






Abstract
Background. This study examined the therapeutic effects of an orally active nonpeptide kinin B1 receptor antagonist, BI113823, in a clinically relevant experimental model of polymicrobial sepsis in rats.
Methods. Sepsis was induced by cecal ligation and puncture (CLP). Animals received treatment with either vehicle or BI113823. The experiment was terminated in the first set of animals 15 hours after CLP. Seven-day survival following CLP was determined in the second set of animals.
Results. Compared with vehicle treatment, administration of BI113823 reduced neutrophil and macrophage infiltration, reduced cytokine production, attenuated intestinal mucosal hyperpermeability, prevented hemodynamic derangement, and improved cardiac output. Furthermore, administration of BI113823 reduced inducible nitric oxide synthase expression and the injury score in the lung and attenuated nuclear factor ĸB activation and apoptosis in the liver. Treatment with BI113823 also reduced plasma levels of cardiac troponin, aspartate aminotransferase, alanine aminotransferase, urea, and lactate, as well as proteinuria. Finally, administration of BI113823 improved the 7-day survival rate following CLP in rats.
Conclusions. Administration of BI113823 reduced systemic and tissue inflammatory responses, prevented hemodynamic derangement, attenuated multiorgan injury, and improved overall survival.
Sepsis is a complex inflammatory response that is often triggered via the innate immune system in response to polymicrobial infection [1, 2]. It is a common and critical complication in patients with recent surgery that often leads to multiple organ failure and death. Despite intensive supportive care, organ failure arising from severe sepsis accounts for nearly 6 million deaths worldwide annually [1, 2]. The release of endotoxins from gram-negative bacteria triggers activation of inflammatory cells, including polymorphonuclear leukocytes, monocytes/macrophages, and lymphocytes, leading to a massive, uncontrollable release of inflammatory cytokines, chemokines, and other inflammatory factors [3, 4]. High levels of inflammatory mediators can increase blood capillary permeability, intravascular coagulation, edema, multiple organ failure, and ultimately death [3, 4]. Thus, early control of the inflammatory response could be critical for the treatment of sepsis. However, controlling the overwhelming inflammatory reaction associated with polymicrobial sepsis remains a prevalent clinical challenge with few treatment options.
Kinins are proinflammatory peptides that are formed in plasma and tissues in response to infection, tissue trauma, and inflammatory factors [5, 6]. Kinins exert a wide range of biological effects, including inflammation, hypotension, vasodilation, increased vascular permeability, pain, edema, smooth muscle contraction, and cell proliferation [5, 6]. These actions are mediated by the stimulation of 2 pharmacologically distinct receptor subtypes, B1 and B2 [5, 6]. The B2 receptor is constitutively expressed, whereas the B1 receptor is strongly upregulated during inflammation, tissue injury, and exposure to bacterial endotoxins, such as lipopolysaccharide, growth factors, cytokines, and its own agonist [5–7]. There is an important difference between the 2 receptor subtypes [8, 9]. B2 receptor activation by binding of bradykinin results in rapid, receptor-mediated ligand internalization accompanied by loss of surface receptors [8, 9]. In contrast, B1 receptor, once induced displays a lack of desensitization and absence of internalization [8, 9]. Furthermore, a receptor subtype switch, from kinin B2 receptors to B1 receptors, is induced under various pathologic conditions [10, 11]. B1R induction elicits persistent inflammatory responses, and its upregulation is further boosted by ligand binding [10, 12]. Transgenic overexpression of B1 receptors in rats and mice lead to increased susceptibility to endotoxic shock [13, 14], whereas loss of B1 receptors prevents endotoxic shock in mice [15]. These observations suggest that the B1 receptor blockade might represent a novel therapeutic strategy in the treatment of sepsis.
Small-molecule, nonpeptide, orally active antagonists of B1 receptors are desired for clinical drug development for a variety of inflammatory diseases. BI113823, a small-molecule, orally active, nonpeptide B1 receptor antagonist, is a potent antiinflammatory agent with a favorable cardiovascular profile [16, 17]. The present study examined the therapeutic potential of BI113823 in a rat model of polymicrobial sepsis induced by cecal ligation and puncture (CLP).
METHODS
Animals
All animal studies were approved by the institutional animal care and use committees of Chonbuk National University and Mount Sinai Medical Center and complied with the Animal Welfare Act. Male Wistar rats weighing 275–325 g were used in all experiments. The rats were housed under controlled light/dark conditions and fed standard rat food and water ad libitum. All animals were observed daily for general health, and all invasive procedures were performed under aseptic conditions.
Induction of Sepsis
Animals were anesthetized with ketamine (60 mg kg−1 intramuscularly) and xylazine (10 mg kg−1 intramuscularly). Sepsis was induced by CLP, as previously described [14]. Briefly, a 2-cm midline abdominal incision was performed. The cecum was then exposed, ligated just distal to the ileocecal valve to avoid intestinal obstruction, punctured 10 times with an 18-gauge needle, and returned to the abdominal cavity. The animals were resuscitated immediately after surgery with 3 mL per 100 g of body weight of subcutaneous normal saline and returned to their cages.
Experimental Protocol
Following CLP, animals were randomly assigned to 2 study groups and received treatment of vehicle (0.5% Natrosol; n = 8) or BI113823 (30 mg kg−1 orally; n = 8) at 1 and 12 hours after CLP. The dose was selected on the basis of preclinical pharmacokinetic data.
Hemodynamic Assessment and Tissue Harvest
At 15 hours after CLP, animals were anesthetized with ketamine, and hemodynamic variables were measured as previously described [18]. At the end of the experiment, specimens of blood, abdominal ascites, urine, and organ tissue were collected for analysis. Peritoneal cell counting was performed using a standard hemocytometer. Differential cell counts were performed on cytospin preparations stained with Giemsa–Wright (Microscopy Hemacolor-Merck; Germany).
Histologic Examination
Lung tissue specimens were fixed in 10% formalin, embedded in paraffin, and sliced into 5-µm-thick sections. After deparaffinization, slides were stained with hematoxylin and eosin. Morphological alterations in the lungs were examined by light microscopy. A previously described scoring system [19] measuring inflammatory infiltration, edema, disorganization of lung parenchyma, and hemorrhage was used to grade the degree of lung injury. In this scoring system, defined as follows, higher scores indicate more-severe lung abnormalities: 0, normal; 1, mild; 2; moderate; 3, severe; and 4, very severe [19]. All histologic studies were performed in a blinded fashion.
Determination of Intestinal Mucosal Permeability
Small intestinal mucosal barrier function was assessed using the ex vivo–isolated ileum sac as previously described [18]. Briefly, isolated ileum sacs were prepared in ice-cold modified Krebs-Henseleit bicarbonate buffer (KHBB; pH 7.4). Fluorescein-isothiocyanate dextran with a molecular weight of 4000 Da (FD4; Sigma, St. Louis, Missouri) was used as a permeability probe. The ileal segment, ligated with silk on one end, was filled with 1.5 mL of KHBB and suspended in a glass beaker of 80 mL of KHBB (containing FD4 [20 μg mL−1]) at 37°C and gassed with 95% O2 and 5% CO2. The beaker was incubated for 30 minutes in a shaker bath at a frequency of 80–100 shakes min−1. After incubation, the fluid was aspirated from within the ileal sac to determine FD4 concentrations on the serosal side. The length and diameter of the ileal sac were then measured. FD4 concentrations on the serosal side, as well as the initial and final levels in the glass beaker, were determined by fluorescence measurements at an excitation wavelength of 480 nm and emission wavelength of 520 nm. Intestinal mucosal permeability was expressed as the mucosal-to-serosal clearance of FD4, as previously described [18].
Biochemical Assay
Enzyme immunoassay kits for cardiac troponin I (CTNI-2-HSP, Life Diagnostics, West Chester, Pennsylvania), tumor necrosis factor α (TNF-α; #438204, BioLegend, San Diego, California), and interleukin 1β (IL-1β; RLB00, R & D Systems, Minneapolis, Minnesota) were used to determine the concentrations of these mediators in the plasma and tissues. Plasma levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), urea (k753, k752, and k375, all from Biovision, Milpitas, California), and lactate (MAK064, Sigma) were determined using assay kits according to the manufacturer's instructions. The urine protein concentration was determined using a Smart BCA Assay Kit (#21 071, Intron Biotechnology, South Korea).
Neutrophil accumulation in the lung and liver was measured on the basis of myeloperoxidase (MPO) activity, according to previously described methods [18]. Nuclear factor κB (NF-κB [p65]) binding activity was measured in liver nuclear protein extracts obtained using the Nuclear Extraction Kit from Cayman Chemical (Ann Arbor, Michigan) [20]. The translocation and binding activities of NF-kB (p65) in the nuclear protein extracts were measured using a colorimetric assay according to the manufacturer's instructions (Cayman Chemical, Ann Arbor, Michigan). Liver tissue apoptosis was detected by the TUNEL assay (G-3250; Promega, Madison, Wisconsin). TUNEL assay–positive cells were quantified by randomly selecting 6 fields (100× original magnification) from each tissue slide and calculating the number of TUNEL assay–positive nuclei.
Immunohistochemical Staining
Immunohistochemical staining for neutrophil elastase, CD68, and inducible nitric oxide synthase (iNOS) was performed to depict neutrophil and macrophage accumulation, as well as iNOS expression in tissue sections, as previously described [19]. Briefly, 5-μm tissue sections were deparaffinized, hydrated, and incubated in 10 mM sodium citrate buffer at 99°C for 20 minutes for antigen retrieval. Sections were incubated with primary antibodies for neutrophil elastase (sc-9518), CD-68 (sc-9139), or iNOS (sc-651; all from Santa Cruz Biotechnology, Santa Cruz, California) antibodies overnight, followed by incubation for 1 hour with fluorescein isothiocyanate–labeled secondary antibody (sc-2012 or Sc-2777; Santa Cruz Biotechnology). Sections were counterstained with Ultra Cruz Mounting Medium with DAPI (sc-24941; Santa Cruz Biotechnology) and covered by a coverslip. Fluorescent images were obtained by a Nikon Eclipse TE2000-U fluorescence microscope (Nikon, Tokyo, Japan) and a Nikon LWD 0.52 digital camera. Fluorescence intensities for neutrophil elastase, CD68, and iNOS were quantified using Image Pro Premier 9.1 software.
Survival Study
Additional groups of animals were used to evaluate the effect of BI113823 on survival after CLP-induced sepsis. Animal preparation was the same as described above for the first series of experiments, with the following exceptions: the cecum was ligated and punctured twice; 20 hours after CLP, the necrotic cecum was excised, and the abdominal cavity was washed twice with 40 mL of warm, sterilized normal saline; the abdominal incision was closed in layers; the cecal excision procedure was performed in animals that underwent CLP, to mimic clinical situations in which the septic focus is typically removed [18]; and the animals were then returned to their cages and allowed to consume food and water. One hour after CLP, animals received daily treatments of vehicle (0.5% Natrosol) or BI113823 (30 mg kg−1 orally twice daily). This experiment was terminated 7 days after CLP.
Statistical Analysis
All data are reported as mean values ± standard errors of the mean. Statistical differences were determined using analysis of variance for repeated measures, followed by the Bonferroni post hoc test, using GraphPad Prism 5. P values of <.05 were considered to indicate statistical significance. Survival rates were estimated by the Kaplan–Meier method (GraphPad Prism 5.01).
RESULTS
Hemodynamic Variables
At 15 hours following CLP, vehicle-treated animals had a significant decrease in arterial blood pressure, left-ventricular systolic pressure, first derivative of left ventricular pressure and cardiac index, and a significant increase in left-ventricular end-diastolic pressure. In contrast, BI113823 treatment significantly improved the arterial blood pressure, left-ventricular systolic pressure, and first derivative of left ventricular pressure and cardiac index and attenuated the elevation of left-ventricular end-diastolic pressure (Table 1).
Hemodynamic Variables in Sham-Operated Control Animals and Septic Animals Treated With Vehicle or BI113823 15 hours After Cecal Ligation and Puncture (CLP)
| Variable | Sham | Vehicle | BI113823 |
|---|---|---|---|
| Heart rate, beats/min | 287 ± 15 | 228 ± 25 | 274 ± 29 |
| Arterial BP, mm Hg | 111 ± 7.0 | 81 ± 7.4a | 113 ± 8.1b |
| LVP, mm Hg | 112 ± 7.2 | 79 ± 7.4a | 116 ± 9.5b |
| LVEDP, mm Hg | 2.3 ± 1.0 | 10.3 ± 1.4a | 5.1 ± 1.1b |
| ±dp/dt max, mm Hg/s) | 1921 ± 123 | 1331 ± 74a | 1872 ± 121b |
| Cardiac index | 147 ± 10.1 | 92 ± 11.2a | 135 ± 11.3b |
| Variable | Sham | Vehicle | BI113823 |
|---|---|---|---|
| Heart rate, beats/min | 287 ± 15 | 228 ± 25 | 274 ± 29 |
| Arterial BP, mm Hg | 111 ± 7.0 | 81 ± 7.4a | 113 ± 8.1b |
| LVP, mm Hg | 112 ± 7.2 | 79 ± 7.4a | 116 ± 9.5b |
| LVEDP, mm Hg | 2.3 ± 1.0 | 10.3 ± 1.4a | 5.1 ± 1.1b |
| ±dp/dt max, mm Hg/s) | 1921 ± 123 | 1331 ± 74a | 1872 ± 121b |
| Cardiac index | 147 ± 10.1 | 92 ± 11.2a | 135 ± 11.3b |
All values are means ± standard errors of the mean (n = 7–8).
Abbreviations: BP, blood pressure; LVEDP, left-ventricular end-diastolic pressure; LVP, left-ventricular systolic pressure; ±dp/dt max, first derivative of left ventricular pressure.
aP < .05 vs the control group.
bP < .05 vs the vehicle group.
c Calculated as milliliters per minute per kilogram.
Hemodynamic Variables in Sham-Operated Control Animals and Septic Animals Treated With Vehicle or BI113823 15 hours After Cecal Ligation and Puncture (CLP)
| Variable | Sham | Vehicle | BI113823 |
|---|---|---|---|
| Heart rate, beats/min | 287 ± 15 | 228 ± 25 | 274 ± 29 |
| Arterial BP, mm Hg | 111 ± 7.0 | 81 ± 7.4a | 113 ± 8.1b |
| LVP, mm Hg | 112 ± 7.2 | 79 ± 7.4a | 116 ± 9.5b |
| LVEDP, mm Hg | 2.3 ± 1.0 | 10.3 ± 1.4a | 5.1 ± 1.1b |
| ±dp/dt max, mm Hg/s) | 1921 ± 123 | 1331 ± 74a | 1872 ± 121b |
| Cardiac index | 147 ± 10.1 | 92 ± 11.2a | 135 ± 11.3b |
| Variable | Sham | Vehicle | BI113823 |
|---|---|---|---|
| Heart rate, beats/min | 287 ± 15 | 228 ± 25 | 274 ± 29 |
| Arterial BP, mm Hg | 111 ± 7.0 | 81 ± 7.4a | 113 ± 8.1b |
| LVP, mm Hg | 112 ± 7.2 | 79 ± 7.4a | 116 ± 9.5b |
| LVEDP, mm Hg | 2.3 ± 1.0 | 10.3 ± 1.4a | 5.1 ± 1.1b |
| ±dp/dt max, mm Hg/s) | 1921 ± 123 | 1331 ± 74a | 1872 ± 121b |
| Cardiac index | 147 ± 10.1 | 92 ± 11.2a | 135 ± 11.3b |
All values are means ± standard errors of the mean (n = 7–8).
Abbreviations: BP, blood pressure; LVEDP, left-ventricular end-diastolic pressure; LVP, left-ventricular systolic pressure; ±dp/dt max, first derivative of left ventricular pressure.
aP < .05 vs the control group.
bP < .05 vs the vehicle group.
c Calculated as milliliters per minute per kilogram.
Peritoneal Neutrophil Accumulation and Intestinal Mucosal Permeability
There was a marked increase in peritoneal neutrophil accumulation at 15 hours following CLP in vehicle-treated rats (Figure 1A). Treatment with BI113823 reduced peritoneal neutrophil infiltration, as well as plasma levels of TNF-α and IL-1β, and significantly attenuated the hyperpermeability of intestinal mucosa, compared with vehicle-treated rats (Figure 1).
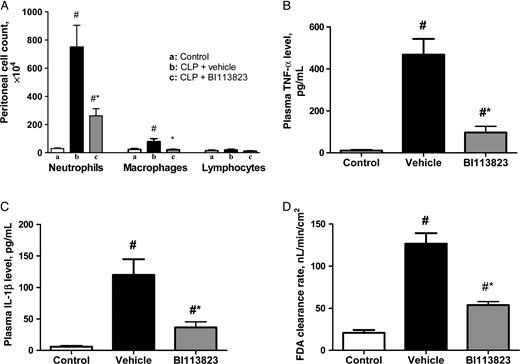
Kinin B1 receptor inhibition with BI113823 reduced cecal ligation and puncture (CLP)–induced peritoneal inflammatory cell accumulation and plasma levels of tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β) and reduced permeability of the intestinal mucosa to the fluorescent macromolecule FD4, compared with vehicle-treated rats, 15 hours following CLP. All values are means ± standard errors of the mean (n = 8). #P < .05 vs the control group; *P < .05 vs the vehicle group.
Lung Inflammatory Injury
The histological analysis by hematoxylin-eosin staining showed that CLP caused destruction of the lung architecture, characterized by interstitial edema and inflammatory cell accumulation at 15 hours following CLP in vehicle-treated rats (Figure 2A). Quantification of lung damage confirmed significantly reduced lung injury scores in rats treated with BI113823, compared with vehicle-treated rats (Figure 2B). Treatment with BI113823 also reduced CD68+ macrophages (Figure 2D and 2E) and neutrophils in lung tissues, as evidenced by reductions in neutrophil elastase-positive staining (Figure 2D and 2E) and MPO activity (Figure 2C). In addition, treatment with BI113823 also reduced lung iNOS expression (Figure 2D and 2E).


Treatment with BI113823 reduced cecal ligation and puncture (CLP)–induced lung inflammatory cell accumulation and lung tissue damage at 15 hours following CLP. A, Representative hematoxylin-eosin (HE) staining of lung sections for neutrophil elastase, CD-68, and inducible nitric oxide synthase (iNOS; original magnification, 20×). B, Histologic score of lung injury. C, Lung myeloperoxidase (MPO) activity. D and E, Representative staining and quantification of lung expression of neutrophil elastase, CD-68, and iNOS. All values are means ± standard errors of the mean (n = 7–8). #P < .05 vs control group; *P < .05 vs the vehicle group.
Liver Inflammatory Responses and Apoptosis
Treatment with BI113823 reduced the inflammatory responses in liver tissues at 15 hours following CLP-induced sepsis, as evidenced by a reduction in MPO activity, CD68+ macrophage infiltration, and TNF-α and IL-1β content (Figures 3 and 4). Severe sepsis also resulted in significant increases in NF-κB activation and NF-κB (p65) nuclear translocation. Administration of BI113823 decreased nuclear NF-κB (p65) binding activity (Figure 3) and reduced cell apoptosis in liver tissues (Figure 4).
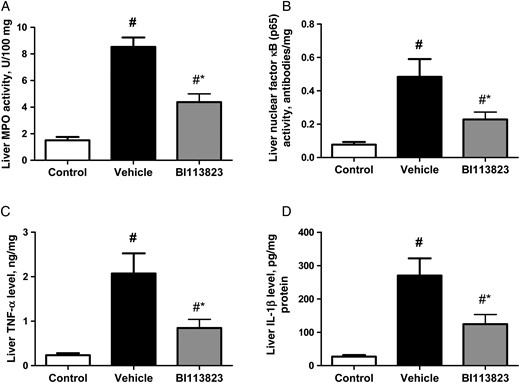
Myeloperoxidase (MPO) activity, nuclear factor κB (p65) binding activity, tumor necrosis factor α (TNF-α) level, and interleukin 1β (IL-1β) level in the liver tissue of sham-operated control rats and septic rats treated with vehicle, compared with those treated with BI113823, 15 hours following cecal ligation and puncture. All values are means ± standard errors of the mean (n = 7–8). #P < .05 vs the control group; *P < .05 vs the vehicle group.
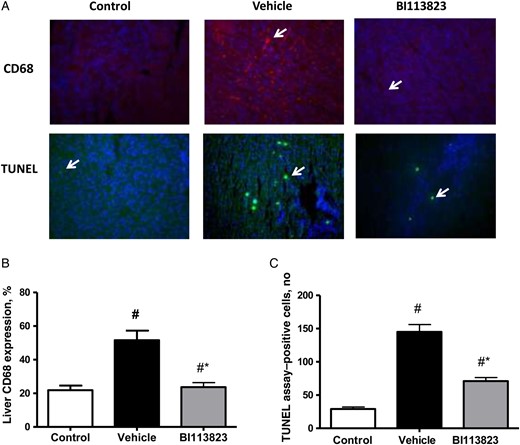
Treatment with BI113823 attenuated iNOS expression and apoptosis at 15 hours following CLP-induced sepsis. A, Representative CD68- and TUNEL-stained liver sections (original magnification, 20×). B, CD68+ macrophages. C, Quantification of TUNEL-positive cells. All values are means ± standard errors of the mean (n = 8). #P < .05 vs the sham control group; *P < .05 vs the vehicle group.
Biomarkers of Multiorgan Injury
Sepsis induced by CLP resulted in significant increases in markers of organ injury, including plasma levels of cardiac-specific troponin-I (a marker of cardiac injury), ALT and AST (markers of liver injury), lactate (a marker for tissue hypoxia), and urea and proteinuria (indicators of impaired kidney function and/or increased catabolism) in vehicle-treated rats at 15 hours following CLP (Figure 5). In contrast, treatment with BI113823 significantly reduced plasma levels of troponin-I, ALT, AST, lactate, urea, and proteinuria following CLP-induced sepsis (Figure 5).
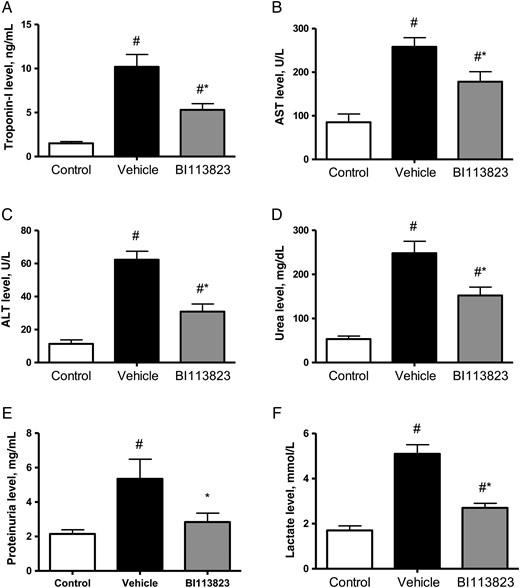
Plasma levels of cardiac troponin-I, aspartate aminotransferase (AST), alanine aminotransferase (ALT), urea, proteinuria, and lactate in sham-operated control animals and septic animals treated with vehicle, compared with those treated with BI113823, 15 hours following cecal ligation and puncture. All values are means ± standard errors of the mean (n = 8). #P < .05 vs the sham control group; *P < .05 vs the vehicle group.
Survival
CLP and cecal excision led to the death of 73% of the vehicle-treated animals (8 of 11), compared with 36% of BI113823-treated animals (4 of 11) at 7 days after the CLP (Figure 6).
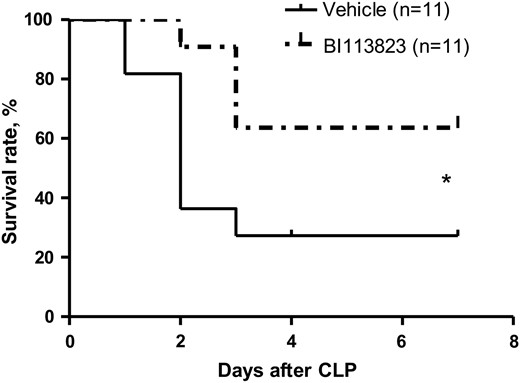
Survival rates 7 days following cecal ligation and puncture (CLP) in septic rats treated with vehicle, compared with those treated with BI113823. *P < .05 vs the vehicle group.
DISCUSSION
This study was undertaken to examine the therapeutic effects of a potent, orally active, small-molecule nonpeptide kinin B1 receptor antagonist, BI113823, in a clinically relevant model of CLP-induced sepsis in rats. We found that oral administration of BI113823 prevented severe sepsis–induced hemodynamic derangement, as well as multiple organ injury, by reducing inflammatory cell infiltration, proinflammatory cytokine production, and cell apoptosis, ultimately resulting in improved survival.
Hemodynamic derangement and cardiac dysfunction are frequent complications associated with severe sepsis and important factors in the high mortality associated with multiorgan failure [21]. Kinins are implicated in the cardiovascular dysfunction induced by endotoxemia [22, 23]. Kinin B2 receptor blockade by CP0127 abolished the development of endotoxin-induced hypotension in rats [22]. In another study, kinin B2 receptor blockade by icatibant blocked the hemodynamic effects of bradykinin but had no beneficial effects on Neisseria meningitidis–induced edema, shock, and inflammation in pigs [24]. Mice deficient in B1 receptors or both B1/B2 receptors are protected from endotoxin-induced hypotension, but mice deficient in B2 receptors are not [23]. Consistent with these findings, the present study found that treatment with BI113823 prevented CLP-induced hemodynamic derangement and improved cardiac output, compared with vehicle treatment. Endotoxin-induced hypotension has been attributed to NO production that is mediated by both inducible and constitutive NOS pathways [25, 26]. Upregulation of iNOS and excessive NO production during endotoxemia contribute to the hypotension seen in endotoxic shock; animals lacking iNOS are protected from this endotoxin-induced hypotension [26]. B1 receptors couple to the Galphai and Galphaq families of G proteins, leading to iNOS-mediated dense and prolonged NO production, thus contributing to hypotension and cellular injury [27]. Consistent with previous reports, treatment with BI113823 reduced CLP-induced upregulation of iNOS in the present study.
A systemic inflammatory response is a pathologic hallmark of severe sepsis and is mediated by innate immune cells, including neutrophils, monocytes, and macrophages [28]. In severe sepsis, uncontrolled recruitment of inflammatory cells into the tissues causes release of inflammatory mediators, including cytokines, oxidants, and proteases, resulting in multiorgan injury [1–3]. Kinin B1 receptors play an important role in mediating the recruitment of leukocytes to sites of inflammation [5–8]. B1 receptors are constitutively expressed in neutrophils, eosinophils, macrophages, and dendritic cells [6–8]. Activation of B1 receptors stimulates leukocyte activation and chemotaxis and synthesis of cytokines and chemokines and increases vascular permeability [5–8]. The excessive and prolonged production of proinflammatory cytokines causes capillary leakage, tissue injury, multiple organ failure, and death in severe sepsis [1–3]. The 5′-flanking region of the B1 receptor gene contains several putative transcriptional regulatory sites, including that for NF-ĸB [29]. NF-κB is a key inflammatory mediator that is activated by the B1 receptor agonist but is also involved in B1 receptor upregulation [30]. B1 receptor inhibition has been shown to inhibit neutrophil accumulation, cytokine production, and NF-ĸB activation; to attenuate leukocyte-endothelial cell interactions; and to reduce tissue inflammatory injury [5–8, 29, 30]. Consistent with these previous reports, the present study found that treatment with BI113823 reduced inflammatory cell infiltration and production of cytokines, including TNF-α and IL-1β. BI113823 treatment also reduced iNOS expression, NF-κB activation, and cell apoptosis; prevented CLP-induced hemodynamic derangement, lung and liver injury, and intestinal mucosal hyperpermeability; and reduced levels of other markers of organ injury, as evidenced by reductions in plasma levels of cardiac troponin-I, ALT, AST, lactate, and urea, as well as proteinuria. Most importantly, treatment with BI113823 improved the survival rate.
In summary, the present study demonstrates that kinin B1 receptor inhibition with BI113823 can protect against severe sepsis–induced hemodynamic derangement, attenuate the inflammatory response, reduce multiorgan injury, and, most importantly, improve the survival rate. These observations support the therapeutic potential of kinin B1 receptor antagonists in the treatment of sepsis.
Notes
Financial support. This work was supported by Boehringer Ingelheim Pharma; the Brain Korea 21 PLUS Project, National Research Foundation of Korea (NRF); and the Korean Ministry of Education, Science and Technology (grant 2012007331 to the Basic Science Research Program, via the NRF).
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}