





Abstract
Secondary sexual traits can convey information on mate quality with the signal honesty maintained by the costly nature of trait expression. Mating signals are also often underpinned by physiological, morphological, and behavioural adaptations, which may require the evolution of novelty, but the genetic basis in many cases is unknown. In orchid bees, males acquire chemical compounds from the environment that act as pheromone-like bouquets (perfumes) during courtship displays. This process could be costly, potentially due to the cognitive demands of learning and the physiological demands of collecting a mix of extrinsic chemical compounds that may require detoxification. Furthermore, a novel trait, a specialized perfume pouch in the hind leg, is required for compound storage. We studied gene expression in the brain, hind leg, and Malpighian tubules—a tissue involved in detoxification—to investigate changes in gene expression following perfume collection. We detected upregulation of genes enriched in functions related to transcription, odorant binding, and receptor activity in the Malpighian tubules. On the other hand, we did not find any evidence for learning processes following perfume collection, or gene expression changes in the hind leg, perhaps due to constitutive expression, or the age of the sampled bees. We did identify high expression of chemosensory proteins in the hind legs, which we suggest could play a role in perfume collection or storage, with further functional studies necessary to determine their binding properties and potential physiological importance.
Los rasgos sexuales secundarios pueden servir como indicadores de calidad de la pareja, y en algunos casos la honestidad de la señal se mantiene por el costo de expresar el rasgo. A menudo las señales sexuales están respaldadas por adaptaciones fisiológicas, morfológicas y de comportamiento por lo tanto pueden requerir la evolución de nuevos rasgos, pero en muchos casos se desconoce la base genética. En las abejas de las orquídeas, los machos recolectan compuestos químicos del medio ambiente, los cuales actúan como feromonas (perfumes) durante el despliegue de cortejo. Este proceso podría ser costoso, posiblemente debido a las demandas cognitivas del aprendizaje y las demandas fisiológicas de recolectar una mezcla de compuestos químicos extrínsecos que pueden requerir desintoxicación. Además, se requiere la evolución de un contenedor para almacenar perfumes en la pata trasera. Para investigar los cambios en la expresión génica después de la recolección de perfume, estudiamos la expresión génica en el cerebro, la pata trasera y los túbulos de Malpighi (tejido involucrado en la desintoxicación). Encontramos varios genes regulados positivamente en los túbulos de Malpighi después de la recolección que están enriquecidos en factores de transcripción, proteínas de fijación de olores, y proteínas con actividad de receptor. Por otro lado, no encontramos ninguna evidencia de procesos de aprendizaje posteriores a la recolección de perfumes, o cambios en la expresión génica en la pata trasera, esto quizás debido a la expresión constitutiva o la edad de las abejas muestreadas. Además, identificamos una alta expresión de proteínas quimio-sensoriales en las patas traseras, que podría desempeñar un papel en la recolección o almacenamiento de perfumes. Más estudios funcionales son necesarios para determinar las propiedades de fijación de las proteínas y su potencial importancia fisiológica.

Introduction
Secondary sexual traits evolve under sexual selection due to competition for access to mates. Sexual selection could result either from direct competition among members of the same sex (intrasexual competition) or via mate choice imposed on members of the opposite sex (intersexual selection) (Darwin, 1871; Shuster & Wade, 2003). This process can result in the evolution of elaborate secondary sexual traits, such as chemical signals, bright colour patterns, and elaborate vocalizations that play a central role in mate choice (Andersson, 1994; Andersson & Simmons, 2006).
Secondary sexual traits can act as honest indicators of mate quality (Andersson, 1986; Zahavi, 1975, 1977). If a signal is costly to produce, this can enforce honesty. The costliness of the signal can be driven by various factors. The trait itself could be metabolically costly to develop (Nijhout & Emlen, 1998) or maintain (Somjee et al., 2018). The trait might require effort or time that could be allocated to other functions, for example, foraging for dietary components of a pheromone signal, or dietary-derived pigment (Conner et al., 1981; Hill, 1992; Hill et al., 2002; Landolt & Phillips, 1997). The cognitive demands of the trait could also be high, for example, if learning is required, as is the case in bird song (Byers & Kroodsma, 2009; Verzijden et al., 2012) and bower construction in bowerbirds (Day et al., 2005). The trait might also have negative impacts on the signaller outside of the mating context, for example, by attracting predators (Legett et al., 2019). Many well-studied examples combine multiple aspects of costliness. For example, bird coloration is linked to that ingestion of dietary-derived carotenoids, ensuring that it is a reliable indicator of nutritional condition, foraging ability, and more (Brush, 1990; Hill, 1992, 2011; Hill et al., 2002).
The evolution of a complex mating signal is underpinned by physiological, morphological, and, in many cases, behavioural adaptations. Some of these traits might evolve through the co-option of existing traits, for example, the ability to forage, while others may require the evolution of novelty. An outstanding question in evolutionary biology is which types of genes are more likely to be involved in the evolution of novelty. For a long time, the idea of a “shared toolkit” has prevailed in evolutionary developmental biology, which emphasizes the diversity of organisms produced by the same developmental regulators across animals (Wilkins, 2013). However, many genes are not shared across clades, and more recently, taxonomically restricted genes have been found to play a crucial role in the evolution of a range of traits (Wu & Lambert, 2023), including the evolution of courtship in Drosophila (Dai et al., 2008).
A fascinating example of an elaborate mating signal that requires a suite of adaptations is that of the orchid bees, a group found in the lowlands of tropical America that acquire their mating signals from the environment. Male bees collect chemical compounds from sources such as flowers and fungi, which they release during mating displays as pheromone-like bouquets (perfumes) (Dressler, 1982; Eltz et al., 1999; Eltz, Sager, et al., 2005; Vogel, 1966). Perfume collection has been directly linked to male reproductive fitness, where males that contain a full set of perfume compounds in hind legs attract more females and sire more offspring than males without perfume access (Henske et al., 2023). Perfume composition differs drastically between species and also exhibits individual-level variation with the same species, potentially contributing to reproductive isolation and mate choice (Eltz et al., 1999; Pokorny et al., 2017; Weber et al., 2016; Zimmermann et al., 2009).
Perfume compound collection involves behavioural, morphological, and physiological adaptations in a suite of specialized tissues. Males first release long-chain aliphatic compounds (lipids) from their labial glands onto the collection surface, such as a flower or fungus (Whitten et al., 1989). The mixture of lipids and volatile compounds is then transferred using fore-tarsal brushes to specialized hind leg pouches (Kimsey, 1984; Vogel, 1966). The hind leg pouches are swollen cuticular pockets filled with a sponge-like structure made of numerous modified hairs that provide a large surface area for compound absorption and retention, which acts as a storage container for chemical compounds (Eltz et al., 1999, 2019; Vogel, 1966). While the perfume compounds remain stored in the hind leg, the labial lipid compounds are actively recycled and relocated to the head glands for reuse (Eltz et al., 2007, 2019). Thus, the male hind leg pouch corresponds to a novel organ that is linked to the unique biology of orchid bees.
While many tests for signal costliness have focused on the metabolic costs of signal production or foraging, perfume collection by male orchid bees is an excellent system in which to test other aspects of signal costliness. By collecting a large variety of volatile compounds, male bees experience high chemical exposure to potential toxins. It has been suggested that compound collection acts as an indicator of male quality by indicating that they can withstand these toxins (Arriaga‐Osnaya et al., 2017; Eltz et al., 1999). The evolution of compound collection and, therefore, the necessity to handle such a wide variety of potentially toxic chemical compounds may have driven the evolution of the detoxification machinery of orchid bees. The P450 gene family encodes the largest group of enzymes involved in exogenous compound detoxification in diverse lineages across the tree of life. No significant expansion in the P450 gene family was observed in the orchid bee Euglossa dilemma (Darragh et al., 2021) relative to bees from other bee families. However, evolution of gene expression rather than gene family size could be associated with perfume collection in orchid bees.
Another challenge of perfume collection, cognitive in nature, is how orchid bees collect and maintain species-specific perfume blends (Weber et al., 2016; Zimmermann et al., 2006). Male bees that belong to the same species exhibit species-specific antennal responses to different compounds, suggesting that innate odour preferences contribute to the species-specific nature of blends (Brandt et al., 2021; Mitko et al., 2016). However, some species also exhibit a large overlap in the compounds to which they are attracted, with multiple unrelated species attracted to the same chemical compound (Ackerman, 1989; Pearson & Dressler, 1985). One mechanism that could assist in the maintenance of species-specific perfumes is if bees learn and retain information on which chemical compounds they collect, resulting in learned avoidance to prevent compound over-collection (Eltz, Roubik, et al., 2005). This avoidance response is only displayed by bees that actively collect compounds, suggesting that this behaviour stimulates a neural mechanism of avoidance. However, the physiology of this mechanism is unclear. For example, the compounds that have been collected could be detected via sensory neurons in the hind leg pocket or in the antennae. If memory formation is involved in the acquisition of species-specific perfumes, we would expect to observe changes in gene expression in the brain in response to learning and memory formation (Geng et al., 2022; Guo et al., 2016).
Sexual selection is thought to be a driving force in shaping the evolution of gene expression (Harrison et al., 2015). In this study, we investigate the hypothesis that perfume collection in orchid bees is costly, both in terms of a physiological response to toxicity and a cognitive response to learning. To do this, we study patterns of gene expression in three different tissues: the brain, the hind leg, and the Malpighian tubules—an internal organ involved in detoxification (Nocelli et al., 2016). We hypothesize that changes in gene expression in the brain are expected to be related to learning, changes in the Malpighian tubules to detoxification, and changes in the hind leg to compound detection, detoxification, and lipid recycling. Furthermore, we investigate the evolution of the unique hind leg organ in Eg. dilemma by examining the patterns of tissue-specific gene expression, and also by comparing the genes that are expressed in the hind leg of Eg. dilemma and those expressed in the legs of the closest bee lineage that lacks this specialized organ, Apis cerana.
Materials and methods
Sample collection
We nest-trapped Eg. dilemma bees around Fort Lauderdale, FL, using small (10 × 8 × 3 cm) wooden boxes. Once females built brood cells in these nests, the boxes were transported to the Campus of the University of Florida (UF) Ft. Lauderdale Research and Education Center in Davie, FL and kept in clear plastic boxes with holes until adults emerged. We checked boxes for newly emerged bees daily and placed them in 1-mm mesh cages of 181 × 181 × 181 cm containing branches for perching. Bees were initially placed on artificial flowers containing a solution of sugar water (sucrose dissolved in water 50% w/v) that was changed daily. Bees were left in the cage for 2 days to acclimate to the conditions prior to experimentation. On the experiment days, male bees were given access to either 1 ml of ethanol or dimethoxybenzene (DMB) (0.1 g/ml dissolved in ethanol) on filter paper attached to the side of the cage. DMB is commonly used, both in Florida and elsewhere, to attract orchid bees, and therefore, we chose it as a compound that is highly likely to induce perfume collection behaviour (Ramírez et al., 2010, 2015). We also placed a small fan to blow air on the filter paper, providing a stream of odour towards the centre of the cage to induce perfume collection. Bees were exposed to DMB for 2 hr from 9 a.m. until 11 a.m. and flash frozen in liquid nitrogen for 24 hr after their final collection, which in all cases occurred between 9 a.m. and 10 a.m. Based on these observations, control bees were exposed to ethanol for 1 hr between 9 a.m. and 10 a.m. and flash frozen at 10 a.m. on the following day. Frozen bees were stored for up to 1 week in a liquid nitrogen dry shipper before being transferred to a −80 °C freezer for storage.
Dissections, RNA extraction, and RNA sequencing
Dissections were carried out on dry ice. First, we removed the cuticle surrounding the tissues of interest and placed the tissues in RNAlater ICE for at least 16 hr at −20 °C. Following the thaw in RNAlater ICE, samples were dissected using dry ice to cool the dissecting station and immediately placed in Trizol solution for RNA extraction. The entire brain was included in the dissection, but the ocelli and retina were removed following previous protocols (Saleh & Ramírez, 2019). A standard Trizol extraction protocol was used to extract RNA, with glycogen added to samples to increase yield. RNA extracts were treated with an Invitrogen Turbo DNA-free kit and quantified using a Qubit. Samples were sent to Novogene for final quality control, library preparation, and sequencing. Our final dataset consisted of 11 brain samples (6 = control, 5 = experimental), 11 hind leg samples (6 = control, 5 = experimental), and 8 Malpighian tubule samples (4 = control, 4 = treatment). Samples were sequenced using 150 bp paired-end reads on a NovaSeq 6000. This generated approximately 51 million reads per library (mean = 50.99 million, SD = 8.97 million, N = 30). The raw sequence data were deposited in the SRA under the BioProject accession PRJNA910557.
Differential expression analysis
We analysed both the data generated for Eg. dilemma and previously published gene expression data (PRJNA499671) from three samples of the legs of 1-day-old A. cerana (Du et al., 2019) using the same pipeline. We trimmed the reads using TrimGalore! (Martin, 2011). We then mapped the Eg. dilemma reads to the Eg. dilemma genome (Brand et al., 2017) with a previously published annotation updated with manual annotations of cytochrome P450s and chemosensory loci (Brand et al., 2015, 2017; Darragh et al., 2021). Apis cerana reads were mapped to the A. cerana reference genome (Park et al., 2015). We ran 2pass mapping using STAR (Dobin et al., 2013). Following mapping, we used featureCounts (Liao et al., 2014) to count the reads. The output from featurecounts was read into R for further analysis. We filtered genes to include those with at least one count per million in at least one library and normalized reads by library size using TMM (trimmed mean of M values) (Robinson & Oshlack, 2010).
First, we tested for differences in gene expression between experimental and control treatments for each tissue in Eg. dilemma. Second, we compared the hind legs of Eg. dilemma to the legs of A. cerana. We searched for 1:1 orthologues between the two species using OrthoFinder (Emms & Kelly, 2019) and filtered our datasets to include only these 4,254 orthologous genes before combining the datasets. Differential gene expression was evaluated using the non-parametric noiseqbio function from the NOISeq package (Tarazona et al., 2011, 2015). We used the default cut-off of q = 0.95 in the degenes function, which is equivalent to an adjusted p-value of .05. We removed differentially expressed genes with a logfold change of <2.
Gene ontology analysis
We used data provided from the Ortholog Data Bank (OrthoDB) to gather gene functional annotations (Zdobnov et al., 2021). We combined these annotations from OrthoDB with further annotations from InterPro2GO and Blast2GO to improve the percentage of genes with gene ontology (GO) annotations in our analysis (Burge et al., 2012; Götz et al., 2008). We imported these functional annotations into R to carry out enrichment analysis with topGO (Alexa & Rahnenfuhrer, 2022). We considered the background to be all genes considered in that specific differential expression analysis after filtering.
Tissue specificity
To calculate tissue specificity of the genes, we first normalized genes using gene length-corrected TMM (GeTMM), an approach that facilitates both inter- and intra-sample comparisons within the same dataset, carried out using R and the package edgeR (Robinson et al., 2010; Smid et al., 2018). We then calculated the tissue mean for each gene across all samples and then transformed the data using log2. Values that were negative were replaced with zero. This removes any values that were <1 before log2 transformation, which removes genes with low support for true expression. We measured the tissue complexity of each tissue by calculating the percentage of total mRNA made up by the 10 most highly expressed genes. To quantify the tissue specificity of each gene, we calculated the τ index for each gene (Yanai et al., 2005) (https://github.com/severinEvo/gene_expression/blob/master/tau.R). The τ index ranges from 0 to 1, with 0 being no specificity to expression and 1 being fully tissue specific. We used a cut-off of 0.8 for a gene to be considered tissue specific.
Selection analyses
The ratio of non-synonymous to synonymous substitutions (dN/dS) is often used to measure the strength and type of selection acting on a gene. To calculate dN/dS for Eg. dilemma genes, we first used OrthoFinder (Emms & Kelly, 2019) to find orthologues between Eg. dilemma and Eufriesea mexicana, the most closely related species with a sequenced genome (Kapheim et al., 2015). We included only genes that had a 1:1 orthologue in each species. We extracted the six thousand three hundred and one 1:1 orthologues from each genome and aligned them to create codon-specific alignments using PRANK (Löytynoja, 2014). We then calculated pairwise dN/dS for each gene using paml (Yang, 2007). We excluded genes with dS < 0.01 and dN > 2 indicating saturation. We tested for differences in mean dN/dS between tissue-specific genes for each tissue using an analysis of variance (ANOVA) test in R. We tested for differences in mean dN/dS between differentially expressed genes and background genes included in the analysis using an ANOVA test in R. These tests can only be carried out for genes with a 1:1 orthology between Eg. dilemma and Ef. mexicana.
Phylostratigraphy analyses
To determine the evolutionary age of each gene in Eg. dilemma, we carried out phylostratigraphy analyses using the R package phylostratr with taxizedb (Arendsee et al., 2019; Chamberlain & Arendsee, 2023). This package searches for homologues of each protein to find the deepest clade in which a homolog is found. This is referred to as the phylostratum of the gene encoding a protein. Five species are included at each ancestral node, which are identified by phylostratr; in addition, we included a standard set of prokaryotes, and we manually added the vinegar fly Drosophila melanogaster, human Homo sapiens, and yeast Saccharomyces cerevisiae. In addition, we included the following bee species representing 3 different bee families: Apis mellifera, Bombus impatiens, Dufourea novaeangliae, Ef. mexicana, Habropoda laboriosa, Lasioglossum albipes, Megachile rotundata, Melipona quadrifasciata, and Osmia bicornis. In total, 167 proteomes were downloaded from UniProt (The UniProt Consortium, 2019), and BLASTp (Altschul et al., 1990) was used to compare the predicted protein sequences from Eg. dilemma against the downloaded proteomes. To test for enrichment between different sets of genes, we used the PlotEnrichment function in the R package myTAI (Drost et al., 2018). PlotEnrichment identifies phylostrata, which are over- or under-represented in the test set when compared with the background using Fisher’s exact tests. We corrected for multiple testing with a false detection rate correction, which controls for the proportion of false positives.
Plotting and data manipulation
Plots were made using cowplot (Wilke, 2020), ggplot2 (Wickham, 2009), and scales (Wickham & Seidel, 2022). Additional packages used for data transformation were dplyr (Wickham et al., 2021), knitr (Xie, 2023), gdata (Warnes et al., 2023), magrittr (Bache & Wickham, 2023), readr (Wickham, Hester, et al., 2023), reshape2 (Wickham, 2007), tibble (Müller & Wickham, 2022), and tidyr (Wickham, Vaughan, et al., 2023). Analyses were carried out in R version 4.1.2 (R Core Team, 2021).
Results
Which genes show expression changes following orchid bee perfume collection?
We found that no genes were differentially expressed between bees that had collected DMB or did not collect DMB in the brain and the hind leg. However, we found 445 genes to be differentially expressed in the Malpighian tubules (Supplementary Data 1) as a result of perfume collection. The majority of these genes (442/445) were upregulated in the bees that collected DMB relative to the control group.
What type of genes show expression changes in the Malpighian tubules following orchid bee perfume collection?
Functional annotation showed that differentially expressed genes were enriched for regulation of several processes, including transcription, transmembrane transport, and detection of chemical stimuli (biological process); DNA binding transcription factors, calcium channels, olfactory receptor activity, and odorant binding (molecular function); and membrane proteins (cellular component) (Figure 1, Supplementary Table S1).
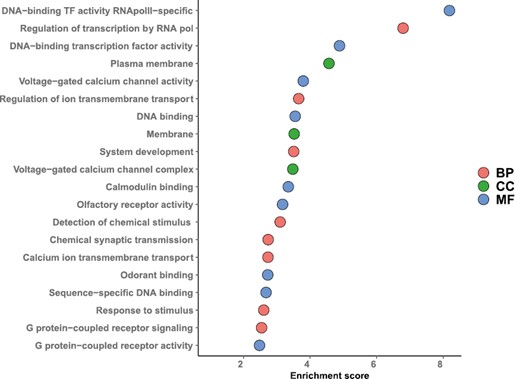
Top 20 GO terms enriched in differentially expressed genes in Malpighian tubules after perfume collection. GO terms are ordered by Fisher’s exact p-value. BP = biological process; CC = cellular component; GO = gene ontology; MF = molecular function.
While functional annotation provides us with information about the potential function of genes, it does not tell us about their evolutionary history, such as their age or whether or not they are under selection. To determine the relative age of genes that change in expression following perfume collection, we compared differentially expressed genes to the background of all genes included in the differential expression analysis after filtering by NOISeq. We found that more recent phylostrata, such as genes specific to Euglossini or Eg. dilemma are over-represented while the oldest phylostrata, such as genes shared with cellular organisms or Eukaryota, are under-represented in the differentially expressed gene set (Figure 2). For example, all the odorant receptors upregulated in the Malpighian tubules in response to perfume collection (OR17, OR19, OR22, OR24, OR30, OR52, OR88, OR77, OR94, and OR140) are shared with Neoptera (winged insects), a phylostratus found to be over-represented in upregulated genes (Figure 2).
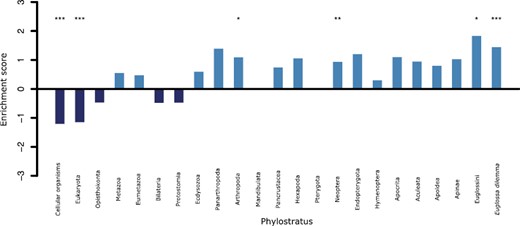
Barplot showing the enrichment of each phylostratus in the genes which are differentially expressed in Malpigihan tubules after perfume collection. Phylostratus which are over-represented have an enrichment score greater than 0. Significantly under- or over-represented phylostratus are highlighted with * to show significance where *p < .05, **p < .01, and ***p < .001 where p-values are adjusted using a false detection rate correction.
We also found that the differentially expressed genes were more likely to be tissue specific (Figure 3A). For example, IR8a, an ionotropic receptor, is upregulated following perfume collection and has a tissue-specificity value of 0.98, meaning that it is almost exclusively expressed in the Malpighian tubules in our dataset. Similarly, Edil_08594, which encodes the P450 CYP9P35, a CYP3 family member, has a tissue-specificity value of 0.96. Many other differentially expressed genes exhibited a tissue-specificity value of 1, meaning that they were only found to be expressed in Malpighian tubules.
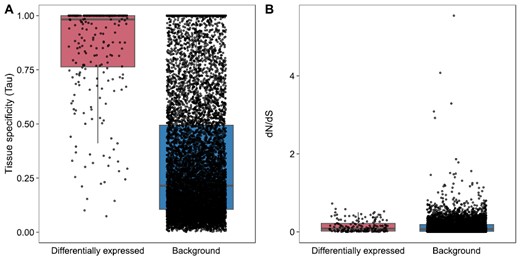
Boxplots showing the tissue-specificity and dN/dS values for genes that are differentially expressed in Malpighian tubules after perfume collection and background genes. Differentially expressed genes are more tissue specific (ANOVA, F(1,9949) = 798.3, p = <2e-16) (A) but do not differ in dN/dS values compared with the background (ANOVA, F(1,5884) = 0.314, p = .576) (B).
On the other hand, we found no difference in the dN/dS values of differentially expressed genes when compared with the background (Figure 3B). This suggests that these genes are not experiencing a different selection pressure to the background expectations (Figure 3B). Overall, we found that the genes that are upregulated in response to collection behaviour are newer, more tissue-specific genes than the background.
What are the patterns of tissue-specific gene expression?
We measured the tissue complexity of each tissue by calculating the percentage of total mRNA made up by the 10 most highly expressed genes. Malpighian tubules and brain have a similar complexity with 14% and 11% of total mRNA made up by the top 10 genes, respectively (Supplementary Data 2). In contrast, the hind leg is a simpler tissue with 39% of mRNA in the top 10 genes. Interestingly, the top two genes expressed in hind legs are CSP7 and CSP3 (chemosensory proteins), and another in the top 10 is a P450 (Edil_06932_b). Two P450s are also in the top 10 most highly expressed in the Malpighian tubules (Edil_09195 and Edil_11059_a).
We also identified tissue-specific genes, or genes that are primarily expressed in one of the three tissues included in this study. Of the 1713 tissue-specific genes we identified, 1227s were brain specific, 281 Malpighian tubule specific, and 205 hind leg specific (Supplementary data 3). Functional annotation showed that brain-specific genes were enriched for G protein-coupled receptors, as well as potassium ion transporters and membrane components (Supplementary Figure S1). Malpighian tubule-specific genes were enriched for genes involved in transmembrane transport, detection of chemical stimuli, and olfactory receptor activity (Supplementary Figure S2). Hind leg-specific genes were enriched for extracellular components, transcription factors, chitin processes, and odorant binding (Supplementary Figure S3; Supplementary Data 4).
We did not find evidence for tissue-specific evolutionary differences. We did not find a difference in mean dN/dS between tissue-specific genes of each tissue (ANOVA, F(2,809) = 0.641, p = .53). Furthermore, we did not find a difference in mean phylostratus between tissue-specific genes of each tissue (ANOVA, F(2,1710) = 1.309, p = .27).
We found a weak positive correlation between the phylostratus of a gene and its tissue specificity (Spearman’s correlation, r = .20, p < 2.2e-16), with newer genes more likely to be more tissue specific. We also found a weak negative correlation between the dN/dS of a gene and its tissue specificity (Spearman’s correlation, r = −.084, p < 2.8e-10), with more tissue-specific genes having a lower dN/dS value and therefore experiencing more purifying selection.
We found that almost a third of the orthologues described between Eg. dilemma hind legs and A. cerana legs were differentially expressed between the two spescies. The results are described in Supplementary Materials (Supplementary Results).
Discussion
Orchid bees provide an excellent system to study the evolution of novel secondary sexual traits and how gene expression changes accompany an elaborate mating signalling system due to their unique and specialized behaviour of perfume collection. We found that the Malpighian tubules, an organ involved in detoxification, shows upregulation of genes enriched in functions related to transcription, odorant binding and receptor activity, and membrane proteins following perfume collection. We did not find any gene expression changes in the hind leg or brain following perfume collection. This could suggest that these tissues do not show physiological responses to perfume collection, or that the newly emerged bees we sampled had not reached behavioural maturity required for learning (Grosso et al., 2018). Finally, we found evidence for evolutionarily young genes in these specialized tissues, and in those genes upregulated in the Malpighian tubules, supporting the hypothesis that taxonomically restricted genes play an important role in the evolution of novelty.
The physiological costs of honest mating signals: detoxification and immune function
It has been suggested that perfume collection in orchid bees indicates male quality by demonstrating that these males can withstand the high chemical exposure to volatile compounds (Arriaga‐Osnaya et al., 2017; Eltz et al., 1999). We hypothesized that if this were true, detoxification genes in the Malpighian tubules, a tissue important for detoxification (Nocelli et al., 2016), would be upregulated following perfume collection. While we did find gene upregulation in response to perfume collection, it is unclear if the upregulated genes are related to detoxification. We were particularly interested in the cytochrome P450s due to their importance for exogenous compound detoxification. Evidence for the upregulation of P450s in response to toxic stress, such as insecticide exposure, is mixed. Some studies have identified P450s as showing very plastic expression patterns (Liang et al., 2015), and others have found tissue-specific changes (Zhao et al., 2011) or that P450s are constitutively expressed, rather than being inducible (Nauen et al., 2022). In our dataset, we found that the most highly expressed CYP P450s in the Malpighian tubules do not show expression differences before and after perfume collection and instead appear to be constitutively expressed. Two CYP9 genes (Edil_08949 and Edil_08594), a family known to metabolize pesticides in other bee species (Beadle et al., 2019; Manjon et al., 2018), are upregulated in response to perfume collection; however, they are expressed at low levels. In summary, our evidence for a detoxification response to perfume collection remains limited.
Another hypothesis is that instead of playing a role in detoxification, it could be that gene expression differences in the Malpighian tubules are related to the immune role of this tissue (McGettigan et al., 2005). Interestingly, olfactory receptor activity, odorant binding, and sensory perception of smell were all found within the most highly enriched GO terms. Odorant binding proteins (OBPs) play a role in red flour beetle defence against essential oil containing many compounds known to be collected by orchid bees (Zhang et al., 2020). While genes from the OBP family are not upregulated in the Malpighian tubules, other genes with odorant binding properties could play a similar role in this context. It remains to be determined if collected compounds can enter the haemolymph from the hind leg and what type of physiological response is triggered in the Malpighian tubules by collection. Further studies will be needed to confirm the link between immune response, detoxification, and perfume collection in orchid bees.
No evidence for changes in gene expression in the brain following perfume collection
We did not detect gene expression differences in the brain following perfume collection. We were surprised by this result, as chemical exposure to different types of compounds has been shown to induce changes in gene expression in bee brains, including pheromones (Grozinger et al., 2003; Ma et al., 2019) and pesticides (Christen et al., 2018; Li et al., 2019). These changes include genes related to detoxification, metabolism, immune function, and chemosensory function, highlighting the diversity of potential downstream effects. In this study, we did not detect expression differences in any genes, perhaps due to the extent of exposure, a single event rather than chronic exposure, the timing of our sample collection, or the use of a single compound rather than the usual complex bouquet a bee would interact with at perfume sources (e.g., flowers, fungi, decaying vegetation).
Our original hypothesis was that changes in gene expression in the brain following perfume collection would reflect transcriptional changes involved in learning and memory. This follows the previous hypothesis that orchid bees learn which compounds they collect to prevent over collecting the same compound (Eltz, Roubik, et al., 2005). Long-term memory formation involves transcriptional changes, which result in protein synthesis (Barondes & Cohen, 1967; Castellucci et al., 1989). In both honey bees (Villar et al., 2020) and Drosophila (Crocker et al., 2016), odour memory formation has been shown to involve transcriptional changes one day or more after a single exposure. Therefore, we expect transcriptional changes in orchid bees, as they have been shown to avoid compounds multiple days after their last collection (Eltz, Roubik, et al., 2005). It is possible that the timing of our experiment was too early to detect changes in gene expression from learning, as honey bees have been noted to have a window of plasticity at a later age (5–8 days after emergence) than we carried out the experiment (Grosso et al., 2018). We also did not sequence peripheral sensory tissues and so could not have captured these potentially quicker regulatory changes including odorant receptor expression (Claudianos et al., 2014). Further experiments at a later life stage combined with antennal expression studies will be necessary to investigate the roles of antennal and brain gene expression changes in orchid bee learning and potentially memory formation.
Old and new genes play a role in the evolution of novelty
The evolution of novelty can be driven by regulatory changes involving “shared toolkits” of genes (Wilkins, 2013), or the evolution of new taxonomically restricted genes that do not have significant sequence similarities to genes in other more distantly related species (Johnson, 2018). Taxonomically restricted genes account for approximately 10%–20% of the genes in the genome of Eg. dilemma and have been suggested in other systems to provide a source of raw material for evolutionary processes such as local adaptation (Khalturin et al., 2009). We found that genes that were differentially expressed in the Malpighian tubules of bees following perfume collection were enriched for evolutionarily younger genes, including those specific to Eg. dilemma. We also found that tissue-specific genes, those primarily expressed in only one of the three tissues, were evolutionarily younger than the background. These findings support the hypothesis that taxonomically restricted genes play a role in the evolution of novelty. Taxonomically restricted genes have previously been found to play a role in the evolution of sociality in the honey bee (Johnson & Tsutsui, 2011), the evolution of the innate defense system and morphological adaptations in Hydra (Khalturin et al., 2009), chemical defense in Tribolium castaneum (Li et al., 2013), and the evolution of courtship in Drosophila (Dai et al., 2008). Our results corroborate that the novel perfume collection behaviour in orchid bees involves taxonomically restricted genes.
In addition to a role for taxonomically restricted genes, we found preliminary evidence for the repurposing of existing gene families in the unique perfume storage pouch on the hind legs of orchid bee males. We expected to detect expression differences in response to perfume collection in the hind legs, such as upregulation of transporters or proteins involved in odorant binding for compound storage. Our study, however, did not find any evidence of gene expression changes, perhaps suggesting that the genes required for perfume collection and storage in the hind legs are already constitutively expressed. While not differentially expressed, one of the most highly expressed genes, CSP3, appears to be multifunctional and context dependent, also known to act as a brood pheromone carrier (Briand et al., 2002), a hydrocarbon transporter (Erban et al., 2016), and a fatty acid transporter (Wu et al., 2017). Thus, its role in the hind legs of Eg. dilemma remains unclear, but it could play a conserved role in gustatory reception, perhaps sugar detection, as suggested by the high levels of expression also found in A. cerana legs (Du et al., 2019). Alternatively, it could be important for a more orchid bee-specific role such as recycling the lipid compounds that are released from the labial glands to facilitate perfume collection (Eltz et al., 2007; Whitten et al., 1989). While the function of CSP3 remains unclear, it provides an interesting candidate to test how shared genes between species can be used for different functions.
Another CSP, CSP7, which is most closely related to CSP1 in Apis, is the most highly expressed gene in Eg. dilemma hind legs but is found in very low levels in A. cerana legs. This gene has been noted to evolve more rapidly than other CSPs in orchid bees (Brand & Ramírez, 2017). In both A. mellifera and A. cerana, CSP1 is highly expressed in the antennae and shows the strongest binding to floral volatiles and pheromone ligands of the CSPs, suggesting a role in chemosensory processes (Li et al., 2016). We hypothesize that CSP7 in orchid bees is adapted for a role specific to perfume collection, such as binding and storage of volatiles, or recycling of labial lipids. While CSPs are an ancient gene family, they may have been re-purposed for perfume collection in orchid bees, with further functional studies required to confirm their role in this process.
Conclusion
Our study describes gene expression changes in the Malpighian tubules in response to perfume collection but a lack of change in both the brain and the hind leg tissues. The upregulation of genes in the Malpighian tubules in response to perfume collection suggests that a physiological response occurs in this tissue, but it remains unclear whether this is related to the detoxification of compounds. While we did not find evidence for a transcriptional response to perfume collection in the hind legs, we identified high expression of chemosensory proteins in this tissue. We hypothesize that these proteins could play a role in labial lipid recycling, perfume collection, or storage. To better understand the role of chemosensory proteins and odorant-binding proteins in the physiological adaptations for perfume collection in orchid bees, functional studies of the roles of these proteins in binding key perfume compounds will be needed.
Data availability
Data and R scripts used for analysis are available from Open Science Framework: https://osf.io/wnm8p/?view_only=57f7bd46ded5439b9df6fa16cd7bd6cc. The raw sequence reads are deposited in the SRA (BioProject PRJNA910557).
Author contributions
Kathy Darragh (Conceptualization [equal], Data curation [lead], Formal analysis [lead], Investigation [lead], Project administration [equal], Visualization [lead], Writing – original draft [lead], Writing – review & editing [equal]) and Santiago Ramirez (Conceptualization [equal], Funding acquisition [lead], Project administration [equal], Resources [lead], Supervision [lead], Writing – review & editing [equal])
Funding
S.R.R. was funded by the David and Lucile Packard Foundation (2014-40378).
Acknowledgments
We thank Brock Harpur, Ana Pinharanda, and Nicholas Saleh for advice regarding RNAseq analysis. We also thank Thomas Eltz and Jonas Henske for scientific guidance regarding the experimental setup and hind leg dissection. We thank Thomas Chouvenc and Aaron Mullins for their support in Florida. We are grateful to the Ramírez lab for their feedback throughout the project.
Conflicts of interest
The authors confirm they have no conflict of interest to declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}