






Abstract
Background. Three full doses of RTS,S/AS01 malaria vaccine provides partial protection against controlled human malaria parasite infection (CHMI) and natural exposure. Immunization regimens, including a delayed fractional third dose, were assessed for potential increased protection against malaria and immunologic responses.
Methods. In a phase 2a, controlled, open-label, study of healthy malaria-naive adults, 16 subjects vaccinated with a 0-, 1-, and 2-month full-dose regimen (012M) and 30 subjects who received a 0-, 1-, and 7-month regimen, including a fractional third dose (Fx017M), underwent CHMI 3 weeks after the last dose. Plasmablast heavy and light chain immunoglobulin messenger RNA sequencing and antibody avidity were evaluated. Protection against repeat CHMI was evaluated after 8 months.
Results. A total of 26 of 30 subjects in the Fx017M group (vaccine efficacy [VE], 86.7% [95% confidence interval [CI], 66.8%–94.6%]; P < .0001) and 10 of 16 in the 012M group (VE, 62.5% [95% CI, 29.4%–80.1%]; P = .0009) were protected against infection, and protection differed between schedules (P = .040, by the log rank test). The fractional dose boosting increased antibody somatic hypermutation and avidity and sustained high protection upon rechallenge.
Discussions. A delayed third fractional vaccine dose improved immunogenicity and protection against infection. Optimization of the RTS,S/AS01 immunization regimen may lead to improved approaches against malaria.
Clinical Trials Registration. NCT01857869.
Early in the new millennium, malaria continues to impart a devastating global morbidity and mortality, with >214 million cases and 438 000 deaths estimated in 2015 [1]. During the past decade, renewed international commitment and increased funding to scale-up malaria control interventions have resulted in morbidity and mortality reductions in several African countries [2], but parasite drug and mosquito insecticide resistance threatens the sustainability of present interventions [3]. The development of a protective malaria vaccine has been identified as a key component of a sustainable malaria control program and an important tool for the malaria elimination technical roadmap [4].
RTS,S/AS01 is a preerythrocytic Plasmodium falciparum malaria candidate vaccine. The vaccine is a self-assembling virus-like particle vaccine containing a fusion protein (RTS) of the NANP repeat and C-terminal portions (R and T, respectively) of the NF54 strain of P. falciparum circumsporozoite protein (CS) and the hepatitis B virus surface antigen (HBsAg; the S portion), together with free HBsAg (S), adjuvanted with AS01 (a liposome formulation with monophosphoryl lipid A [MPL] and QS-21 [Quillaja saponaria Molina, fraction 21; licensed by GlaxoSmithKline [GSK] from Antigenics, a wholly owned subsidiary of Agenus] immunostimulants). Phase 3 evaluation of RTS,S/AS01 showed a vaccine efficacy (VE) against malaria of 55.1% (95% confidence interval [CI], 50.5%–59.3%) over 12 months after vaccination when delivered according to a 0-, 1-, and 2-month schedule in children aged 5–17 months at first vaccination [5]. In July 2015, the European Medicines Agency's Committee for Medicinal Products for Human Use adopted a positive scientific opinion on the risk-benefit balance of RTS,S/AS01 under an Article 58 regulatory procedure [6].
Efforts toward development of vaccine strategies providing higher protection are ongoing. The prominent role of humoral responses in RTS,S/AS01-induced protection was highlighted in a recent study, where the replacement of the first dose of RTS,S/AS01 by a CS-expressing recombinant adenovirus resulted in an increased CS-specific T-cell response but lower antibody titers and led to a lower point estimate of VE against controlled human malaria parasite infection (CHMI) [7]. Here, based on a previous observation, we hypothesized that a change in immunization regimen might lead to improved humoral immunogenicity. In the first RTS,S CHMI trial, which evaluated 3 different adjuvant formulations, dose 3 was reduced in 2 of the study groups, following reactogenicity concerns after dose 2. Six of 7 subjects who received RTS,S/AS02 (a formulation closely related to RTS,S/AS01) on a 0-, 1-, and 7-month schedule, with a one fifth fractional (0.1 mL) third dose, were protected against CHMI (VE, 86% [95% CI, .02–.88]; P < .005) [8]. The initial reactogenicity concern was alleviated in subsequent studies with 3 identical vaccine doses, which showed lower estimates of efficacy [9]. It was interpreted that the initially high point estimate of VE was most likely a chance finding in the context of a small sample size, and the fractional third dose schedule was not reevaluated until the current study. We explored the possibility for a RTS,S/AS01 immunization regimen with a delayed fractional third dose to improve immunogenicity and protection against malaria parasite infection.
METHODS
Study Design
This was a phase 2A, open-label, controlled, single-center, single-country (United States) study.
The protocol was approved by the Walter Reed Army Institute of Research (WRAIR) Institutional Review Board and the Western Institutional Review Board. The trial was undertaken in accordance with International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines and good clinical practice. Written informed consent was obtained from each subject before study procedures were initiated (Clinical Trials.gov identifier: NCT01857869).
Study Subjects
Eligibility criteria included males and nonpregnant females aged 18–50 years who were free of any serious acute or chronic illness, as determined by clinical or physical examination, medical history records, or laboratory screening tests of hematologic, renal, and hepatic function; did not have a history of malaria; were seronegative for HBsAg, hepatitis C virus, and human immunodeficiency virus (HIV); and had the ability to comply with the study protocol (Supplementary Materials).
Study Vaccines and Vaccination
RTS,S/AS01 is manufactured by GSK (Rixensart, Belgium). Each 0.5-mL RTS,S/AS01 dose tested in this study contained 50 µg of RTS,S and AS01B, an adjuvant system containing 50 µg of MPL and 50 µg of QS-21 [9]. Vaccine was administered intramuscularly in the deltoid muscle of the nondominant arm.
Randomization
The target enrollment was 65 volunteers with consecutive (nonrandomized and open) allocation to study groups: the first 34 volunteers were to be immunized according to a 0-, 1-, and 7-month schedule with a fractional third dose (Fx017M), the subsequent 17 volunteers were to be immunized with 3 full doses according to a 0-, 1-, and 2-month schedule (012M), and 14 subjects were to be allocated to the infectivity control group (IC).
In a study follow-up phase for investigation of fractional dose boosting and protection against a second CHMI 8 months after the first one, consenting subjects were allocated to study groups, depending on their protection status against first CHMI. Subjects initially not protected (NP) were not randomized but were allocated to receive a fractional boost in either the Fx017M booster (Fx017M NP-Bo) or 012M booster (012M NP-Bo) subgroup, depending on their primary vaccination schedule. Protected (P) subjects from the Fx017M and 012M groups in the first CHMI were randomized to receive a booster (Bo) or no booster (No-Bo), respectively, leading to 4 additional subgroups (Fx017M P-No-Bo, Fx017M P-Bo, 012M P-No-Bo, an 012M P-Bo). The second CHMI also included 6 newly enrolled infectivity controls.
Randomization was generated at GSK, using Material Excellence, a program developed for use in Statistical Analysis System (SAS Institute; Cary, North Carolina) by GSK. Allocation of the subject to a study group at the investigator site was managed using a randomization system on the Internet (SBIR).
Efficacy Assessments
First and second CHMIs were conducted as previously described [8, 10], through the bite of 5 P. falciparum (3D7, a clone of the NF54 strain)–infected Anopheles stephensi mosquitoes. The first CHMI occurred 3 weeks after dose 3, and the second CHMI occurred 8 months after the first one. Parasitemia level was monitored by daily blood slide reading and polymerase chain reaction (PCR), as previously described [11], from days 5 through 19 after CHMI and then every 2 days thereafter, up to 28 days after CHMI. Malaria parasite–infected volunteers were treated with chloroquine phosphate or other Food and Drug Administration–approved antimalarials if clinically indicated.
Safety Assessments
Local injection site and general solicited adverse events (AEs) were monitored over 7 days after each vaccination (days 0–6) and were graded as mild, moderate, or severe (grades 1, 2, or 3, respectively). All other AEs (unsolicited) were recorded over a 30-day period after each vaccination. Serious AEs (SAEs) were captured by passive case detection throughout the study. All injection site AEs were considered causally related to vaccination; the causality of all other AEs was assessed by the investigator. Hematological and biochemical tests for safety assessment were conducted at various time points during the primary and booster phases. See the Supplementary Materials for additional details on safety assessments.
Immunogenicity Assessments
Immunology assessments are described in detail in the Supplementary Materials. Briefly, antibody levels against CS repeat region were measured by standard enzyme-linked immunosorbent assays (ELISAs), using plate-adsorbed R32LR antigen [NVDP(NANP)15]2LR as previously described [12]. Antibodies against the full-length recombinant CS protein and the C-terminal pf16 peptide were measured by ELISA, as further described in the Supplementary Materials. By using 4M urea as a chaotropic reagent, ELISA-based avidity assays were conducted to assess antibody binding to the full-length and C-term CS antigens and to an (NANP)6 repeat antigen. The results are reported as an avidity index (determined as the ratio of the concentration with to the concentration without the chaotropic agent). Antibodies against HBsAg were measured by a chemiluminometric immunoassay (Siemens Centaur XP CLIA).
To assess antibody maturation, the variable regions of heavy and light chain messenger RNA from peripheral blood mononuclear cell (PBMC)–sorted plasmablasts sampled 7 days after the third immunization were sequenced. For each sequenced B cell, the number of somatic mutations was determined by aligning the heavy and light chain sequences to predicted germ line sequences and counting the number of nucleotides in the alignment that differing from that of the best-matching germ line. V(D)J assignment, mutation identification, and plasmablast isolation were performed as previously described, with minor modifications [13, 14].
Cell-mediated immunogenicity was assessed using standard intracellular cytokine staining of antigen-stimulated PBMCs fluorescently labeled with CD40L, interleukin 2, tumor necrosis factor α, and interferon γ monoclonal antibodies.
Statistical Analysis
All analyses were conducted according to a predefined analysis plan. Statistical analyses were conducted using SAS, version 8, and expanded details are available in the Supplementary Materials. All safety analyses were performed on the intent-to-treat (ITT) population. Analyses for efficacy and immunogenicity were performed on the according-to-protocol (ATP) population. The primary end points of the study were the occurrence of P. falciparum parasitemia, defined by a positive blood slide, comparing each schedule group to infectivity controls. VE was defined as 100*[1 − relative risk of P. falciparum parasitemia after sporozoite challenge]. The Fisher exact test was used to compare the proportion of subjects infected after CHMI, and Kaplan–Meier analysis and log-rank statistics were used to compare the time to onset of P. falciparum parasitemia (the prepatent period was defined as the interval between challenge and detection of parasitemia). All statistical tests were 2-tailed at 5% significance level.
RESULTS
A total of 63 subjects were enrolled and included in the ITT population (34 in the Fx017M group, 17 in the 012M group, and 12 in the IC group). Figure 1 summarizes subject participation during the course of the study. In total, 58 subjects underwent a first CHMI (ATP population, 30 in the Fx017M group, 16 in the 012M group, and 12 IC group), and 37 subjects underwent a second CHMI (ATP population, 7 in the Fx017M P-NoBo group, 10 in the Fx017M P-Bo group, 2 in the Fx017M NP-Bo group, 5 in the 012M P-No-Bo group, 4 in the 012M P-Bo group, 3 in the 012M NP-Bo group, and 6 in the IC group).

Consolidated Standard of Reporting Trials flow diagram. The total cohort includes all subjects who were enrolled in the study, and the intent-to-treat (ITT) challenge group includes all subjects with documented receipt of at least 1 vaccine dose. The according-to-protocol (ATP) challenge group denotes the population analyzed for efficacy and immunogenicity and comprises all subjects included in the ITT population who received all vaccinations according to protocol procedures within the protocol specified intervals, met all eligibility criteria, did not use any medication or blood products forbidden by the protocol, did not report any underlying medical condition influencing immune responses, and underwent Plasmodium falciparum challenge. The ITT rechallenge group includes subjects consenting to the rechallenge phase. The ATP rechallenge group denotes the population analyzed for efficacy and immunogenicity in the rechallenge phase and comprises all subjects included in the ATP population for efficacy and immunogenicity who consented to rechallenge. Abbreviations: Con med, concomitant medication; control, infectivity challenge/rechallenge controls; FU, follow-up; Fx017M, delayed fractional dose, comprising a 0.5-mL dose of RTS,S/AS01B followed 1 month later by a 0.5-mL dose of RTS,S/AS01B, followed 6 months later by a 0.1-mL dose of RTS,S/AS01B; 012M, 0- , 1-, and 2-month standard schedule comprising 3 doses of 0.5 mL of RTS,S/AS01B given 1 month apart; NP-Bo, volunteers who were not protected following the primary phase and received a booster dose prior to rechallenge; P-Bo, volunteers who were protected following the primary phase and received a booster dose prior to rechallenge; P-No-Bo, volunteers who were protected following the primary phase and did not receive a booster dose prior to rechallenge.
At screening, the mean age of subjects was 33.6 years. The ratio of females to males was 15:19, 6:11, and 7:5 in the Fx017M, 012M, and IC groups, respectively.
As presented in Table 1, while all volunteers in the IC group developed parasitemia after CHMI, 26 of 30 subjects in the Fx017M group (VE, 86.7% [95% CI, 66.8%–94.6%]; P < .0001, by the Fisher exact test) and 10 of 16 subjects in the 012M group (VE, 62.5% [95% CI, 29.4%–80.1%]; P = .0009) were protected. Protection in the Fx017M group represented an increase in VE over that for the 012M group, assessed as a reduction in the proportion of unprotected subjects, of 64.4% (95% CI, –7.9%–88.3%; P = .0741, by the Fisher exact test). The time to parasitemia is shown in a Kaplan–Meier survival plot (Figure 2), with evidence for a difference in survival time between the Fx017M and 012M groups (P = .040, by the log-rank test). The mean prepatent period from challenge to infection in the Fx017M, 012M, and IC groups was 17, 15, and 13 days, respectively. One subject from the Fx017M group had parasitemia on day 26 after CHMI only, a very long, unusual delay. PCR testing for infection provided earlier evidence of infection but produced similar VE estimates (Supplementary Figure 1).
Occurrence of Parasitemia and Vaccine Efficacy (VE) Against First and Second Controlled Human Malaria Parasite Infection (CHMI) in the According-to-Protocol Population
| Variable | Protected/Challenged, No. | VE, % (95% CI) | P Valuea | P Valueb |
|---|---|---|---|---|
| First CHMI results, by primary immunization groupc | ||||
| Fx017M schedule | 26/30 | 86.7 (66.8–94.6) | < .0001 | <.0001 |
| 012M schedule | 10/16 | 62.5 (29.4–80.1) | .0009 | .0002 |
| Infectivity controls | 0/12 | … | ||
| Second CHMI results, by primary immunization groupd | ||||
| Fx017M schedule | ||||
| Not protected, fractional boost | 2/2 | 100 (−127–100) | .0357 | .0257 |
| Protected, fractional boost | 9/10 | 90 (36–98) | .0009 | <.0001 |
| Protected, no fractional boost | 3/7 | 43 (−9–70) | .1923 | .0084 |
| 012M schedule | ||||
| Not protected, fractional boost | 2/3 | 67 (−65–93) | .0833 | .0369 |
| Protected, fractional boost | 1/4 | 25 (−32–57) | .4000 | .0420 |
| Protected, no fractional boost | 1/5 | 20 (−24–48) | .4545 | .0168 |
| Infectivity controls | 0/6 | … | … | … |
| Variable | Protected/Challenged, No. | VE, % (95% CI) | P Valuea | P Valueb |
|---|---|---|---|---|
| First CHMI results, by primary immunization groupc | ||||
| Fx017M schedule | 26/30 | 86.7 (66.8–94.6) | < .0001 | <.0001 |
| 012M schedule | 10/16 | 62.5 (29.4–80.1) | .0009 | .0002 |
| Infectivity controls | 0/12 | … | ||
| Second CHMI results, by primary immunization groupd | ||||
| Fx017M schedule | ||||
| Not protected, fractional boost | 2/2 | 100 (−127–100) | .0357 | .0257 |
| Protected, fractional boost | 9/10 | 90 (36–98) | .0009 | <.0001 |
| Protected, no fractional boost | 3/7 | 43 (−9–70) | .1923 | .0084 |
| 012M schedule | ||||
| Not protected, fractional boost | 2/3 | 67 (−65–93) | .0833 | .0369 |
| Protected, fractional boost | 1/4 | 25 (−32–57) | .4000 | .0420 |
| Protected, no fractional boost | 1/5 | 20 (−24–48) | .4545 | .0168 |
| Infectivity controls | 0/6 | … | … | … |
Abbreviations: CI, confidence interval; Fx017M, delayed fractional dose, comprising a 0.5-mL dose of RTS,S/AS01B followed 1 month later by a 0.5-mL dose of RTS,S/AS01B, followed 6 months later by a 0.1-mL dose of RTS,S/AS01B; 012M, 0- , 1-, and 2-month standard schedule comprising 3 doses of 0.5 mL of RTS,S/AS01B given 1 month apart.
a By the Fisher exact test, compared with infectivity controls.
b By the log-rank test, compared with infectivity controls.
c Fx017M vs 012M, P = .074, by the Fisher exact test; P = .040, by the log-rank test.
d “Protected” denotes subjects who were protected after the first CHMI. “Not protected” denotes subjects who were not protected after the first CHMI.
Occurrence of Parasitemia and Vaccine Efficacy (VE) Against First and Second Controlled Human Malaria Parasite Infection (CHMI) in the According-to-Protocol Population
| Variable | Protected/Challenged, No. | VE, % (95% CI) | P Valuea | P Valueb |
|---|---|---|---|---|
| First CHMI results, by primary immunization groupc | ||||
| Fx017M schedule | 26/30 | 86.7 (66.8–94.6) | < .0001 | <.0001 |
| 012M schedule | 10/16 | 62.5 (29.4–80.1) | .0009 | .0002 |
| Infectivity controls | 0/12 | … | ||
| Second CHMI results, by primary immunization groupd | ||||
| Fx017M schedule | ||||
| Not protected, fractional boost | 2/2 | 100 (−127–100) | .0357 | .0257 |
| Protected, fractional boost | 9/10 | 90 (36–98) | .0009 | <.0001 |
| Protected, no fractional boost | 3/7 | 43 (−9–70) | .1923 | .0084 |
| 012M schedule | ||||
| Not protected, fractional boost | 2/3 | 67 (−65–93) | .0833 | .0369 |
| Protected, fractional boost | 1/4 | 25 (−32–57) | .4000 | .0420 |
| Protected, no fractional boost | 1/5 | 20 (−24–48) | .4545 | .0168 |
| Infectivity controls | 0/6 | … | … | … |
| Variable | Protected/Challenged, No. | VE, % (95% CI) | P Valuea | P Valueb |
|---|---|---|---|---|
| First CHMI results, by primary immunization groupc | ||||
| Fx017M schedule | 26/30 | 86.7 (66.8–94.6) | < .0001 | <.0001 |
| 012M schedule | 10/16 | 62.5 (29.4–80.1) | .0009 | .0002 |
| Infectivity controls | 0/12 | … | ||
| Second CHMI results, by primary immunization groupd | ||||
| Fx017M schedule | ||||
| Not protected, fractional boost | 2/2 | 100 (−127–100) | .0357 | .0257 |
| Protected, fractional boost | 9/10 | 90 (36–98) | .0009 | <.0001 |
| Protected, no fractional boost | 3/7 | 43 (−9–70) | .1923 | .0084 |
| 012M schedule | ||||
| Not protected, fractional boost | 2/3 | 67 (−65–93) | .0833 | .0369 |
| Protected, fractional boost | 1/4 | 25 (−32–57) | .4000 | .0420 |
| Protected, no fractional boost | 1/5 | 20 (−24–48) | .4545 | .0168 |
| Infectivity controls | 0/6 | … | … | … |
Abbreviations: CI, confidence interval; Fx017M, delayed fractional dose, comprising a 0.5-mL dose of RTS,S/AS01B followed 1 month later by a 0.5-mL dose of RTS,S/AS01B, followed 6 months later by a 0.1-mL dose of RTS,S/AS01B; 012M, 0- , 1-, and 2-month standard schedule comprising 3 doses of 0.5 mL of RTS,S/AS01B given 1 month apart.
a By the Fisher exact test, compared with infectivity controls.
b By the log-rank test, compared with infectivity controls.
c Fx017M vs 012M, P = .074, by the Fisher exact test; P = .040, by the log-rank test.
d “Protected” denotes subjects who were protected after the first CHMI. “Not protected” denotes subjects who were not protected after the first CHMI.
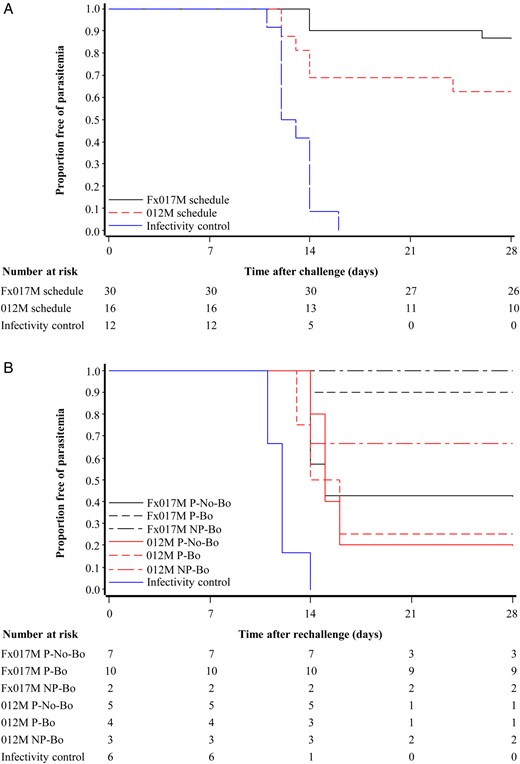
Proportion of subjects remaining free of parasitemia after the first (A) and second (B) controlled human malaria parasite infection (CHMI) in the according-to-protocol population. Thirty subjects from the group that received a 0-, 1-, and 7-month regimen, including a fractional third dose (Fx017M), 16 subjects from the group that received the 0-, 1-, and 2-month full-dose regimen (012M group), and 12 infectivity controls took part in the first CHMI. Of these, 26, 10, and 0, respectively, were protected against the first CHMI. There was evidence for a true difference in protection against the first CHMI between schedules (P = .074, by the Fisher exact test; P = .04, by the log-rank test). Thirty-seven subjects, including 6 infectivity controls, participated in the second CHMI. Abbreviations: NP-Bo, volunteers who were not protected following the primary phase and received a booster dose prior to rechallenge; P-Bo, volunteers who were protected following the primary phase and received a booster dose prior to rechallenge; P-No-Bo, volunteers who were protected following the primary phase and did not receive a booster dose prior to rechallenge.
The number of subjects undergoing a second CHMI and protection data are presented in Figure 2, Table 1, and Supplementary Figure 1. Three of 7 subjects from the Fx017M group who were initially protected and received no booster were protected against a second challenge, indicating that there is waning efficacy after a primary regimen that includes a fractional third dose. In contrast, high protection could be maintained in the Fx017M group by the administration of an additional fractional dose before the second CHMI, with 9 of 10 subjects protected. Furthermore, 4 of 5 subjects (2 in the Fx017M group and 2 in the 012M group) who were infected after the first CHMI were protected against a second CHMI after having received a fractional boost.
No new safety concern associated with the reduction of the third dose or overall emerged. One SAE (alcohol related) was reported in a subject from the Fx017M group, but it was considered to be unrelated to vaccination. Local and general reactogenicity to the first 2 doses were as reported in past RTS,S/AS01 studies (Supplementary Figure 2). Reactogenicity following full and fractional third doses were similar. There was no apparent imbalance in the occurrence of unsolicited AEs considered to be related to vaccination between the Fx017M and 012M groups (Supplementary Table 2). Unsolicited AEs following the third fractional dose were reported in 63.3% of subjects (95% CI, 43.9%–80.1%), compared with 93.8% (95% CI, 69.8%–99.8%) who received a third full dose (Supplementary Table 3). The incidence of individual symptoms reported as causally related to vaccination was similar after dose 3 in both the Fx017M and 012M groups (Supplementary Table 4).
Abnormal laboratory findings were infrequent, and none were grade 3 (data not shown).
Anti-CS humoral responses are presented in Figure 3. Subjects from the 012M group who were protected against first CHMI had, on the day of challenge, slightly higher anti-CS (R32LR) antibody concentrations than those who were NP (012M pooled, 100.1 EU/mL [95% CI, 63.5–157.8 EU/mL]; protected, 139.0 EU/mL [95% CI, 94.0–205.7 EU/mL]; and nonprotected, 57.9 EU/mL [95% CI, 19.5–171.8 EU/mL]), in line with what has been reported previously [15]. Anti-CS (full length) or anti-CS (C-term) antibody concentrations did not differ significantly according to protection status.
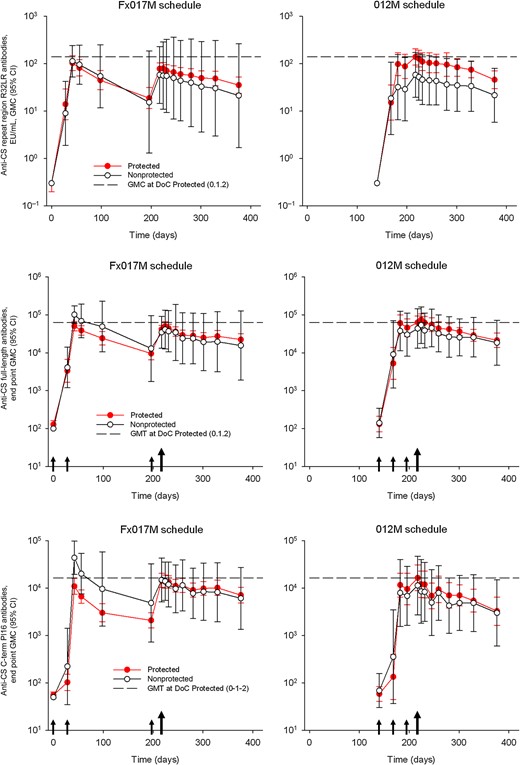
Anti–Plasmodium falciparum circumsporozoite protein (CS) antibody response, by primary schedule and protection status against the first controlled human malaria parasite infection in the according-to-protocol population. Geometric mean antibody concentrations (GMCs) are shown with 95% confidence intervals (CIs) for antibody responses against the CSP repeat region (R32LR), the CSP full-length antigen, and the CSP C-term Pf16 peptide. Antibody responses in subjects protected at challenge are indicated by red bullets, and white bullets show the antibody responses in subjects not protected at challenge. Horizontal hatched lines show GMCs on the day of CHMI in the protected subjects in the group that received the 0-, 1-, and 2-month full-dose regimen. Small arrows represent vaccinations, and the big arrow represents the day of the first CHMI. Abbreviations: DoC, day of challenge; Fx017M, delayed fractional dose, comprising a 0.5-mL dose of RTS,S/AS01B followed 1 month later by a 0.5-mL dose of RTS,S/AS01B, followed 6 months later by a 0.1-mL dose of RTS,S/AS01B; 012M, 0- , 1-, and 2-month standard schedule comprising 3 doses of 0.5 mL of RTS,S/AS01B given 1 month apart.
Anti-CS (R32LR) antibody concentrations were slightly lower in subjects from the Fx017M group (75.2 EU/mL; 95% CI, 57.8–97.8 EU/mL) and similar for anti-CS full length and C-term antibodies. Considering the Fx017M group, no significant differences in antibody concentrations were found when comparing the 4 nonprotected subjects and the 26 protected subjects.
In contrast, antibody avidity was higher on the day of challenge in the Fx017M group as compared to the 012M group when considering all CS antigen targets (Table 2). There was a trend in both schedule groups for higher avidity associated with protection when considering anti-NANP but not anti-CS full-length or C-term assays.
Anti–Plasmodium falciparum Circumsporozoite Protein (CS) Antibody Avidity Index on the Day of Challenge in the According-to-Protocol Population
| Variable | Subjects, No. | Anti-CS Antibody Antigen Target | ||
|---|---|---|---|---|
| Repeat region (NANP) | Full length | C-term Pf16 | ||
| Overall | ||||
| Fx017M schedule | 30 | 0.58 ± 0.16 | 0.68 ± 0.10 | 0.72 ± 0.08 |
| 012M schedule | 16 | 0.48 ± 0.13 | 0.55 ± 0.08 | 0.50 ± 0.13 |
| P Valuea | .0367 | <.0001 | <.0001 | |
| Fx017M scheduleb | ||||
| Protected | 26 | 0.59 ± 0.16 | 0.68 ± 0.10 | 0.72 ± 0.08 |
| Not protected | 4 | 0.50 ± 0.13 | 0.70 ± 0.08 | 0.71 ± 0.11 |
| 012M scheduleb | ||||
| Protected | 10 | 0.51 ± 0.14 | 0.55 ± 0.08 | 0.51 ± 0.13 |
| Not protected | 6 | 0.44 ± 0.09 | 0.55 ± 0.06 | 0.48 ± 0.12 |
| Variable | Subjects, No. | Anti-CS Antibody Antigen Target | ||
|---|---|---|---|---|
| Repeat region (NANP) | Full length | C-term Pf16 | ||
| Overall | ||||
| Fx017M schedule | 30 | 0.58 ± 0.16 | 0.68 ± 0.10 | 0.72 ± 0.08 |
| 012M schedule | 16 | 0.48 ± 0.13 | 0.55 ± 0.08 | 0.50 ± 0.13 |
| P Valuea | .0367 | <.0001 | <.0001 | |
| Fx017M scheduleb | ||||
| Protected | 26 | 0.59 ± 0.16 | 0.68 ± 0.10 | 0.72 ± 0.08 |
| Not protected | 4 | 0.50 ± 0.13 | 0.70 ± 0.08 | 0.71 ± 0.11 |
| 012M scheduleb | ||||
| Protected | 10 | 0.51 ± 0.14 | 0.55 ± 0.08 | 0.51 ± 0.13 |
| Not protected | 6 | 0.44 ± 0.09 | 0.55 ± 0.06 | 0.48 ± 0.12 |
Data are mean values ±SDs, unless otherwise indicated. Challenge was performed approximately 3 weeks after the last dose.
Abbreviations: Fx017M, delayed fractional dose, comprising a 0.5-mL dose of RTS,S/AS01B followed 1 month later by a 0.5-mL dose of RTS,S/AS01B, followed 6 months later by a 0.1-mL dose of RTS,S/AS01B; 012M, 0- , 1-, and 2-month standard schedule comprising 3 doses of 0.5 mL of RTS,S/AS01B given 1 month apart.
a By the 2-sample Student t test, for comparisons for each antibody target.
b “Protected” denotes subjects who were protected after the first CHMI. “Not protected” denotes subjects who were not protected after the first CHMI.
Anti–Plasmodium falciparum Circumsporozoite Protein (CS) Antibody Avidity Index on the Day of Challenge in the According-to-Protocol Population
| Variable | Subjects, No. | Anti-CS Antibody Antigen Target | ||
|---|---|---|---|---|
| Repeat region (NANP) | Full length | C-term Pf16 | ||
| Overall | ||||
| Fx017M schedule | 30 | 0.58 ± 0.16 | 0.68 ± 0.10 | 0.72 ± 0.08 |
| 012M schedule | 16 | 0.48 ± 0.13 | 0.55 ± 0.08 | 0.50 ± 0.13 |
| P Valuea | .0367 | <.0001 | <.0001 | |
| Fx017M scheduleb | ||||
| Protected | 26 | 0.59 ± 0.16 | 0.68 ± 0.10 | 0.72 ± 0.08 |
| Not protected | 4 | 0.50 ± 0.13 | 0.70 ± 0.08 | 0.71 ± 0.11 |
| 012M scheduleb | ||||
| Protected | 10 | 0.51 ± 0.14 | 0.55 ± 0.08 | 0.51 ± 0.13 |
| Not protected | 6 | 0.44 ± 0.09 | 0.55 ± 0.06 | 0.48 ± 0.12 |
| Variable | Subjects, No. | Anti-CS Antibody Antigen Target | ||
|---|---|---|---|---|
| Repeat region (NANP) | Full length | C-term Pf16 | ||
| Overall | ||||
| Fx017M schedule | 30 | 0.58 ± 0.16 | 0.68 ± 0.10 | 0.72 ± 0.08 |
| 012M schedule | 16 | 0.48 ± 0.13 | 0.55 ± 0.08 | 0.50 ± 0.13 |
| P Valuea | .0367 | <.0001 | <.0001 | |
| Fx017M scheduleb | ||||
| Protected | 26 | 0.59 ± 0.16 | 0.68 ± 0.10 | 0.72 ± 0.08 |
| Not protected | 4 | 0.50 ± 0.13 | 0.70 ± 0.08 | 0.71 ± 0.11 |
| 012M scheduleb | ||||
| Protected | 10 | 0.51 ± 0.14 | 0.55 ± 0.08 | 0.51 ± 0.13 |
| Not protected | 6 | 0.44 ± 0.09 | 0.55 ± 0.06 | 0.48 ± 0.12 |
Data are mean values ±SDs, unless otherwise indicated. Challenge was performed approximately 3 weeks after the last dose.
Abbreviations: Fx017M, delayed fractional dose, comprising a 0.5-mL dose of RTS,S/AS01B followed 1 month later by a 0.5-mL dose of RTS,S/AS01B, followed 6 months later by a 0.1-mL dose of RTS,S/AS01B; 012M, 0- , 1-, and 2-month standard schedule comprising 3 doses of 0.5 mL of RTS,S/AS01B given 1 month apart.
a By the 2-sample Student t test, for comparisons for each antibody target.
b “Protected” denotes subjects who were protected after the first CHMI. “Not protected” denotes subjects who were not protected after the first CHMI.
Subjects in the Fx017M and 012M groups had similar anti-HBV antibody responses (Supplementary Figure 3).
Messenger RNA sequencing of heavy and light chain variable regions and evaluation of nucleotide changes relative to germ line sequences showed that subjects in the Fx017M group had higher median levels of somatic hypermutation than those in the 012M group (P < .01, by the Wilcoxon rank sum test; Figure 4). Average germ line divergence in B-cell heavy and light chain sequences were correlated with anti-CS avidity, when considering both schedule groups together (r = 0.54; P = .0001).
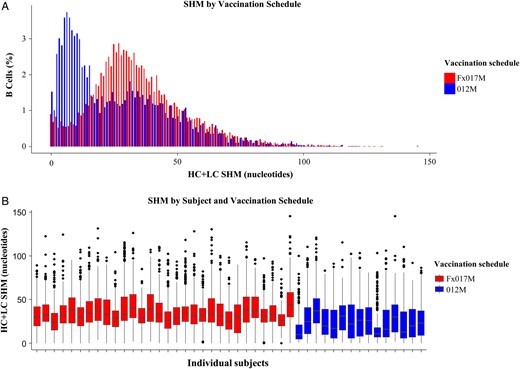
Somatic hypermutation (SHM), by schedule group. A, Bar chart of SHMs in B cells, colored by vaccination schedule. The x-axis is the total number of heavy chain (HC) and light chain (LC) mutated nucleotides, and the y-axis is the fraction of B cells having that level of mutation. For each sequenced B cell, the number of mutations was determined by aligning the HC and LC sequences to predicted germ line sequences and then counting the number of nucleotides in the alignment that differed from those in the best-matching germ line. Because different numbers of B cells were sequenced from different subjects, the height of the bar chart represents the sum of the percentage of B cells for a given level of mutation, normalized by the number of subjects in the arm. B, Box plots of total SHMs, by individual subject, colored by schedule group, showing the range of SHMs for each individual. The x-axis is the subject identifier, and the y-axis is the number of mutations. Each box plot gives the median (horizontal line), interquartile range (IQR; height of rectangle), largest and smallest values within 1.5 times the IQR (vertical lines), and outliers outside of 1.5 times the IQR (points). The difference in the median level of SHMs between the 2 schedule groups is statistically significant (P < .01, by the Wilcoxon rank sum test). Abbreviations: Fx017M, delayed fractional dose, comprising a 0.5-mL dose of RTS,S/AS01B followed 1 month later by a 0.5-mL dose of RTS,S/AS01B, followed 6 months later by a 0.1-mL dose of RTS,S/AS01B; 012M, 0- , 1-, and 2-month standard schedule comprising 3 doses of 0.5 mL of RTS,S/AS01B given 1 month apart.
CS-specific polyfunctional CD4+ T-cell responses were similar in the Fx017M and 012M groups, with no evidence for an association with protection (Supplementary Figure 4). CS-specific CD8+ T-cell responses were low or not detectable.
DISCUSSION
While RTS,S/AS01 is currently being evaluated for World Health Organization policy recommendation for use on the basis of a demonstrated reduction of malaria-related disease burden in children during phase 3 evaluation [16], efforts toward vaccine strategies generating higher protection are ongoing. A number of complex approaches are under consideration, including antigen combinations, a heterologous prime-boost regimen, DNA vaccine platforms, and intravenous injection of mosquito-dissected, liquid nitrogen-stored, irradiated sporozoites [17]. Here, we demonstrate that a change in immunization schedule that includes a delayed administration and dose reduction of the third RTS,S/AS01 vaccination, results in a significant increase in protection against CHMI without an increase in the absolute antibody concentration after vaccination. The low number of unprotected subjects in the Fx017M group does not allow a statistically robust analysis of correlates of protection, but qualitative evaluation of immune responses showed that the third dose spacing and dose reduction increased somatic hypermutation in immunoglobulin genes and avidity. Our working hypothesis, considering germinal-center and B-cell biology [18], is that competitive antigen binding in germinal centers led to survival and expansion of B cells, with surface immunoglobulins showing the highest antigen avidity. Work is ongoing to assess the fine specificity of the B-cell repertoire and other immune functions, such as follicular T-cell help. It will be important to delineate the CS- and HBV-specific findings.
The study design did not support evaluation of the respective role of third dose spacing and fractioning or of the respective role of fractioning the third dose antigen or adjuvant content. Although vaccine dose spacing has been classically described as supporting immune maturation [19], short vaccine schedules can also generate protective responses against a number of diseases [20, 21, 22]. A 0-, 7-, and 28-day RTS,S/AS02 immunization schedule was shown to generate anti-CS antibody titers and protection against CHMI similar to that observed using longer schedules [23]. In one past study of RTS,S/AS01 administered as 3 identical pediatric doses on a 0-, 1-, and 2-month or a 0-, 1-, and 7-month schedule to infants in coadministration with other vaccines in conditions of natural malaria exposure, third dose spacing alone did not increase VE against clinical malaria captured by passive case detection; avidity indices after dose 3 did not differ according to schedule, and there was no association between avidity after dose 3 and protection [24]. It is possible that a third dose fractioning alone was the driver of increased protection, and a study evaluating a 0-, 1-, and 2-month schedule including a third fractional RTS,S/AS01 dose is presently ongoing (NCT02252640). While fractional dose immunization has been evaluated as a dose-sparing strategy for a number of vaccines [25, 26, 27], this is to our knowledge the first demonstration of within-schedule successive dose-level reduction leading to higher protection.
Past studies have shown the relevance of the CHMI model as being indicative of protection under conditions of natural exposure [28]. As an important next step, the fractional dose regimen needs to be evaluated in malaria-endemic countries. Duration of protection will be critical to the potential role of this schedule improvement on vaccine-based malaria control and elimination efforts. In this study, which included a second CHMI 8 months after the first one, we demonstrated that, in the absence of a booster dose, protection in the fractional dose regimen was reduced after 8 months, but administration of a fractional boost maintained high protection. Furthermore, fractional dose boosting was shown to reverse susceptibility to infection in 4 of 5 subjects who were infected after the first CHMI but not after the second one. Evaluation of the fractional dose regimen in malaria-endemic countries will determine whether this strategy, including boosting, may reach the second-generation malaria vaccine target of 75% efficacy and the potential to significantly reduce malaria transmission [29]. The impact of other schedule- and dose-associated changes on further improvements in immunogenicity and protection needs to be evaluated, as well as the role of dose spacing and reduction in other vaccine disease areas.
Notes
Acknowledgments. We thank the study participants, for their participation; Linda Murray and Doug Stagnaro, for operational support; Sarah Benns and Ulrike Krause, for editorial assistance; Sophie Timmery, for manuscript review coordination; Philippe Moris, Nathalie Baudson, and Ariane Meurée, for immunological assay support; Xiaomu Chen, Kevin Williamson, Dongkyoon Kim, Brittany D. Chilson, Yann Chong Tan, Gregg Espiritu Santo, and Eldar Giladi, for contribution to B-cell transcriptomics; Nancy Richie and the Walter Reed Army Institute of Research (WRAIR) Malaria Serology Laboratory; Saba Alemayehu and the WRAIR Department of Malaria Diagnostics; Lindsey Garver, Megan Dowler, Tatyana Savransky, and Kathryne Walker, for human malaria parasite infection (CHMI) support; and the WRAIR microscopy team (Elizabeth H. Duncan, Evelina Angov, Whitney Cabrera, Fouzia Farooq, Wathsala K. Wijayalath, Lucia Gerena, Christina M. McCray, Alexander V. Pichugin, Jessica E. Heavin, Jessica Bolton, Irina V. Roman, Anuj Sharma, Robert J. Schwenk, Joanne M. Lumsden, Emily C. Smith, Rebecca L. Danner, and Andrea K. Strein).
J. C., R. B., M. L., J. V., E. J., D. M., R. W., C. F. O., A. J. B., C. K. L., U. W.-R., D. K., J. B., J. R., K. M. P., S. B. C., N. C. W., B. Y.-R., W. W., D. E., and W. H. R. were involved in the study conception and design. J. C., R. B., M. L., J. V., E. J., D. M., R. W., C. F. O., A. J. B., C. K. L., U. W.-R., D. C. K., J. W. B., J. A. R., A. K. K., K. M. P., J. E. M., J. L. K., S. B. C., P. T., A. Q., N. C. W., K. H., M. W., J. L., A. T. K., M. G., C. M., S. A. D., B. Y.-R., R. M. P., W. W., D. E., W. H. R., P. E. W., and S. A. D. were involved in the collection, supervision, analysis, and/or interpretation of the data. R. B., J. V., D. M., C. F. O., R. W., U. W.-R., A. J. B., C. K. L., D. C. K., J. W. B., J. A. R., A. K. K., K. M. P., J. L. K., S. B. C., N. C. W., C. M., B. Y.-R., R. M. P., and P. E. W. were involved in the administrative, technical, and logistical support of the study (ie, acquisition of funding, choice and coordination of centers, and supervision of the study). The authors had access to data from the study and took responsibility for the decision to submit for publication.
GlaxoSmithKline (GSK) Biologicals was involved in the study design, data collection, analysis and interpretation. PATH Malaria Vaccine Initiative was involved in study design and data interpretation. The Department of Defense Military Malaria Research Program (MMRP) was involved in study design and data interpretation and provided laboratory and clinical research infrastructure and capability.
Disclaimer. The opinions or assertions contained herein are the private views of the authors, and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense.
Financial support. This work was supported by GSK Biologicals, the Department of Defense MMRP, the PATH Malaria Vaccine Initiative (to GSK Vaccines and the MMRP), and the Bill and Melinda Gates Foundation (to the Malaria Vaccine Initiative and Atreca).
Potential conflicts of interest. R. B., M. L., E. J., and D. M. are GSK employees. J. V. was a GSK employee at the time of the study and manuscript development. J. C. is an independent consultant to GSK. W. V. and D. E. are employees of Atreca. W. H. R. is a consultant to and is on the board of directors of Atreca. J. C., R. B., M. L., J. V., E. J., D. M., and S. B. C.'s spouse own GSK stock/stock options. W. V., D. E., and W. H. R. own Atreca stock. J. C. is a named inventor on patents or patent applications, pending or issued, in the field of malaria vaccine, but no compensation or royalties are received. R. B. has a pending patent application. All other authors report no potential conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
Author notes
Presented in part: 64th Annual Meeting of the American Society of Tropical Medicine and Hygiene, Philadelphia, Pennsylvania, 25–29 October 2015.


{kind=link}
{kind=link}
{kind=link}
{kind=link}