






Abstract
Streptococcus pneumoniae causes high mortality as a major pneumonia-inducing pathogen. In pneumonia, control of innate immunity is necessary to prevent organ damage. We assessed the role of microRNAs (miRNAs) as regulators in pneumococcal infection of human macrophages. Exposure of primary blood-derived human macrophages with pneumococci resulted in transcriptional changes in several gene clusters and a significant deregulation of 10 microRNAs. Computational network analysis retrieved miRNA-146a as one putatively important regulator of pneumococci-induced host cell activation. Its induction depended on bacterial structural integrity and was completely inhibited by blocking Toll-like receptor 2 (TLR-2) or depleting its mediator MyD88. Furthermore, induction of miRNA-146a release did not require the autocrine feedback of interleukin 1β and tumor necrosis factor α released from infected macrophages, and it repressed the TLR-2 downstream mediators IRAK-1 and TRAF-6, as well as the inflammatory factors cyclooxygenase 2 and interleukin 1β. In summary, pneumococci recognition induces a negative feedback loop, preventing excessive inflammation via miR-146a and potentially other miRNAs.
Pneumonia constitutes one of the most devastating diseases worldwide, particularly affecting children and elderly individuals [1]. Among children aged <5 years, pneumonia poses the leading cause of death, killing more children in this age group than malaria, AIDS, and measles together [1, 2].
The gram-positive diplococcus Streptococcus pneumoniae is the most common cause of bacterial pneumonia [1, 3]. Moreover, due to increasing resistance to antimicrobial agents, the morbidity due to pneumococci and, hence, the mortality rates of patients remain high. Multidrug-resistant pneumococcus serotypes constitute a major challenge because their spread poses a serious public health problem [4, 5].
Resident alveolar macrophages are critical for initiating the rapid and coordinated innate immune response to local S. pneumoniae infection [6]. Upon recognition of the pathogen through Toll-like receptors (TLRs) [7], signaling cascades including the TLR adaptor protein MyD88, the serine/threonine kinases IRAK-1, -2, and -4, and the E3 ubiquitin ligase TRAF-6 are induced, culminating in the nuclear translocation of the transcription factor NF-κB [8]. Consequently, macrophage-derived cytokines such as tumor necrosis factor α (TNF-α), interleukin 1α (IL-1α), and interleukin 1β (IL-1β) trigger the recruitment of leukocytes, as well as the activation of lung epithelial cells, both leading to bacterial clearance [6, 9, 10].
Fighting bacterial growth has to be regulated carefully to preserve lung function and to prevent barrier failure, which would result in lung injury and sepsis. For instance, in the United States, pneumonia followed by severe sepsis represents the most common cause of acute respiratory distress syndrome, characterized by respiratory failure and lung injury [11].
During the past 2 decades, microRNAs (miRNAs) have emerged as pivotal posttranscriptional effectors of gene expression [12]. In macrophages, modulated miRNA expression in response to external stimuli is implicated in regulating cell signaling, the expression of inflammatory cytokines, receptor morphology, and cell cycle progression [13, 14]. Thus, miRNA-induced changes in gene expression affect the differentiation of macrophages toward distinct polarized phenotypes [15, 16].
In this study, we comprehensively studied the transcriptome and miRNA profile of human macrophages challenged with pneumococci. Among the deregulated miRNAs with immunomodulatory potential, we found miR-9, miR-124a, miR-155, and miR-146a. Exemplarily, we analyzed the induction and function of miRNA-146a. We found that its expression depends on the TLR-2–MyD88 axis and establishes a negative feedback loop to the proximal mediators of TLR-2, thereby restricting proinflammatory gene expression.
METHODS
Chemicals and Antibodies
Details are shown in the Supplementary Data.
Cell Cultures
Details are shown in the Supplementary Data.
Bacterial Cultivation and Infection
For bacterial infection of cells, the encapsulated S. pneumoniae serotype 2 strain D39 or clinical isolates of other S. pneumoniae serotype strains—serotype 1 (ST1), serotype 3 (ST3), serotype 4 (TIGR4), serotype 7F (ST7F), and serotype 8 (ST8)—were used. Serotype 2 strain D39 and serotype 4 strain (TIGR4) were kindly provided by Prof Sven Hammerschmidt (Ernst-Moritz-Arndt University Greifswald, Germany). Serotype strains ST1, ST3, ST7F, and ST8 were kindly provided by the National Reference Center for Streptococci, Germany [17]. Pneumococci were plated on blood agar plates overnight and then cultured in Todd Hewitt broth supplemented with 5% yeast extract to mid-log phase (OD600, 0.2–0.3), harvested by centrifugation, and resuspended in cell culture medium without supplements. Heat inactivation of D39 was performed for 1 hour at 56 °C. UV inactivation of D39 was performed by irradiation with UV light at maximal intensity for 10 minutes. Infections were performed for 16 hours at the indicated multiplicities of infection (MOIs).
RNA Extraction and Analysis by Microarray and Real-Time Polymerase Chain Reaction (PCR) Analysis
RNA from cells was extracted by Isol-RNA Lysis Reagent (5Prime) according to the manufacturer′s protocol. Global analysis of miRNA expression was performed using the Taqman Low Density Array (TLDA; Set A, v. 2.0, human, Life Technologies). Three independent experiments were performed for this purpose. TLDAs were executed on a 7900HT real-time PCR system (Applied Biosystems). Details of RNA analysis by real-time PCR and respective primers are given in the Supplementary Data.
Microarray analyses were performed as described before [18, 19], using an Agilent SurePrint G3 Human Gene Expression 8 × 60 K v2 Microarray kit with 200 ng of RNA per sample. Control and infected samples were measured in biological triplicate.
Enzyme-Linked Immunosorbent Assay (ELISA)
Secretion of IL-8, IL-1β, and TNF-α in the supernatants of stimulated primary human blood-derived macrophages (BDMs) was analyzed using commercial ELISA kits from Becton Dickinson, according to the supplier's protocol. All measurements were done at least in duplicates.
miRVana Inhibitor and Mimic Transfection
For miRVana inhibitor and mimic transfection, primary human BDMs at 2 × 105 cells/mL were transfected with miRVana control inhibitor, miRVana control mimic, miRVana miR-146a inhibitor, or miRVana miR-146a mimic (4464076, 4464058, MH10722, or MC10722, respectively; Life Technologies) by Viromer BLUE transfection reagent (Lipocalyx) according to the manufacturer's instructions.
Bioinformatics Analysis
Details are shown in the Supplementary Data.
Statistical Analysis
The Mann–Whitney U test was used to compare the miRNA and messenger RNA (mRNA) expression between 2 groups. If >2 groups were involved, the Kruskal–Wallis test followed by a post hoc test (Mann–Whitney U test) was used. P values generated in this study correspond with the 2-sided asymptotic significance. P values of <.05 were considered statistically significant.
RESULTS
S. pneumoniae Induces Complex Changes in the Transcriptional Network of Human Macrophages
To assess the regulatory role of miRNAs in pneumococcal infection, we first analyzed changes in mRNA and miRNA expression in human BDMs after infection with S. pneumoniae serotype 2 wild-type strain D39. Cytotoxic effects were moderate with the chosen time- and dose-frame as assessed by microscopy (data not shown) and the LDH assay (Supplementary Data).
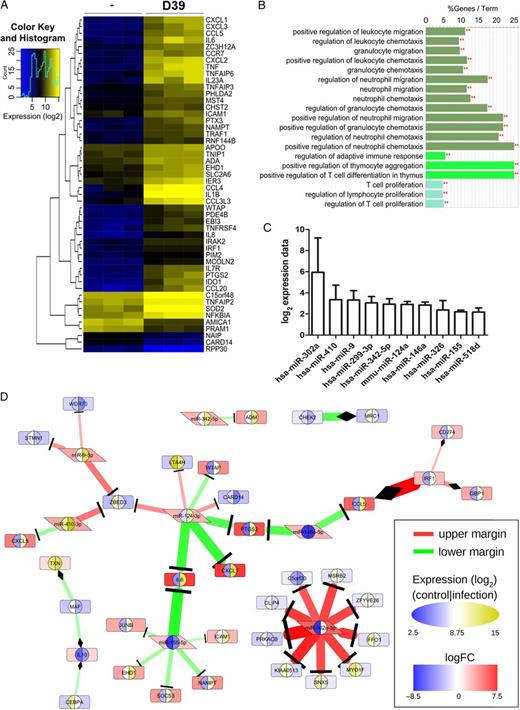
Analysis of high-throughput data from infected macrophages reveals microRNA-146a (miRNA-146a), miR-9, and miR-155 as potential cooperative regulators of the transcription factor NF-κB. Furthermore, miR-146a and miR-124 appear to be both regulators of cyclooxygenase 2 (PTGS2) expression. A, Heat map of differential gene expression in primary human blood-derived macrophages (BDMs) stimulated with Streptococcus pneumoniae. BDMs from different donors were left untreated or stimulated with S. pneumoniae serotype 2 strain (D39; multiplicity of infection, 0.1) for 16 hours in 3 independent experiments. RNA was isolated and subjected to microarray analysis. The 50 most significantly regulated genes are shown. (B) GO term enrichment analysis of the genes from panel A reflects a focus on leukocyte differentiation and migration and a connection to the T-cell branch of the adaptive immune system. The analysis was performed in Cytoscape with ClueGO against a preselected background of genes involved in immune system processes. At a significance threshold of .001, 3 GO term clusters (distinguishable by bar colors) were identified. C, Ranked presentation of log2 expression data ± standard error of the mean for significantly regulated miRNAs in the same samples as in panel A. The expression of 377 miRNAs was analyzed by the Taqman Low Density Array kit. Significantly regulated miRNAs (P < .05) were normalized to the internal reference, U6. D, Part of an interaction network (Supplementary DataC) that was reconstructed based on validated interactions (extracted from miRTarBase) between the genes and miRNAs selected in the high-throughput data analysis. We extracted highly perturbed interactions from the network with ExprEssence. The expression level of each molecule is shown twice: the inner left and right semicircle represent absolute expression in control and infected conditions, respectively (blue-white-yellow elliptical scale), while the background color of the enclosing shape illustrates the difference between infected and control (blue-white-red rectangular scale). We considered only interactions that were either below the 10th percentile (lower margin; green) or above the 90th percentile (upper margin; red) of ExprEssence's LinkScore. **P < .001.
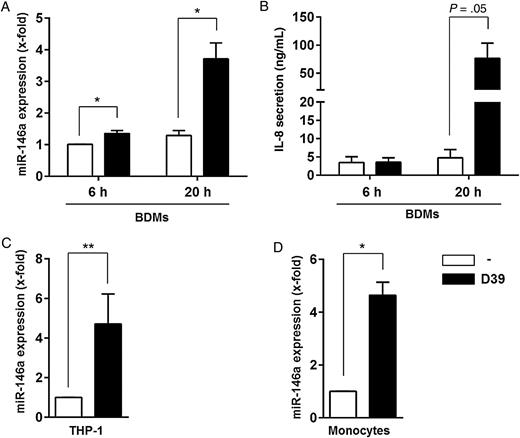
Streptococcus pneumoniae induces expression of the gene encoding microRNA-146a (miRNA-146a) in different immune cells. Primary human blood-derived macrophages (BDMs; A and B), THP-1 cells (C), or monocytes (D) were left untreated or infected with S. pneumoniae serotype 2 strain D39 (multiplicity of infection, 0.1) for 6 hours (A and B) or 20 hours (A, B, C, and D), respectively. RNA was isolated, and MIR146A expression was analyzed by quantitative reverse transcription–polymerase chain reaction (A, C, and D). B, Interleukin 8 (IL-8) secretion by BDMs was determined by enzyme-linked immunosorbent assay. Data are shown as mean ± standard error of the mean of 4 (A), 3 (B and C), or 2 (D) independent experiments performed in duplicates (C) or triplicates (D). Statistical analysis was performed as described in “Methods” section. *P < .05 and **P < .01. Abbreviation: THP-1, human monocytic leukemia cell line.
S. pneumoniae–Induced Expression of MIR146A Depends on TLR-2 and Bacterial Structural Integrity
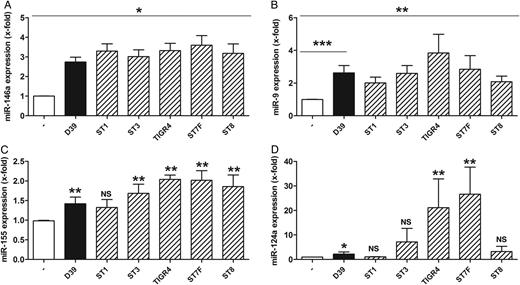
Clinical isolates of Streptococcus pneumoniae induce microRNA-146a (miRNA-146a), miR-9, miR-155, and miR-124a release in human macrophages. Primary human blood-derived macrophages were left untreated or stimulated with S. pneumoniae serotype 2 strain (D39; multiplicity of infection, 0.1) or other S. pneumoniae serotype strains for 16 hours. RNA was isolated, and expression of the indicated miRNAs (A–D) was analyzed by quantitative reverse transcription–polymerase chain reaction. The following S. pneumoniae strains were used: D39, ST1, ST3, TIGR4, ST7F, and ST8. Data are shown as mean ± standard error of the mean of at least 4 independent experiments. The Mann–Whitney U test was used. *P < .05, **P < .01, and ***P < .001. Abbreviation: NS, not significant.
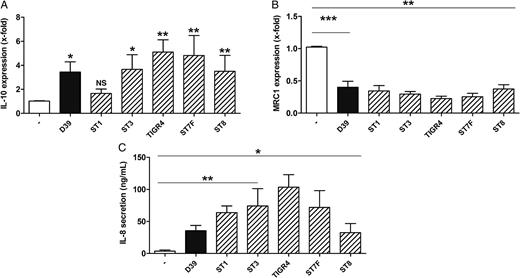
Clinical isolates of Streptococcus pneumoniae differentially regulate IL10 and MRC1 expression in human macrophages. Primary human blood-derived macrophages (BDMs) were left untreated or stimulated with S. pneumoniae serotype 2 strain (D39; multiplicity of infection, 0.1) or other S. pneumoniae serotype strains for 16 hours. RNA was isolated, and expression was analyzed by quantitative reverse transcription–polymerase chain reaction (A and B). Interleukin 8 (IL-8) secretion by BDMs was determined by enzyme-linked immunosorbent assay (C). Data are shown as mean ± standard error of the mean of at least 4 independent experiments. The following S. pneumoniae strains were used: D39, ST1, ST3, TIGR4, ST7F, and ST8. For statistical comparison between uninfected control and stimulated samples, the Mann–Whitney U test was used. *P < .05, **P < .01, and ***P < .001. Abbreviation: NS, not significant.
![Induction of microRNA-146 (miRNA-146a) release after Streptococcus pneumoniae infection of primary human blood-derived macrophages (BDMs) depends on Toll-like receptor 2 (TLR-2) and its downstream mediator MyD88. A, Stimulation with heat-killed pneumococci reduces miRNA-146a induction in human macrophages. BDMs were left untreated or stimulated either with S. pneumoniae serotype 2 strain (D39; multiplicity of infection [MOI], 0.1) or heat-inactivated D39 (h.i.; MOI, 1) or UV-inactivated D39 (UV i.; MOI, 1) for 16 hours. RNA was isolated and MIR146A expression was analyzed by quantitative reverse transcription–polymerase chain reaction (RT-PCR). Data are shown as mean ± standard error of the mean (SEM) of 6 independent experiments. Statistical analysis was performed as described in “Methods” section. B, TLR-2/-1 agonist Pam3CysK4 induces miRNA-146a expression in BDMs. Cells were left untreated or stimulated either with S. pneumoniae serotype 2 strain (D39; MOI. 0.1) or different concentrations of Pam3CysK4 for 16 hours. Data are shown as mean ± SEM of 3 independent experiments. RNA was isolated, and MIR146A expression was analyzed by quantitative RT-PCR. For statistical comparison between uninfected control and stimulated samples, the Mann–Whitney U test was used. C, Blocking of TLR-2 inhibits the induction of miRNA-146a release. BDMs were incubated with isotype control (10 µg/mL) or α-TLR-2 blocking antibody (10 µg/mL) 30 minutes prior to stimulation. Then, cells were left untreated or stimulated with S. pneumoniae serotype 2 strain (D39; MOI, 0.1), Pam3CysK4 (10 ng/mL), or lipopolysaccharide (LPS; 100 ng/mL) for 16 hours. RNA was isolated, and MIR146A expression was analyzed by quantitative RT-PCR. Data are shown as mean ± SEM of 3 or 4 independent experiments performed in duplicates (uninfected vs infected). Statistical analysis was performed as described in “Methods” section. D, S. pneumoniae–induced MIR146A expression is regulated in a MyD88-dependent manner in THP-1 cells. Lentiviral stably transfected control vector (pLKO.1) or MyD88- (pLKO.1_MyD88) knockdown THP-1 cells were stimulated with S. pneumoniae serotype 2 strain (D39; MOI, 0.1) for 16 hours. RNA was isolated, and MIR146A expression was analyzed by quantitative RT-PCR. Data are shown as mean ± SEM of 5 independent experiments performed in duplicates or triplicates. Statistical analysis was performed as described in “Methods” section. *P < .05, **P < .01, and ***P < .001. Abbreviations: Ab, antibody; NS, not significant; Pam3CysK4, TLR-2/-1 agonist; pLKO.1, lentiviral control vector; pLKO.1_MyD88, lentiviral vector for knock down of MyD88.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/214/2/10.1093_infdis_jiw109/2/m_jiw10905.jpeg?Expires=1750289683&Signature=rI9bUSwqbqgJXZ1bUYvn5tIGdhQVoqrRtmua1TSuUfn0kUeLE7l7o5V5nfoSEr6QB9I8jTFEGuD7-4queP3jv9vuP2~2zPH-n970Gd-4hzv9Yg9bImp3F-OgiT0nmwwdq~XYvPf~5eTBQnYkRqXcaxBUY4tUZc8oCOqnADdRVoMmEzZvIqcX4PMSBQVVpZvgpeNfcTCQOWJqAiYJ85Ilmh1iaYj9v5LEnXBRW2YBgAGjtaZZjvggO5JmCi8Q~L69RRPDT2XajeZgV6tcZ5hUHMfJRkDeXvY~sCOb6~MjZFKTkCsP74lnrm-gOh~14VBdgfIwWlxqNx0dHPkQPedNMA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Induction of microRNA-146 (miRNA-146a) release after Streptococcus pneumoniae infection of primary human blood-derived macrophages (BDMs) depends on Toll-like receptor 2 (TLR-2) and its downstream mediator MyD88. A, Stimulation with heat-killed pneumococci reduces miRNA-146a induction in human macrophages. BDMs were left untreated or stimulated either with S. pneumoniae serotype 2 strain (D39; multiplicity of infection [MOI], 0.1) or heat-inactivated D39 (h.i.; MOI, 1) or UV-inactivated D39 (UV i.; MOI, 1) for 16 hours. RNA was isolated and MIR146A expression was analyzed by quantitative reverse transcription–polymerase chain reaction (RT-PCR). Data are shown as mean ± standard error of the mean (SEM) of 6 independent experiments. Statistical analysis was performed as described in “Methods” section. B, TLR-2/-1 agonist Pam3CysK4 induces miRNA-146a expression in BDMs. Cells were left untreated or stimulated either with S. pneumoniae serotype 2 strain (D39; MOI. 0.1) or different concentrations of Pam3CysK4 for 16 hours. Data are shown as mean ± SEM of 3 independent experiments. RNA was isolated, and MIR146A expression was analyzed by quantitative RT-PCR. For statistical comparison between uninfected control and stimulated samples, the Mann–Whitney U test was used. C, Blocking of TLR-2 inhibits the induction of miRNA-146a release. BDMs were incubated with isotype control (10 µg/mL) or α-TLR-2 blocking antibody (10 µg/mL) 30 minutes prior to stimulation. Then, cells were left untreated or stimulated with S. pneumoniae serotype 2 strain (D39; MOI, 0.1), Pam3CysK4 (10 ng/mL), or lipopolysaccharide (LPS; 100 ng/mL) for 16 hours. RNA was isolated, and MIR146A expression was analyzed by quantitative RT-PCR. Data are shown as mean ± SEM of 3 or 4 independent experiments performed in duplicates (uninfected vs infected). Statistical analysis was performed as described in “Methods” section. D, S. pneumoniae–induced MIR146A expression is regulated in a MyD88-dependent manner in THP-1 cells. Lentiviral stably transfected control vector (pLKO.1) or MyD88- (pLKO.1_MyD88) knockdown THP-1 cells were stimulated with S. pneumoniae serotype 2 strain (D39; MOI, 0.1) for 16 hours. RNA was isolated, and MIR146A expression was analyzed by quantitative RT-PCR. Data are shown as mean ± SEM of 5 independent experiments performed in duplicates or triplicates. Statistical analysis was performed as described in “Methods” section. *P < .05, **P < .01, and ***P < .001. Abbreviations: Ab, antibody; NS, not significant; Pam3CysK4, TLR-2/-1 agonist; pLKO.1, lentiviral control vector; pLKO.1_MyD88, lentiviral vector for knock down of MyD88.
S. pneumoniae–Induced Expression of MIR146A Does Not Involve Autocrine IL-1β or TNF-α Regulation
![Induction of microRNA-146a (miRNA-146a) release is not influenced in an autocrine manner by interleukin 1β (IL-1β) or tumor necrosis factor α (TNF-α) in human macrophages stimulated with Streptococcus pneumoniae. Primary human blood-derived macrophages (BDMs) were left untreated or stimulated with S. pneumoniae serotype 2 strain (D39; multiplicity of infection [MOI], 0.1) for 16 hours. A and C, Secretion of IL-1β (A) and TNF-α (C) was measured by enzyme-linked immunosorbent assay. B and D, BDMs were left untreated or stimulated either with the IL-1 receptor antagonist anakinra (Kineret; 1 µg/mL; B) or the TNF-α antagonist adalimumab (Humira; 1 µg/mL; D) 1 hour prior to stimulation. After that, BDMs were left untreated or were stimulated with either S. pneumoniae strain D39 (MOI, 0.1), IL-1β (10 ng/mL; B), or TNF-α (10 ng/mL; D) for 16 hours. RNA was isolated, and MIR146A expression was analyzed by quantitative reverse transcription–polymerase chain reaction. Data are shown as mean ± standard error of the mean of 4 (A), 5 (C), and 3 (B and D) independent experiments. Statistical analysis was performed as described in “Methods” section. *P < .05 and **P < .01. Abbreviation: NS, not significant.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/214/2/10.1093_infdis_jiw109/2/m_jiw10906.jpeg?Expires=1750289683&Signature=UKITXnu0CbkQk5XRe2mqrkmlpujHqKPXB0-BTSPrFUr581TRcOTIHMskXhd1v3kCz-i7Np~DRKS757dqsh5vrhmJDsE5EnCrnbTdjxj8mtFyJqZ~ToutTGAYkZg0rGhWB1Yvdm0lw1cPq3JOxt2fKY1tgzJLvv2H7qpZTW~cCsu1V5WOlQAThvsZBwk6iboPXqg8ZaJ9ZRGdgd5M1sdzCJONmB7mMCRgAQ54TsXcjvfAnqQpPUFlpHrLd0g0qELWX1DjsKgngwvYG~5pfgXQI0LcSVl7X~h6o1u05w3DBTstcIfzZVWOCzV1upNLLxI4dX2f8jbr3rE9CVFMSlVlgw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Induction of microRNA-146a (miRNA-146a) release is not influenced in an autocrine manner by interleukin 1β (IL-1β) or tumor necrosis factor α (TNF-α) in human macrophages stimulated with Streptococcus pneumoniae. Primary human blood-derived macrophages (BDMs) were left untreated or stimulated with S. pneumoniae serotype 2 strain (D39; multiplicity of infection [MOI], 0.1) for 16 hours. A and C, Secretion of IL-1β (A) and TNF-α (C) was measured by enzyme-linked immunosorbent assay. B and D, BDMs were left untreated or stimulated either with the IL-1 receptor antagonist anakinra (Kineret; 1 µg/mL; B) or the TNF-α antagonist adalimumab (Humira; 1 µg/mL; D) 1 hour prior to stimulation. After that, BDMs were left untreated or were stimulated with either S. pneumoniae strain D39 (MOI, 0.1), IL-1β (10 ng/mL; B), or TNF-α (10 ng/mL; D) for 16 hours. RNA was isolated, and MIR146A expression was analyzed by quantitative reverse transcription–polymerase chain reaction. Data are shown as mean ± standard error of the mean of 4 (A), 5 (C), and 3 (B and D) independent experiments. Statistical analysis was performed as described in “Methods” section. *P < .05 and **P < .01. Abbreviation: NS, not significant.
miRNA-146a Reduces Expression of the Genes Encoding IRAK-1, TRAF-6, COX-2, and Pro–IL-1β in S. pneumoniae–Exposed Macrophages
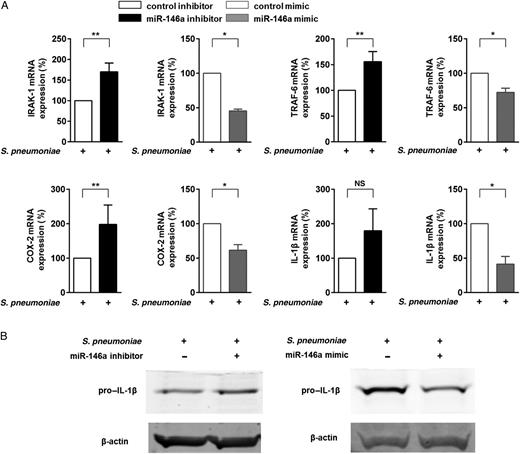
MicroRNA-146a (miRNA-146a) regulates the antiinflammatory activity in human macrophages after stimulation with Streptococcus pneumoniae. Primary human blood-derived macrophages (BDMs) were transfected with miRVana control inhibitor, miRVana miRNA-146a inhibitor, miRVana control mimic, or miRVana miRNA-146a mimic. After 24 hours, BDMs were left untreated or stimulated with S. pneumoniae serotype 2 strain (D39; multiplicity of infection, 0.1) for 15 hours. A, RNA was isolated, and IRAK-1, TRAF-6, COX-2, and interleukin 1β (IL-1β) messenger RNA (mRNA) expression was analyzed by quantitative reverse transcription–polymerase chain reaction, using GAPDH expression for normalization. Graphs represent the quantification of GAPDH-normalized mRNA levels in relation to untreated control, expressed as a percentage of infected control samples. Data represent mean ± standard error of the mean of 5 (control inhibitor and miRNA-146a inhibitor experiments) or 3 (control mimic and miRNA-146a mimic experiments) independent experiments. B, Levels of expression of the gene encoding pro–interleukin 1β (pro-IL-1β) were detected by Western blot. β-actin was the internal control. *P < .05 and **P < .01. Abbreviation: NS, not significant.
DISCUSSION
In this study, we have found that S. pneumoniae D39 infection of primary human macrophages elicits a complex transcriptional response in terms of mRNA and miRNA. This includes in general a classical macrophage activation, as well as immunomodulatory expression of IL10. miR-146a–mediated dampening of the proinflammatory response, either directly by targeting COX-2 and IL-1β or via a miRNA-146a–dependent negative feedback loop, abrogates TLR-2–dependent signaling. Our results suggest that macrophages activation exhibits built-in negative feedback via a miRNA-146a–dependent mechanism to prevent overshooting inflammation upon infection with S. pneumoniae.
Previous studies have shown that human macrophages seem to be strongly involved in pulmonary immune defense against S. pneumoniae [20]. In a murine model of pneumococcal pneumonia, depletion of alveolar macrophages correlated with a strongly increased mortality [21]. However, the transcriptional response of primary human macrophages to exposure with S. pneumoniae has not been studied so far. Therefore, we initially characterized the changes in protein-coding RNA expression after pneumococcal infection of human macrophages and identified several gene clusters with strong bias toward immune defense, cellular activation, chromatin modulation, and apoptosis (data not shown). Markers of classical macrophage activation, such as IL-1β, TNF, and IRAK-2, featured prominently in the upregulated genes. Conversely, the observed IL-10 upregulation upon pneumococcal infection argues for a mechanism that counteracts classical macrophage activation. It has been shown before that pneumococci induce IL10 in lung epithelial cells [22]. Here, we show that this mechanism also seems to be active in macrophages.
In an early study, Rogers et al exposed undifferentiated THP-1 cells for 3 hours to the same D39 strain used in this study [23]. They found 117 mRNAs to be upregulated, including IL8, MCP3, IL2RB, and PTGES3, and 64 mRNAs to be downregulated, many of them depending on pneumolysin expression. Our analysis confirmed the upregulation of IL8 and found that 3 other differentially expressed genes were also dependent on pneumolysin expression (IL15RA, CASP4, and TXN), whereas none were independent. The small size of the overlap in genes might be due to several factors, including differences in cell subtype, the infection paradigm, and the statistical approach of the analysis. Still, there is an obvious consensus with regard to the pathways both infections perturbed (eg, cytokine signaling, eicosanoid metabolism, antibacterial defense, and apoptosis).
In a next step, we screened 377 miRNAs in S. pneumoniae–exposed human primary macrophages and identified 10 to be significantly upregulated. We could extent this finding by showing exemplarily that genes encoding miR-9, miR-124a, and miR-155 were also induced by different S. pneumoniae serotypes, with the magnitude of upregulation partly mirroring the induction of IL8. This indicates that the different pneumococci strains activate macrophages with individual intensity, as, for example, the TIGR4 strain, which is among the most potent inducers. In addition, MIR124A was not significantly upregulated by the ST1 and ST8 strains. These 2 strains have been described to carry a nonhemolytic pneumolysin allele and are associated with outbreaks of invasive disease [24]. miRNA regulation in pneumococcus- or streptococcus-exposed human macrophages has not been studied before. However, a set of preselected miRNAs has been analyzed in murine polymorphonuclear leukocytes after intratracheal infection with Escherichia coli or S. pneumoniae. Only 2 miRNAs were regulated by pneumococci in this study [25]. MIR155 has been found to be induced by S. pneumoniae infection of murine peritoneal macrophages or sputum leukocytes. Its deficiency impaired the recruitment of macrophages and the induction of protective T-helper type 17 immunity against colonization with S. pneumoniae but did not influence bacterial invasion, murine survival, or the release of IL-6 or TNF-α [26]. More generally, some other pathogens, such as Mycobacterium tuberculosis [27], Pseudomonas aeruginosa [28], and Francisella tularensis [29], have been shown to alter miRNA expression in human macrophages. Altogether, the miRNA profile we found differed clearly from the ones reported for infections with other bacteria.
Such a modeling approach has not been undertaken before. Previous studies have addressed important isolated aspects of pneumococcus-associated signaling in the lung, like receptor engagement [22, 30], stress signaling [31], and the inflammasome [32, 33], as well as regulation of the transcription factors NF-κB, AP-1, KLF4, and Nrf2 [22, 31, 34–36], but mainly in lung epithelial cells. Further efforts are necessary to bring these 2 layers together.
On the basis of our signaling map, we selected MIR146A for further analysis and found that it was induced by pneumococcal exposure also in undifferentiated primary human monocytes, the differentiated myeloid cell line THP-1, and in PBMCs. It has been shown before that miR-146a can negatively influence the production of proinflammatory cytokines and that its promoter is a target of NF-kB [37]. Different serotypes of S. pneumoniae are associated with disease severity and mortality [38]. Interestingly, exposure of BDMs to different clinically important pneumococci serotypes that differ with respect to factors such as the hemolytic potential of their pneumolysins resulted in varying levels of IL-8 release but similar expression levels of miRNA-146a. Recently, some serotype 1 and 8 bacteria have been shown to differ significantly in their potential to activate inflammasomes [17]. In conclusion, both observations indicate that induction of MIR146A by pneumococci is a stable and conserved event in myeloid cells.
Recognition of pneumococci or their components has been shown to involve several pattern-recognition receptors on the cell surface, in vesicles, or in the cytosol [22, 30]. Our results suggest that surface engagement of TLR-2 by intact bacteria is the major component necessary for MIR146A induction in our model. As exposure to pneumococci resulted in a release of IL-1β and TNF-α by BDMs, and both cytokines were able to induce MIR146A expression, we examined and ruled out an autocrine mechanism with blocking antagonists.
After elucidating the prerequisites of MIR146A expression, we addressed the functional relevance of miRNA-146a in pneumococcus-dependent macrophage activation. For that, we selected and investigated COX-2 and IL-1β from our network model.
COX-2 has been found to be upregulated in lung tissue during human and murine pneumococcal pneumonia, as well as during experimental pneumococcus infection in human lung epithelial cells and alveolar macrophages [39, 40]. IL-1β secretion has been described in pneumococcus-infected human lung tissue and PBMCs [17]. In addition, administration of IL-1β at certain time points improved survival in a murine model of pneumococcal pneumonia [41]. We could confirm induction of the genes encoding both COX-2 and pro–IL-1β in S. pneumoniae–exposed BDMs by Western blot (data not shown).
In pneumococcus-challenged primary human macrophages, we observed that inhibition of miRNA-146a enhanced induction of COX2 and IL1B mRNA, as well as protein, whereas increased levels of miRNA-146a had the opposite effects. It has been reported previously that miRNA-146a binds to the 3′ untranslated region of COX-2 and suppresses Helicobacter pylori–induced COX-2 expression in a gastric epithelial cell line [42]. In addition, miRNA-146 has been observed to reduce silicon dioxide–induced IL1B expression in rat macrophages [43]. Taken together, pneumococcus-induced miRNA-146a dampens the induction of central proinflammatory mediators in human macrophages.
In addition, we wanted to rule out, whether miRNA-146a might also be able to negatively regulate pneumococcus-induced TLR-2 signaling in these cells. It has been shown that the TLR-2-downstream mediators IRAK-1 and TRAF-6 are involved in pneumococcus-induced inflammation in lung epithelial A549 cells and murine dendritic cells, respectively [44, 45]. The mRNAs of both factors have been shown to be direct targets of miRNA-146a [46]. Accordingly, we found that inhibition of miRNA-146a enhanced expression of IRAK1 and TRAF6 in pneumococcus-exposed primary macrophages, whereas increased levels of miRNA-146a had the opposite effects. Thereby, we could show that pneumococcus-induced proinflammatory activation of human macrophages concurrently initiates a miRNA-146a–dependent negative feedback loop.
The observation of a miRNA-146a–driven antiinflammatory network in pneumococcal infection may be of particular clinical relevance, as overwhelming inflammation and sepsis is still a common cause of death due to pneumococcal pneumonia [47]. Many interventions to reduce mortality have failed, in part because of late recognition of inflammatory deterioration and their nonselective application to heterogeneous patient groups [48]. A recent genome-wide association study has identified common genetic variants that are associated with a lower risk of death from sepsis due to pneumonia, giving rise to potential novel therapeutic targets or potential biomarkers [49]. Nevertheless, early risk stratification for pneumonia remains a crucial clinical challenge [50]. Therefore, miRNA networks that dampen the inflammatory reaction of macrophages in pneumococcal infections may be altered in susceptible patients or point toward promising targets for intervention, but comprehensive clinical data are still missing.
Our study provides for the first time a comprehensive analysis of mRNA-miRNA networks in primary human macrophages exposed to S. pneumoniae and characterizes one miRNA-dependent regulatory circuit in more detail. However, its influence remains to be tested in an in vivo model of pneumococcal pneumonia, and many more regulatory subnetworks have to be validated and analyzed in depth.
In summary, we report here a transcriptional network of mRNA and miRNA interactions in S. pneumoniae–infected primary human macrophages that includes miRNA-mediated negative feedback dampening the proinflammatory activation. Besides the molecular characterization of host-pathogen interaction, clinical implications for pneumococcal pneumonia require further investigations.
Notes
Acknowledgments. We thank Bastian Opitz, Kathrin Mühlemann, and Mark van der Linden, for kindly providing different S. pneumoniae serotypes; and Mara Wittig, for kindly providing the lentivirally transduced THP-1 cell lines for MyD88. Part of this work will be included in the doctoral thesis of K. G.
Financial support. This work was supported by the German Research Foundation (GRK1673 and SFB/TR-84 to B. S., N. S., and S. H.), the German Federal Ministry of Education and Research (e:bio miRSys–FKZ 0316175B/0316175A and e:Med CAPSYS–FKZ 01ZX1304E/01ZX1304F to B. S. and J. V. and PROGRESS–FKZ 01KI1010K to B. S.), and the Hessen State Ministry of Higher Education, Research, and the Arts (LOEWE Medical RNomics–FKZ 519/03/00.001-[0003] to B. S.).
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}