residing within the subepithelial mucosa, giving rise to HRV-specific inflammatory responses.
Another active area of HRV research is its cell type and receptor tropism [40] and the susceptibility of human airway epithelial cells to HRV [40–43]. Previous studies on the airway epithelial cell types targeted by HRV yielded inconsistent results. Using primary human bronchial epithelial cells, Jakiela et al concluded that HRV almost exclusively targets ciliated cells [44]. On the other hand, Lachowicz-Scroggins et al claimed that HRV preferentially infects goblet cells, using a model of differentiated primary tracheal cells [7]. Using double-immunofluorescence staining, we observed that HRV almost exclusively infected ciliated cells but not goblet and basal cells at their primary, nasal infection site (Figure 1C–E). Basal cells were not infected despite signs of viral bidirectional release, implying an inherent characteristic of the basal cells that prevented infection. Taken together, HRV infection of hNECs induced preferential loss of ciliated cells.
In addition to their physical protective roles, hNECs were found to not only combat the infection intracellularly, but also to contribute to subsequent innate and even adaptive responses through release of cytokines and chemokines. In view of airway connectivity, the initial responses at the nasal epithelium may initiate an immune cascade affecting the entire airway, leading us to explore factors that trigger downstream antiviral signaling [45–47]. We demonstrated that hNECs use TLR7 and RIG-I as major viral sensors to stimulate downstream antiviral mechanisms via type I and III IFNs. Subsequently, expression of cytokines and chemokines was dominated by type 1 immune responses, where CXCL9, CXCL11, IP-10, and RANTES were further secreted, promoting type 1 immunity in the airway. A noteworthy finding was the secretion of IP-10 by the nasal epithelium, which is proposed as a serum biomarker for asthma exacerbation [44, 48]. These observations suggest that the nasal epithelium plays roles in the initiation of these responses, which vary across individuals at the nasal level [22, 23, 45]. Type 2 immunity genes were also found to be moderately expressed, albeit less than that of type 1 immunity. This implies that under specific conditions, the nasal epithelium may play a role in initiating type 2 immunity, predisposing an individual to inflammatory conditions. Another interesting factor found to be expressed and secreted by the nasal epithelium is FGF2, implying that HRV infection induces nasal epithelial remodeling and may explain the bidirectional viral release. Furthermore, the finding of differential HRV responses (differences of up to 2 logs from the median) from nasal epithelia of multiple individuals was important because it revealed that the differential responses may in part be traced back to the nasal epithelium, likely contributing to differential HRV susceptibility and predisposition to its complications.
In addition to the study of direct HRV effects on the nasal epithelium, an indirect way of elucidating the differential disease progression of HRV is by comparison with other respiratory viruses. When compared with the generally more severe IAV, the key differences were the milder responses against HRV, the absence or low level of apoptosis, and the presence of HRV bidirectional release. HRV-infected hNECs displayed minimal cytopathic effect, but viral load was found to persist for up to 14 days after infection (data not shown), in contrast to the extensive cell death caused by IAV infection [23]. HRV is well known to cause mild cellular damage during infection, which explains the minimal apoptosis. Instead, HRV may undergo cell-to-cell spread via virus-carrying microvesicles, as suggested in a recent review [46]. Hence, the relatively mild responses and minimal apoptosis, which are less detrimental to the cells, coupled with bidirectional viral release may be differentiating factors of HRV disease progression as compared to other respiratory viruses. In addition to the milder overall responses, the extent of activation of type 1 immune genes in the nasal epithelium may also contribute to the differential pathology between HRV and IAV infections. Thus, HRV infection is milder but causes inflammatory complications due to moderate type 1 response activation, whereas IAV infection is relatively more damaging because of stronger type 1 activation promoting cell death. Additionally, type 1 cytokine levels may also determine the degree of suppression of type 2 cytokines, since the balance of type 1 and 2 cytokines contributes to differences in disease progression and complications between HRV and IAV infections [33, 47]. Our findings revealed that the balance between type 1 and 2 cytokines following HRV infection was initiated from the nasal epithelial level at its native state to be preferential for moderate type 1 responses. This observation further suggests that complications such as rhinosinusitis and asthma exacerbation may arise from the weaker type 1 response of the nasal epithelium. This alteration in expression pattern in the nasal epithelium may affect downstream immune cells, culminating in chronic complications over time. In particular, repeated infections may further modify antiviral responses and tilt the balance further to type 2 responses [49].
In conclusion, by systematically analyzing HRV infection in hNECs, we elucidated potential roles of the nasal epithelium and have identified factors significant in initiating the inflammatory cascade in the airway. This model also provides a viable platform for the reproducible study of these nasal epithelial factors in greater detail under airway inflammatory conditions in different individuals without necessitating repeated sample collection. This may be especially important given that the host immune responses vary across individuals and may represent the determinants of the disease burden and pathology more than viral load in different patients, as evidenced by a recent study [50].
Notes
Acknowledgments. We thank the surgeons and staff in the Department of Otolaryngology, National University Hospital, Singapore; and M. C. Phoon and S. H. Lau, for technical assistance.
Financial support. This work was supported by the National Medical Research Council, Singapore (grants NMRC/CIRG/1362/2013 and NMRC/CIRG/1458/2016).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Presented in part: Nature Conference on Viral Infection and Immune Response, Wuhan, China, 21–24 October 2016; European Academy of Allergy and Clinical Immunology Conference, Helsinki, Finland, 17–21 June 2017; International Union of Microbiological Societies Congress, Singapore, 17–21 July 2017.
References
Author notes
K. S. T., H. H. O., Y. Y., and J. L. contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}