





Abstract
Mutator alleles, which confer increased mutation rates, are known to spontaneously emerge and “hitchhike” to fixation in evolving asexual populations. Theory predicts that in an evolving asexual mutator population, a second mutator allele may spontaneously arise and hitchhike to fixation. Here, we describe an empirical test of the hypothesis of repeated hitchhiking. The starting population was a clonal strain of mutL−Escherichia coli whose mutation rate was 100-fold higher than wild type. We exposed the mutL− strain to a series of three antibiotics in increasing order of selective strength: fosfomycin, rifampicin, and streptomycin. Two independent replicates of the experiment were performed. As predicted, elevated mutation rates and enrichment for multilocus mutators (which bear more than one mutator allele) were observed in the end point populations of both experiments. DNA sequencing revealed an identical spontaneous 1-bp insertion in the mutator gene mutT in both end point populations. In the multilocus mutators, the causal relationship between the mutT− mutations and the increase in mutation rate was supported with mutT+ plasmid complementation tests. Surprisingly, when the experiment was repeated with the antibiotics deployed in decreasing order of selective strength, enrichment for multilocus mutators was not observed. Our data support the likelihood that the mutT− mutations rose to fixation in both populations, consistent with the hypothesis of repeated mutator hitchhiking. The escalation of mutation rates in asexual populations is relevant to multiple biological scenarios, including antibiotic resistance, host–pathogen interactions, and carcinogenesis.
Introduction
Genomic mutation rates are affected by numerous genetic loci involved in replication, proofreading, repair, and other aspects of nucleic acid metabolism (Lynch, 2010) and thus can evolve through the action of natural selection and other evolutionary forces (Hershberg, 2015; Lynch, 2010; Raynes & Sniegowski, 2014). Because the phenotypic effects of mutations are usually deleterious (Fisher, 1930), natural selection is generally expected to favour a consistently low genomic mutation rate (Hershberg, 2015; Lynch, 2011; Sturtevant, 1937). When genetic linkage is tight, however, alleles that raise the mutation rate (mutators) may hitchhike to fixation with beneficial mutations (Chao & Cox, 1983; Good & Desai, 2016; Johnson, 1999; Sniegowski et al., 1997; Taddei et al., 1997). In some scenarios where adaptation is hindered by mutational supply, higher mutation rates are especially likely to spread in populations because mutators are more likely to generate the rare beneficial mutations that are necessary for adaptation (Denamur & Matic, 2006). Numerous studies have explored the relationship between mutator hitchhiking and adaptive evolution in asexual populations (Denamur et al., 2002; Gross & Siegel, 1981; LeClerc et al., 1996; Oliver et al., 2000; Raynes & Sniegowski, 2014). Evolutionary reductions in genomic mutation rate following mutator-hitchhiking events are expected in well-adapted populations and have been observed in some experimental systems (Turrientes et al., 2013; Wielgoss et al., 2013).
Theory, however, predicts that mutation rate evolution in an asexual population undergoing continual and strong adaptive evolution can be consistently biased towards higher values as a result of successive mutator-hitchhiking events (André & Gerrish et al., 2006; Godelle, 2006). This theoretical prediction is thus far supported empirically by a limited number of experimental studies. It has been demonstrated that co-propagation of engineered “single-mutator” and “double-mutator” strains of E. coli resulted in the substitution of the double mutator (with its higher mutation rate) into large populations, if the double mutator was initially above a critical number relative to the single mutator (Gentile et al., 2011). Furthermore, computational simulations suggest that mutator hitchhiking in asexual populations may not even depend on initial mutator frequency; mutators may always be favoured, relative to their likelihood of fixing in the population via genetic drift (Raynes & Weinreich, 2019). Additionally, it has been shown that in a single-mutator E. coli population evolving in a limited glucose environment for hundreds of generations, a second mutator allele spontaneously emerged in at least one clone, though it was not present across the entire population (Kinnersley et al., 2014). In a mutation accumulation experiment of double-mutator E. coli, several replicate populations went extinct, and of these, most of them had evolved even higher mutation rates prior to extinction (Singh et al., 2017). However, the surviving populations had mostly lowered mutation rates. It should be noted that there was extreme bottlenecking that intensified the accumulation of mutations. Accordingly, there may have been pronounced selective pressure to decrease the mutational load, and the effective population sizes were so small that the mutators had only minimal advantage at quickly gaining beneficial mutations, reducing the likelihood of mutator hitchhiking (Singh et al., 2017).
It remains unaddressed whether a spontaneously arising mutator, initially present in a single or very few individuals, could hitchhike into a population already fixed for one mutator allele. Here, we report a test of that question. We propagated mutator populations of E. coli through three independent rounds of strong selection mediated by lethal concentrations of antibiotics, investigating the genomic mutation rates of the evolved bacteria via genetic and genomic analysis (Figure 1). We observed the evolution of a multilocus (double-mutator) genotype in two independent populations. Interestingly, the second mutator allele arose in the exact same site in the same genetic locus in both populations. We discuss the implications of our findings and avenues for further investigation.
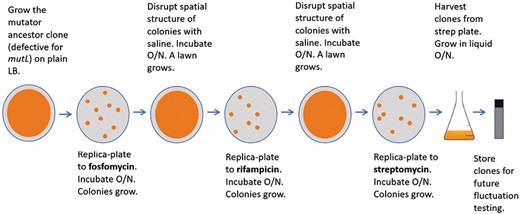
Overview of the antibiotic selection experimental protocol.
Materials and methods
Ancestral strain
We acquired E. coli K-12 strain ES568 (= PS2533) from the Yale University E. coli Genetic Stock Center, which we used as the ancestor for the selection experiments. This strain is defective for mismatch repair: It bears the mutL13 allele containing the point mutation A120T. We have previously estimated that the genomic mutation rate of strain ES568 is approximately 100-fold higher than that of wild-type E. coli (Gentile et al., 2011).
Exposure to successive rounds of lethal selection
We exposed populations of the ancestral strain to lethal concentrations of fosfomycin (100 µg/ml), rifampicin (100 µg/ml), and streptomycin (100 µg/ml) on lysogeny broth (LB) agar plates (Miller, 1972) in succession as follows (Figure 1): we inoculated the ancestor from frozen stock into liquid LB (without antibiotics) and incubated it overnight at 37 °C with shaking at 120 rpm. We then spread 100 µl (~108 cells) of the resulting culture on a single LB agar plate and incubated it overnight at 37 °C, yielding a lawn. We replica-plated the lawn to an LB plate containing fosfomycin, which we incubated overnight at 37 °C. We suspended the resulting fosfomycin-resistant colonies in 150 µl of sterile saline diluent pipetted directly onto the plate, and we spread the cell suspension across the same plate. We incubated the plate overnight at 37 °C again, resulting in a lawn of fosfomycin-resistant cells. We then replica-plated this lawn to an LB–rifampicin plate, and the process was repeated as described above, with the resulting lawn of rifampicin-resistant cells being replica-plated to an LB–streptomycin plate. In our main experiment, we used the three antibiotics in increasing order of their observed selective strength (spontaneous acquisition of fosfomycin resistance is more common than rifampicin resistance, which is more common than streptomycin resistance) (Supplementary Table S4). We chose this order to maximize the numbers of surviving resistant mutants after the first two rounds of selection. In a subsequent experiment, we deployed antibiotics in decreasing order of selective strength: streptomycin, rifampicin, and fosfomycin. Resistances to these three antibiotics are conferred through biochemically distinct modes of action (Goldstein, 2014; Nilsson et al., 2003; Springer et al., 2001).
We randomly sampled colonies representing resistant clones arising at all stages of the experiments from the antibiotic plates (if a given plate contained very few colonies, all colonies were sampled). We inoculated the colonies into liquid LB, incubated them overnight, and archived the resulting cultures in 15% glycerol at −80 °C.
We confirmed the relative selective strengths for each of the three antibiotics (Supplementary Table S4) as follows: We dispensed 100 µl of liquid LB ancestral culture on each LB agar plate. We plated each subsequent lawn on a single-antibiotic plate, with three replicate plates for each of the three antibiotics. We counted one quadrant of each fosfomycin plate, due to the dense but relatively even distribution of colonies. We then multiplied counts for each fosfomycin plate quadrant by 4 to obtain the numbers in Supplementary Table S4.
Mutation rate estimation
We measured mutation rates to nalidixic acid resistance via a modified Luria–Delbrück fluctuation assay (Gerrish, 2008; Luria & Delbrück, 1943) in the evolved clones, the ancestor, and clones transformed with plasmids. Resistance to nalidixic acid can be conferred through mutations in gyrA and parC (Nakamura et al., 1989; Saenz et al., 2003), which prevent nalidixic acid from binding to DNA gyrase and topoisomerase IV (Saenz et al., 2003). As a control, we always measured the mutation rate of the ancestor alongside those of the evolved and transformed clones. For each assay, we inoculated 10 ml of liquid LB from a frozen stock of the clone of interest, which we incubated overnight at 37 °C with shaking at 120 rpm. We then diluted the resulting culture 100,000-fold, and we transferred 100-µl aliquots (representing approximately 1,000 cells) of this dilution to each of a set of replicate flasks (three replicates for the evolved and transformed clones and six replicates for the ancestor), each containing 30 ml of LB. We incubated the resulting cultures for 48 hr at 37 °C with shaking at 120 rpm. To enumerate mutants, we spread 100 µl from each 30-ml culture on an LB plate containing 25 µg/ml of nalidixic acid. To estimate the total population sizes of the cultures, we diluted another 100 µl from each culture 1,000,000-fold, and we spread 300 µl of this dilution on an LB plate without nalidixic acid. We incubated all plates at 37 °C. We enumerated colonies on both sets of plates at 24 hr. To account for the possibility of slowly growing resistant mutants, we incubated the LB–nalidixic acid plates for another 24 hr, after which we again enumerated the colonies. The inferred total number of mutants in each culture never approached the number expected if a pre-existing mutant had been seeded into the culture (see Supplementary Table S3), indicating that all observed mutants originated from mutations that occurred during culture growth as assumed by the fluctuation assay.
We calculated maximum likelihood mutation rates and associated 95% confidence limits from the 48-h LB–nalidixic acid counts and the 24-h permissive LB counts via a programme kindly provided by Dr. Philip Gerrish (Gerrish, 2008; Sniegowski, 2022). We inferred the significance of mutation rate differences from non-overlap of 95% confidence limits (Zheng, 2015).
Genomic sequencing
We extracted genomic DNA for sequencing from two random clones (designated FRS13 and FRS16, where “FRS” refers to “fosfomycin, rifampicin, streptomycin”) that survived exposure to all three antibiotics in the first replicate series (concluding with streptomycin) using the QIAGEN DNeasy Blood & Tissue Kit (69504) following the manufacturer’s instructions. The University of Pennsylvania’s Next Generation Sequencing Core performed library preparations and genomic sequencing using the Illumina Nextera XT DNA library preparation kit (FC-131-109), the Illumina Nextera XT index kit (FC-131-1002), and Illumina NextSeq (150 paired-end) for a mean 325-fold coverage. We detected mutations using the computational pipeline breseq version 0.27 (Deatherage & Barrick, 2014). We aligned the sequences of the evolved clones against the reference sequence, E. coli K-12 MG1655 (NC_000913.3, downloaded from https://www.ncbi.nlm.nih.gov/nuccore/NC_000913.3?report=gbwithparts&log$=seqview on July 15, 2016).
Complementation tests
We rendered cell competent as follows: liquid inocula of the single-mutator ancestor, the evolved clone FRS13, and the evolved clone FRS16 were centrifuged for 10 min at 13,000 rpm at 4 °C. We discarded the supernatants, resuspended the cells in 100 mM MgCl2, and incubated them on ice for 20–30 min. We then centrifuged cells for 10 min at 7,000 rpm at 4 °C. We discarded the supernatants and resuspended the cells in 100 mM CaCl2 with 15% glycerol, before freezing them at −80 °C.
After rendering the cells competent, we transformed the cells, plus a positive control (ThermoFisher Scientific E. coli Subcloning Efficiency DH5α Competent Cells), with a mutT+ampR plasmid and an empty ampR plasmid, yielding a total of eight transformant groups. We isolated the mutT+ plasmid pSK25 (Bhatnagar & Bessman, 1988) from a strain of REL606 E. coli (Jeong et al., 2009) via a QIAprep spin miniprep kit from QIAGEN (product number 27104). We purchased the empty plasmid pBR322 from ThermoFisher Scientific (product number SD0041).
We conducted the transformations as follows: Competent cells were thawed on ice and incubated in the presence of plasmid DNA for 30 min, then heat-shocked for 20 s in a 42 °C water bath, and then incubated on ice for 2 min. We added 950 µl of liquid LB to each transformation reaction before incubating the reactions at 37 °C for 1 hr at 120 rpm. We plated the transformation reactions on LB containing ampicillin at a concentration of 50 µg/ml. We harvested two colonies from each ampicillin plate, inoculated the colonies into liquid LB-amp, and incubated them overnight. The resulting cultures were archived in 15% glycerol at −80 °C. We measured the mutation rates of the transformants via fluctuation assays using nalidixic acid as the selective agent; all LB-based media contained ampicillin.
Polymerase chain reaction and Sanger sequencing
We extracted genomic DNA for sequencing from all surviving clones of the FRS-triple selection series using the QIAGEN DNeasy Blood & Tissue Kit (69504) according to the manufacturer’s instructions. We quantified DNA concentration with a Qubit fluorometer. We designed primers for the mutT repeat region of interest as follows: forward, GCGCACATGGCGAATAAAC; reverse, TTCATTGGCTGGCGGAAA. We designed primers for the mutL gene as follows: forward, CAGCAACAACAGCGAAGAA; reverse, CGGCCCCATCAAAAAAAT. We assembled each polymerase chain reaction (PCR) as follows: 25 µl iProof master mix, 18 µl nuclease-free water, 1 µl 10 µM respective (mutT or mutL) forward primer, 1 µl 10 µM respective reverse primer, and 5 µl of 10 ng/µl DNA extraction. The mutT PCR thermocycler program was 98 °C for 3 min, then 30 rounds of the following: 98 °C at 10 s, 55 °C at 30 s, and 72 °C at 10 s; the final extension period was 72 °C for 5 min. The mutL program was 98 °C for 3 min, then 30 rounds of the following: 98 °C for 10 s, 54 °C for 30 s, and 72 °C for 2 min 5 s; the final extension period was 72 °C for 5 min.
After PCR was completed, we assembled each PCR sample clean-up as follows: 17 µl of PCR sample, 0.3 µl Exonuclease I, 0.3 µl Antarctic phosphatase, and 2 µl nuclease-free H2O, which we ran in a PCR thermocycler for 15 min at 37 °C, then 15 min at 80 °C. We then submitted cleaned-up PCR samples for Sanger sequencing and viewed the chromatograms with the software Chromas.
Results
As shown in Figure 2A, genomic mutation rates were approximately 10 times higher than those of the mutator ancestor in all clones sampled from two experimental E. coli populations that were exposed successively to lethal levels of fosfomycin, then rifampicin, and then streptomycin. Surviving clones were sampled from the final plates containing streptomycin: In one population, numerous clones survived on the final plate and nine random clones were sampled, and in the second population, only one clone survived on the final plate and this was sampled (see Supplementary Table S1 for mutation rate values). Genomic sequencing of two random clones (FRS13 and FRS16, where “FRS” refers to the antibiotic order “fosfomycin, rifampicin, streptomycin”) from the first population revealed new mutations that were mostly non-synonymous (Supplementary Figure S1, Supplementary Tables S2), including a second mutator allele in addition to the original mutL mutation: A frameshift single-base insertion of cytosine in a 6-bp cytosine repeat region of mutT, a strong mutator locus (Bhatnagar & Bessman, 1988) (see Supplementary Figure S2). As shown in Figure 2B, the expression of wild-type MutT in these two clones restored the ancestral mutation rate, confirming the role of the new mutT allele in causing the increased mutation rates. Subsequent PCR and Sanger sequencing of all remaining clones sampled across the two experimental populations showed that each had a 1-bp cytosine insertion in the same repeat region of mutT. The original mutL− mutator mutation was retained in all 10 surviving clones sampled across the two experimental populations.
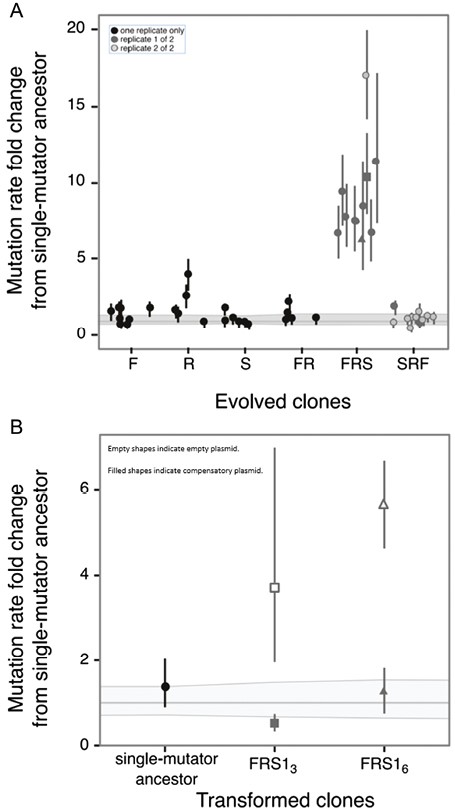
(A, B) Mutation rates to nalidixic acid resistance for all measured clones. The thick horizontal grey line represents the ancestral mutation rate to nalidixic acid resistance relative to itself; the thin lines above and below it represent the 95% confidence limits on the ancestral mutation rate. Error bars on the individual points represent 95% confidence limits on the mutation rates obtained from fluctuation assays, normalized by dividing each by the ancestral mutation rate. (See Supplementary Table S1 for absolute mutation rate and confidence limit values.) (A) Mutation rates in clone postantibiotic exposure, plotted relative to the mutation rate of the single-mutator ancestor. F = fosfomycin exposure only; R = rifampicin only; S = streptomycin only; FR = fosfomycin, then rifampicin; FRS = fosfomycin, then rifampicin, then streptomycin; SRF = streptomycin, then rifampicin, then fosfomycin. The data include numerous different groups of fluctuation assays in which the ancestor was always measured alongside the evolved clones. Although the ancestral mutation rate estimate was approximately constant, its confidence intervals varied across assays. For each treatment (F, R, S, etc.), the 95% confidence limits plotted for the ancestral mutation rate represent the highest and lowest values obtained from the relevant group of assays. For FRS and SRF, the shading difference indicates clones isolated from two different replicate experiments. In FRS, the square and triangle represent two clones (FRS13 and FRS16, respectively; see Appendix) whose genomes were sequenced. (B) Transformation with a mutT+ plasmid restores the ancestral mutation rate in clones that had evolved elevated mutation rates. Here the thick grey line represents the mutation rate of the ancestor transformed with an empty control plasmid. Filled circle represents mutation rate in the ancestral clone transformed with a mutT+ plasmid. Squares and triangles represent mutation rates (relative to the ancestral rate) in two evolved clones with elevated mutation rates that were both found to harbour a mutT frameshift. Filled square and triangle represent mutation rates after transformation with mutT+ plasmid; open square and triangle represent mutation rates after transformation with empty control plasmid.
To test experimentally to what degree one round of lethal selection resulted in further increases in mutation rate, we characterized the mutation rates of randomly sampled clones surviving exposure to fosfomycin only (eight clones), rifampicin only (five clones), and streptomycin only (six clones). The sampled clones represent a fraction of the survivors on the respective plates (refer to Supplementary Table S4). Two of these 19 single-exposure clones (both from the single-rifampicin exposure) displayed statistically significant increases in mutation rate compared with the mutator ancestor (Figure 2A; Supplementary Table S1). To test to what degree two rounds of lethal selection resulted in increased mutation rates, we characterized the mutation rates of five random clones surviving exposure to fosfomycin and then rifampicin. One clone showed a statistically significant increase in mutation rate compared with the mutator ancestor (Figure 2A; Supplementary Table S1). Finally, to test whether the order of exposure to all three antibiotics affected the prevalence of increased mutation rates, we characterized mutation rates of randomly sampled clones surviving sequential exposure to the same three antibiotics, but in reverse order, from greatest to least selective strength: streptomycin, rifampicin, and fosfomycin. This reverse-order series was repeated for a total of two experimental replicates. Nine clones were randomly sampled from the final plate of the first replicate, and two clones were randomly sampled from the final plate of the second replicate. (Both of the final plates contained hundreds of clones, so the sampled clones represent a fraction of the total survivors.) One of these 11 reverse-order clones displayed a statistically significant change in mutation rate compared with the mutator ancestor (Figure 2A; Supplementary Table S1). We conclude that exposing an asexual mutator population to a series of antibiotics in increasing order of selective strength can result in the enrichment of spontaneously arising multilocus mutators. However, if the antibiotic order is reversed, enrichment for multilocus mutators fails to occur.
Discussion
We sought to test the existing theoretical prediction of repeated mutator hitchhiking in an asexual evolving mutator population, which we hypothesized would occur under multiple rounds of strong selection via lethal concentrations of antibiotics. We initially deployed the antibiotics in increasing order of selective strength to maximize the number of survivors after the first two rounds of selection. Unexpectedly, the order of the antibiotics affected the outcome of enrichment of multilocus mutators. In the experiments in which antibiotics were administered in increasing order of selective strength, we observed the evolution of a multilocus mutator (bearing more than one mutator allele), consistent with the theoretical prediction. The most likely explanation for the observed enrichment of multilocus mutators is that spontaneous mutT− mutator mutants (multilocus mutators) became successively enriched in the populations through their higher probability of producing mutants resistant to the three antibiotics. If the antibiotic with the lowest selective strength, fosfomycin, is first in the series, it will lead to a large initial pool of survivors, with a correspondingly higher chance for additional mutator alleles and antibiotic resistance mutations. However, if the first antibiotic has the highest selective strength, streptomycin, then the initial pool of survivors will be smaller.
Overall, our results indicate that it is indeed possible for successive rounds of lethal selection to result in the enrichment of an asexual mutator population with genotypes bearing additional mutator alleles. Due to the stochastic nature of the emergence of mutators and the limited size of our experiment, it remains to be seen whether there would be consistent enrichment for additional mutator alleles each time the starting population is exposed to the selective gauntlet. Very strong selective pressures, such as the lethal concentrations of antibiotics in our experiments, can favour mutators (Matic, 2019; Raynes & Sniegowski, 2014), in this case, due to the higher likelihood of antibiotic resistance mutations emerging in the genome. Though the use of lethal selection in our experiments may seem to be an artificially strong circumstance favouring mutators, there are reasonable scenarios in which natural populations will encounter repeated lethal or very strong selection—most notably, through host–pathogen interactions and coevolution (Graves et al., 2013; Pal et al., 2007). Whether any natural populations under such strong selection have accumulated multiple mutator alleles remains an interesting avenue for further study. Theoretical work, moreover, predicts that there is a genomic mutation rate above which fitness should decline due to the overwhelming burden of prevailingly deleterious mutations (Bull & Wilke, 2008; Gerrish et al., 2013; Sprouffske et al., 2018), and results of some recent experimental evolution work are broadly consistent with this prediction (Sprouffske et al., 2018). However, again, observations of this kind are lacking in natural populations.
Our investigation explored the effects of lethal selection on repeated mutator hitchhiking, where the need to elude antibiotics outweighs the fitness defects typically associated with drug resistance mutations (Matic, 2019). However, lethal selection is not the only evolutionary scenario worth considering. Further investigation of repeated mutator hitchhiking is needed in scenarios where there is less of a pressing need for bacteria to acquire highly specific alleles, e.g., mutations that confer antibiotic resistance. In ongoing work, we are testing whether non-lethal selection can cause a rapidly adapting asexual mutator population to substitute additional spontaneously originated mutators that reach fixation, rather than remaining as merely polymorphisms (Kinnersley et al., 2014). A logical further step would be to test whether rapidly and continually adapting populations that are initially fixed for low, wild-type mutation rates can substitute multiple mutator alleles, as predicted by theory (Gerrish et al., 2007). Simulations of this process under non-lethal selection, however, suggest that it is likely to take a prohibitively long time to unfold in the laboratory (Gerrish et al., 2007). Using repeated rounds of lethal selection would be quicker but is potentially limited by practical population sizes and the number of lethal selective conditions to which cells would not exhibit cross-resistance.
Interestingly, the frameshift producing the new mutT mutator in our populations lies in the same cytosine string as a mutT mutation that spread in a bacterial population that had evolved experimentally for 20,000+ generations (Barrick et al., 2009). Additionally, an indel in the same repeat region of mutT emerged in a mutation accumulation experiment (Lee et al., 2012). These parallels are consistent with the higher mutation rate expected in such repeated sequences (Shaver & Sniegowski, 2003). One potential explanation for our observations is that hypermutable repeat regions in bacterial mutator loci are maintained by natural selection as an adaptive mechanism for readily switching between high and low mutation rates. This hypothesis has been discussed previously (Field et al., 1999; Moxon et al., 1994), but it is not the sole potential explanation for these results, as genetic drift has been proposed as an alternate explanation (Shaver & Sniegowski, 2003).
Our results may have some biomedical implications in addition to their relevance to the general understanding of mutation rate evolution and the fate of asexual populations. Mutator lineages have been identified in pathogenic bacterial populations (Colque et al., 2020; LeClerc et al., 1996; Lindgren et al., 2016; Oliver et al., 2000; Rees et al., 2019) and are also implicated in certain forms of cancer, both in clinical studies (Loeb & Loeb, 2000) and mathematical models (Solé & Deisboeck, 2004). Because the spread of cancer can be conceived of as a process of clonal evolution (Sprouffske et al., 2012), hitchhiking of mutator alleles is a plausible explanation for these observations. Both therapeutic intervention and the lethal, relentlessly specific host immune response in pathogen infections and cancers could conceivably result in the substitution of additional mutator alleles into such populations, as shown by our results. There are two contrasting implications of such hypothetical progressive elevation of the mutation rate in clinical contexts. On one hand, pathogens or tumours with extremely high mutation rates might be highly treatment resistant because they easily acquire further resistances (Sprouffske et al., 2012). On the other hand, additional mutagenesis could potentially lead to the demise of such populations via the influx of deleterious mutations. The latter possibility has been considered for RNA viruses, which already tend to have extraordinarily high genomic mutation rates (Drake & Holland, 1999; Vignuzzi et al., 2005).
Data availability
The data that support the findings of this study are available in Dryad at https://datadryad.org/stash/share/Ba3C1EdEBSe-ljjV9nAPBw3zuoAC5Cib1kIcSvUpbG0.
Author contributions
Mitra Elgrail (Conceptualization [Lead], Formal analysis [Lead], Investigation [Lead], Methodology [Lead], Visualization [Lead], Writing—original draft [Lead]), Kathleen Sprouffske (Formal analysis [Supporting], Methodology [Supporting], Visualization [Supporting], Writing—review & editing [Supporting]), Jude Dartey (Investigation [Supporting], Methodology [Supporting]), and Arlene Garcia (Investigation [Supporting])
Funding
This work was supported by National Aeronautics and Space Administration (NASA) (grant NNA15BB04A).
Acknowledgments
We gratefully acknowledge Paul Sniegowski for providing funding, mentorship, research materials, workspace, and comments on the manuscript. We also thank Andreas Wagner for sharing the ancestral genomic sequence (PS2533), Philip Gerrish for quantitative discussions, and Matthew Culyba for comments on the manuscript.
Conflicts of interest
K.S. is to disclose employed at Novartis. M.E., J.D., and A.G. have no conflicts of interest.

{kind=link}
{kind=link}
{kind=link}