





Abstract
Feminizing adrenocortical carcinoma (ACC) is rare. The source of estrogen production and the underlying mechanism remain unclear.
In the current study, we investigated the source and the molecular mechanism of estrogen production in feminizing ACC.
A total of 46 consecutive patients with a diagnosis of ACC were recruited in our center. We described the clinical characteristics and steroid hormone profile of the peripheral and adrenal vein. In both feminizing ACC tissues and cell lines, we investigated the expression of steroidogenic biomarkers and β-catenin pathways by quantitative PCR and immunohistochemical staining. The effects of Wnt inhibitors on steroidogenesis were also analyzed in NCI-H295R cells.
A total of 46 consecutive patients with ACC were analyzed, and 25 had functional ACC. Four patients received a diagnosis of feminizing ACC based on feminizing manifestations, high levels of estradiol that were normalized after surgery, and histological Weiss score. Gonadal steroidogenic biomarkers including CYP19A1, HSD17B3, and LHCGR were markedly elevated in the feminizing ACC tissues. Adrenal vein sampling and liquid chromatography–tandem mass spectrometry suggested high CYP19A1 activity in the adrenal mass. β-catenin expression was also elevated. When treated with niclosamide and PNU-74654, the H295R cell line showed a decrease in β-catenin expression, cell proliferation, and steroid secretion. All steroid hormone enzymes were inhibited, whereas CYP19A1, HSD17B3, and LHCGR mRNA increased.
Feminizing ACC can express high levels of CYP19A1, thus ectopically producing estrogens. Wnt pathway activation and dedifferentiation toward common adrenal-gonadal precursor cells may be the underlying mechanisms.
Adrenocortical carcinoma (ACC) is rare and highly malignant, with a poor prognosis (1). Despite aggressive surgical and adjunctive therapy, the 5-year survival rate ranges from 16% to 38% (2). Approximately 60% of ACCs are hormone secreting, and the steroid profile often displays a pattern of disorganized steroidogenesis, further contributing to the morbidity associated with ACCs. Most commonly ACCs produce cortisol and androgen. Rarely, ACCs produce estrogens (3, 4).
Feminizing ACC was first reported in 1919 in a 26-year-old man who presented with gynecomastia (5). Since then, ~80 cases have been reported, mainly as case reports. Feminizing ACCs tend to be larger and with worse prognosis compared with nonfeminizing ACCs (6). The source and mechanism of estrogen overproduction in patients with ACC remain unclear. Physiologically, the adrenal cortex has little capacity to produce estrogens because of the lack of aromatase. Previous studies showed high aromatase cytochrome P450 (CYP19A1) mRNA and aromatase activity in feminizing ACCs (7–10), which may contribute to the ectopic estrogen production in ACC. High CYP19A1 expression may be caused by dedifferentiation of ACCs because combined steroidogenic characters of fetal adrenal and Leydig cells have been found in androgen-producing ACC tissues (11).
The Wnt/β-catenin pathway is known to be important in adrenal cortex development (12). Activating mutations in CTNNB1 were identified in 16% of ACCs (13, 14), whereas abnormal cytoplasmic or nuclear β-catenin expression was demonstrated in ≤85% of ACC (15, 16). Wnt/β-catenin activation has been associated with tumorigenesis and progression of ACC. It is an independent factor associated with shorter overall and disease-free survival (17). Wnt pathway inhibitors induce durable anticancer activity through reduction of epithelial-to-mesenchymal transition and β-catenin levels (18). However, their association with steroidogenesis and ACC dedifferentiation remains unclear.
In the current study, we investigated the source and mechanisms of estrogen production in feminizing ACCs.
Materials and Methods
Patients
All patients with ACC were enrolled at Ruijin Hospital from 2002 to 2017. ACC was histologically diagnosed according to the Weiss criteria (19). The preoperative endocrine workup was performed according to the European Network for the Study of Adrenal Tumors (ENSAT) methods (1). Cortisol secreting was defined as elevated 24-hour urinary free cortisol level, suppressed ACTH, unsuppressed low-dose dexamethasone suppression test, and normalized serum hormones after surgery. Aldosterone secreting was defined as a failure of suppression of postinfusion aldosterone levels to 10 ng/dL in hypertensive patients with an elevated aldosterone/renin ratio >30 and normalized serum hormones after surgery. Androgen and estrogen secreting were defined as high serum levels of androgens and estradiol (E2) before surgery and normalized serum hormones after surgery. We excluded patients with recurrent diseases or lack of sex hormone evaluation before operation. Tumor staging was recorded based on the ENSAT classification. ACC was assessed according to pathological examination, and tumors with Weiss scores of ≥3 were considered. Tumor tissues from localized (stage I to II) and local advanced stage (stage III) ACCs were collected for further immunohistochemical staining. Informed consent was obtained from all participants. All the protocols were approved by the Ethics Committee of Ruijin Hospital.
Routine hormone assessments
All tests were performed in a College of American Pathologists (no. 7217913) accredited laboratory. Serum aldosterone, free testosterone (T), 17a-hydroxyprogesterone, androstenedione, and plasma renin activity were measured by radioimmunoassay; plasma and urinary free cortisol were measured on Access Immunoassay Systems (Beckman Coulter Inc, Fullerton, CA); and ACTH levels were determined with an ELSA-ACTH immunoradiometric assay (Cisbio Bioassays, Codolet, France). Other sex hormones, such as dehydroepiandrosterone sulfate (DHEAS), SHBG, LH, FSH, E2, progesterone, and T. were analyzed via chemiluminescence immunoassay (Abbott Laboratories, Abbott Park, IL). Serum LH, FSH, E2, progesterone, and T concentrations for patient 3 were measured by chemiluminescent assay on an Access autoanalyzer (Beckman Coulter Inc).
Measurements of steroid metabolites by liquid chromatography–tandem mass spectrometry
For sample preparation, all hormones were extracted from 100-μL samples with 1 mL extraction buffer [50% N-hexane + 50% ethyl acetate + internal standard (Toronto Research Chemicals, Ontario, Canada) at a final concentration of 0.5 ng/mL], followed by evaporation under constant nitrogen flow and redissolution with 100 μL methanol/deionized water (1:9, v/v). For protein precipitation and extraction of DHEAS, 40.0 μL ZnSO4 1 mM/methanol (1:9, v/v) was added to 100-μL samples before configuration. Then, 100 μL supernatant was extracted and diluted with 100 μL deionized water.
We measured 15 steroid metabolites in the steroidogenic pathway (pregnenolone, 17-hydropregnenolone, progesterone, 17-hydroprogesterone, androstenedione, estrone, E2, estriol, 11-deoxycorticosterone, corticosterone, aldosterone, 11-deoxycortisol, cortisol, cortisone, T, and DHEAS) by using an ABSciex triple-quadrupole mass spectrometer 4500 equipped with an Eksigent ultraHPLC system (ABSciex, Ontario, Canada). The column used was a Kinetex 2.6-μm XB-C18 (2.1 mm × 50 mm; Phenomenex, Torrance, CA) maintained at 45°C. The mobile phases were aqueous formic acid solution (0.1%) with methanolic formic acid solution (0.1%) at a flow rate of 0.5 mL/min, and the final injection volume of each sample was 20 μL. All acquisitions were performed in positive ion mode. Quantitative analyses were performed in multiple reaction monitoring mode. Each steroid was identified by comparing retention time and two mass transitions with a deuterated reference compound. The intra-assay and interassay coefficients of variation of these assays were <10.0% and 14.5%, respectively.
RNA extraction, reverse transcription, and quantitative PCR
RNA extraction was prepared by Qiagen miRNeasy mini kit (Qiagen, Hilden, Germany). Reverse transcription total RNA was converted into first-strand complementary DNA with a reverse transcription kit (Promega, Madison, WI) according to the manufacturer’s instructions. Quantitative real-time PCR was then performed with a Quantstudio Dx Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA). Actin was used as a normalization control. Genes encoding the key steroidogenic enzymes were amplified by specific primers (Supplemental Table 1).
Immunohistochemistry and Western blot
Immunohistochemistry and Western blot were performed according to the standard histopathological procedures (20, 21). Antibodies against CYP19A1 (rabbit polyclonal, 1:400, Sigma, SAB4500606), LHCGR (rabbit polyclonal, 1:400, Sigma, SAB2700732), AKR1C3 (rabbit polyclonal, 1:400, Abcam, ab84327), CYB5A (mouse monoclonal clone 1A8, 1:400, Sigma, SAB1403717), β-catenin (rabbit polyclonal, 1:400 for immunohistochemistry and 1:1000 for Western blot, Cell Signaling Technology, #9562), and p53 (mouse monoclonal, 1:200, Dako, DO-7) were used. Primary antibody binding was demonstrated with a standard avidin-biotin-peroxidase complex technique (Vector pk-7200; Vector Laboratories, Inc., Burlingame, CA).
Abnormal β-catenin staining was defined as focal cytoplasmic staining (<30% but strong staining), diffuse cytoplasmic staining (30% to 70% and at least moderate staining, >70% regardless of staining intensity) or focal nuclear staining (<5% of nuclei but strong staining), or diffuse nuclear staining (>5% and at least moderate staining).
Cell culture and reagents
ACC cell lines H295R and SW13 were obtained from the National Platform of Experimental Cell Resources for Sci-Tech (Beijing, China). Cells were maintained in DMEM F12 medium (Gibco, Life Technologies) supplemented with 2.5% NuSerum (BD Biosciences, Bedford, MA) and 1% insulin-transferrin-selenium culture supplement (BD Biosciences). Niclosamide (2ʹ,5-dichloro-4ʹ-nitrosalicylanilide) and PNU-74654 were purchased from Selleck and dissolved in dimethylformamide and dimethyl sulfoxide at concentrations of 0.25 to 1 μM, 1 to 10 μM, and 10 to 100 μM.
Cell proliferation assay
The H295R cells were seeded in 96-well plates at 8000 cells per well. The cell proliferation assays were performed with a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Kunamoto, Japan) according to the manufacturer’s instructions. The relative cell number was determined by measuring light absorbance at a wavelength of 450 nm (Gen5, BioTek, Winooski, VT).
Results
Patient characteristics and endocrine evaluation
As shown in Table 1, the median age at diagnosis was 56 years, and male patients accounted for 46% of subjects. The median size on CT scan was 8 cm. The preoperative endocrine workup found 25 tumors (54.35%) that produced one or more hormones, defined as functional ACCs (Supplemental Table 2). Compared with nonfunctional ACCs, functional ACCs were associated with advanced clinical stage (P = 0.043) (Table 2). Ten functional ACCs produced sex hormones and four produced estrogens in three male and one female patient. All three male patients complained of painless gynecomastia and gradually worsening hypaphrodisia. Genitourinary examination found no testicular masses. The female patient was 53 years old and presented with postmenopausal bleeding. All four patients showed concurrent hypercortisolism but without classic cushingoid features (Table 3). We performed preoperation adrenal vein sampling in one male patient. The patient had a mass on the left adrenal, and the left adrenal vein showed high E2 and cortisol levels (Supplemental Table 3). The patients underwent surgery and achieved normalized serum estrogen and cortisol levels and phenotypes.
Overview of Clinical Features of Patients With ACC
| Clinicopathological Features | All Patients (n = 46) |
|---|---|
| Age at diagnosis, ya | 56.0 (44.5, 61.5) |
| Sex, male/female | 21/25 |
| Primary tumor size, cma | 8.00 (5.80, 10.59) |
| Staging (ENSAT), n | |
| I–II | 21 |
| III–IV | 25 |
| Hormonal status, n | |
| Pure cortisol secretion | 13 |
| Cosecretion of cortisol and androgens | 6 |
| Cosecretion of cortisol and estrogens | 4 |
| Cosecretion of cortisol and aldosterone | 2 |
| Nonfunctioning | 21 |
| Clinicopathological Features | All Patients (n = 46) |
|---|---|
| Age at diagnosis, ya | 56.0 (44.5, 61.5) |
| Sex, male/female | 21/25 |
| Primary tumor size, cma | 8.00 (5.80, 10.59) |
| Staging (ENSAT), n | |
| I–II | 21 |
| III–IV | 25 |
| Hormonal status, n | |
| Pure cortisol secretion | 13 |
| Cosecretion of cortisol and androgens | 6 |
| Cosecretion of cortisol and estrogens | 4 |
| Cosecretion of cortisol and aldosterone | 2 |
| Nonfunctioning | 21 |
Data are expressed as medians and interquartile ranges.
Overview of Clinical Features of Patients With ACC
| Clinicopathological Features | All Patients (n = 46) |
|---|---|
| Age at diagnosis, ya | 56.0 (44.5, 61.5) |
| Sex, male/female | 21/25 |
| Primary tumor size, cma | 8.00 (5.80, 10.59) |
| Staging (ENSAT), n | |
| I–II | 21 |
| III–IV | 25 |
| Hormonal status, n | |
| Pure cortisol secretion | 13 |
| Cosecretion of cortisol and androgens | 6 |
| Cosecretion of cortisol and estrogens | 4 |
| Cosecretion of cortisol and aldosterone | 2 |
| Nonfunctioning | 21 |
| Clinicopathological Features | All Patients (n = 46) |
|---|---|
| Age at diagnosis, ya | 56.0 (44.5, 61.5) |
| Sex, male/female | 21/25 |
| Primary tumor size, cma | 8.00 (5.80, 10.59) |
| Staging (ENSAT), n | |
| I–II | 21 |
| III–IV | 25 |
| Hormonal status, n | |
| Pure cortisol secretion | 13 |
| Cosecretion of cortisol and androgens | 6 |
| Cosecretion of cortisol and estrogens | 4 |
| Cosecretion of cortisol and aldosterone | 2 |
| Nonfunctioning | 21 |
Data are expressed as medians and interquartile ranges.
Clinicopathological Parameters of Functional ACC
| Clinicopathological Features | Functional ACC (n = 25) | Nonfunctional ACC (n = 21) | P |
|---|---|---|---|
| Age at diagnosis, ya | 57.0 (44.0, 63.0) | 54.0 (42.5, 58.5) | 0.76 |
| Sex, male/female | 13/12 | 8/13 | 0.35 |
| Primary tumor size, cma | 9.23 (5.89, 10.95) | 7.90 (5.80, 10.21) | 0.70 |
| ENSAT stage, n | |||
| Stage I–II | 8 | 13 | <0.05 |
| Stage III–IV | 17 | 8 |
| Clinicopathological Features | Functional ACC (n = 25) | Nonfunctional ACC (n = 21) | P |
|---|---|---|---|
| Age at diagnosis, ya | 57.0 (44.0, 63.0) | 54.0 (42.5, 58.5) | 0.76 |
| Sex, male/female | 13/12 | 8/13 | 0.35 |
| Primary tumor size, cma | 9.23 (5.89, 10.95) | 7.90 (5.80, 10.21) | 0.70 |
| ENSAT stage, n | |||
| Stage I–II | 8 | 13 | <0.05 |
| Stage III–IV | 17 | 8 |
P < 0.05 was considered statistically significant.
Data are expressed as medians and interquartile ranges.
Clinicopathological Parameters of Functional ACC
| Clinicopathological Features | Functional ACC (n = 25) | Nonfunctional ACC (n = 21) | P |
|---|---|---|---|
| Age at diagnosis, ya | 57.0 (44.0, 63.0) | 54.0 (42.5, 58.5) | 0.76 |
| Sex, male/female | 13/12 | 8/13 | 0.35 |
| Primary tumor size, cma | 9.23 (5.89, 10.95) | 7.90 (5.80, 10.21) | 0.70 |
| ENSAT stage, n | |||
| Stage I–II | 8 | 13 | <0.05 |
| Stage III–IV | 17 | 8 |
| Clinicopathological Features | Functional ACC (n = 25) | Nonfunctional ACC (n = 21) | P |
|---|---|---|---|
| Age at diagnosis, ya | 57.0 (44.0, 63.0) | 54.0 (42.5, 58.5) | 0.76 |
| Sex, male/female | 13/12 | 8/13 | 0.35 |
| Primary tumor size, cma | 9.23 (5.89, 10.95) | 7.90 (5.80, 10.21) | 0.70 |
| ENSAT stage, n | |||
| Stage I–II | 8 | 13 | <0.05 |
| Stage III–IV | 17 | 8 |
P < 0.05 was considered statistically significant.
Data are expressed as medians and interquartile ranges.
Steroid Hormones in Four Patients With Feminizing ACCs
| Patient 1, Male, 33 y | Patient 2, Male, 27 y | Patient 3, Male, 36 y | Patient 4, Female, 53 y | Normal Range | |
|---|---|---|---|---|---|
| LH, mIU/mL | <0.07 | <0.07 | 0.69 | 7.24 | Pts 1 and 2: 0.57–12.07; Pt 3: 1.10–8.20; Pt 4: 5.16–61.99 |
| FSH, mIU/mL | <0.05 | <0.05 | 1.40 | 7.51 | Pts 1 and 2: 0.90–12.00; Pt 3: 1.50–11.50; Pt 4: 26.7–133.4 |
| E2, pg/mL | 237 | 85 | 240.3 | 120 | Pts 1 and 2: 11.00–44.00; Pt 3: <60; Pt 4: <10–28 |
| Progesterone, ng/mL | 1 | 0.58 | 18.58 | <0.1 | <0.10–0.20 |
| T, ng/mL | 0.27 | 0.38 | 3.36 | 0.28 | Pts 1 and 2: 1.43–9.23; Pt 3: 2.50–10.51; Pt 4: 0.13–1.08 |
| Free T, pg/mL | 0.61 | 1.96 | — | 1.62 | Pts 1 and 2: 8.69–54.69; Pt 4: 0.29–3.18 |
| SHBG, nmol/L | 48.4 | 58.5 | — | 61.1 | Pts 1 and 2: 13.50–71.40; Pt 4: 19.80–155.20 |
| DHEAS, μg/dL | 490.4 | 222.0 | — | 96.2 | Pts 1 and 2: 136.20–591.90; Pt 4: 56.20–511.70 |
| Dihydrotestosterone, pg/mL | 77.19 | 74.60 | — | 37.76 | Pts 1 and 2: 17.90–579.00; Pt 4: 15.60–142.00 |
| 17-OH progesterone, ng/mL | 3.41 | 1.70 | — | — | Pts 1 and 2: 0.50–2.40 |
| Androstenedione, ng/mL | 1.56 | 2.93 | — | — | Pts 1 and 2: 0.30–2.63 |
| Cortisol, 0800 h, μg/dL | 12.76 | 8.79 | 45.3 | 14.51 | 6.70–22.60 |
| 24-h urine free cortisol, μg/24 h | 250.20 | 171.38 | 1046.9 | 365.25 | 21–111 |
| Patient 1, Male, 33 y | Patient 2, Male, 27 y | Patient 3, Male, 36 y | Patient 4, Female, 53 y | Normal Range | |
|---|---|---|---|---|---|
| LH, mIU/mL | <0.07 | <0.07 | 0.69 | 7.24 | Pts 1 and 2: 0.57–12.07; Pt 3: 1.10–8.20; Pt 4: 5.16–61.99 |
| FSH, mIU/mL | <0.05 | <0.05 | 1.40 | 7.51 | Pts 1 and 2: 0.90–12.00; Pt 3: 1.50–11.50; Pt 4: 26.7–133.4 |
| E2, pg/mL | 237 | 85 | 240.3 | 120 | Pts 1 and 2: 11.00–44.00; Pt 3: <60; Pt 4: <10–28 |
| Progesterone, ng/mL | 1 | 0.58 | 18.58 | <0.1 | <0.10–0.20 |
| T, ng/mL | 0.27 | 0.38 | 3.36 | 0.28 | Pts 1 and 2: 1.43–9.23; Pt 3: 2.50–10.51; Pt 4: 0.13–1.08 |
| Free T, pg/mL | 0.61 | 1.96 | — | 1.62 | Pts 1 and 2: 8.69–54.69; Pt 4: 0.29–3.18 |
| SHBG, nmol/L | 48.4 | 58.5 | — | 61.1 | Pts 1 and 2: 13.50–71.40; Pt 4: 19.80–155.20 |
| DHEAS, μg/dL | 490.4 | 222.0 | — | 96.2 | Pts 1 and 2: 136.20–591.90; Pt 4: 56.20–511.70 |
| Dihydrotestosterone, pg/mL | 77.19 | 74.60 | — | 37.76 | Pts 1 and 2: 17.90–579.00; Pt 4: 15.60–142.00 |
| 17-OH progesterone, ng/mL | 3.41 | 1.70 | — | — | Pts 1 and 2: 0.50–2.40 |
| Androstenedione, ng/mL | 1.56 | 2.93 | — | — | Pts 1 and 2: 0.30–2.63 |
| Cortisol, 0800 h, μg/dL | 12.76 | 8.79 | 45.3 | 14.51 | 6.70–22.60 |
| 24-h urine free cortisol, μg/24 h | 250.20 | 171.38 | 1046.9 | 365.25 | 21–111 |
Abbreviation: Pt, patient.
Steroid Hormones in Four Patients With Feminizing ACCs
| Patient 1, Male, 33 y | Patient 2, Male, 27 y | Patient 3, Male, 36 y | Patient 4, Female, 53 y | Normal Range | |
|---|---|---|---|---|---|
| LH, mIU/mL | <0.07 | <0.07 | 0.69 | 7.24 | Pts 1 and 2: 0.57–12.07; Pt 3: 1.10–8.20; Pt 4: 5.16–61.99 |
| FSH, mIU/mL | <0.05 | <0.05 | 1.40 | 7.51 | Pts 1 and 2: 0.90–12.00; Pt 3: 1.50–11.50; Pt 4: 26.7–133.4 |
| E2, pg/mL | 237 | 85 | 240.3 | 120 | Pts 1 and 2: 11.00–44.00; Pt 3: <60; Pt 4: <10–28 |
| Progesterone, ng/mL | 1 | 0.58 | 18.58 | <0.1 | <0.10–0.20 |
| T, ng/mL | 0.27 | 0.38 | 3.36 | 0.28 | Pts 1 and 2: 1.43–9.23; Pt 3: 2.50–10.51; Pt 4: 0.13–1.08 |
| Free T, pg/mL | 0.61 | 1.96 | — | 1.62 | Pts 1 and 2: 8.69–54.69; Pt 4: 0.29–3.18 |
| SHBG, nmol/L | 48.4 | 58.5 | — | 61.1 | Pts 1 and 2: 13.50–71.40; Pt 4: 19.80–155.20 |
| DHEAS, μg/dL | 490.4 | 222.0 | — | 96.2 | Pts 1 and 2: 136.20–591.90; Pt 4: 56.20–511.70 |
| Dihydrotestosterone, pg/mL | 77.19 | 74.60 | — | 37.76 | Pts 1 and 2: 17.90–579.00; Pt 4: 15.60–142.00 |
| 17-OH progesterone, ng/mL | 3.41 | 1.70 | — | — | Pts 1 and 2: 0.50–2.40 |
| Androstenedione, ng/mL | 1.56 | 2.93 | — | — | Pts 1 and 2: 0.30–2.63 |
| Cortisol, 0800 h, μg/dL | 12.76 | 8.79 | 45.3 | 14.51 | 6.70–22.60 |
| 24-h urine free cortisol, μg/24 h | 250.20 | 171.38 | 1046.9 | 365.25 | 21–111 |
| Patient 1, Male, 33 y | Patient 2, Male, 27 y | Patient 3, Male, 36 y | Patient 4, Female, 53 y | Normal Range | |
|---|---|---|---|---|---|
| LH, mIU/mL | <0.07 | <0.07 | 0.69 | 7.24 | Pts 1 and 2: 0.57–12.07; Pt 3: 1.10–8.20; Pt 4: 5.16–61.99 |
| FSH, mIU/mL | <0.05 | <0.05 | 1.40 | 7.51 | Pts 1 and 2: 0.90–12.00; Pt 3: 1.50–11.50; Pt 4: 26.7–133.4 |
| E2, pg/mL | 237 | 85 | 240.3 | 120 | Pts 1 and 2: 11.00–44.00; Pt 3: <60; Pt 4: <10–28 |
| Progesterone, ng/mL | 1 | 0.58 | 18.58 | <0.1 | <0.10–0.20 |
| T, ng/mL | 0.27 | 0.38 | 3.36 | 0.28 | Pts 1 and 2: 1.43–9.23; Pt 3: 2.50–10.51; Pt 4: 0.13–1.08 |
| Free T, pg/mL | 0.61 | 1.96 | — | 1.62 | Pts 1 and 2: 8.69–54.69; Pt 4: 0.29–3.18 |
| SHBG, nmol/L | 48.4 | 58.5 | — | 61.1 | Pts 1 and 2: 13.50–71.40; Pt 4: 19.80–155.20 |
| DHEAS, μg/dL | 490.4 | 222.0 | — | 96.2 | Pts 1 and 2: 136.20–591.90; Pt 4: 56.20–511.70 |
| Dihydrotestosterone, pg/mL | 77.19 | 74.60 | — | 37.76 | Pts 1 and 2: 17.90–579.00; Pt 4: 15.60–142.00 |
| 17-OH progesterone, ng/mL | 3.41 | 1.70 | — | — | Pts 1 and 2: 0.50–2.40 |
| Androstenedione, ng/mL | 1.56 | 2.93 | — | — | Pts 1 and 2: 0.30–2.63 |
| Cortisol, 0800 h, μg/dL | 12.76 | 8.79 | 45.3 | 14.51 | 6.70–22.60 |
| 24-h urine free cortisol, μg/24 h | 250.20 | 171.38 | 1046.9 | 365.25 | 21–111 |
Abbreviation: Pt, patient.
Comparison of gonadal steroidogenic biomarkers between feminizing ACC, functional ACC without feminization, and nonfunctional ACC
We first quantified the mRNA expression of steroidogenic tissue-specific enzymes (Fig. 1). Compared with the normal adrenal cortex, nonfunctional ACCs, and functional but not feminizing ACCs, feminizing ACCs showed remarkably elevated gonadal markers including CYP19A1, HSD17B3, and LHCGR. However, the expression of adrenocortical tissue-specific markers (StAR, HSD3B2, CYP11B1, CYB5A) was slightly decreased.

Gene expression profile of steroidogenesis markers measured in normal adrenal cortex (green), nonfunctional ACC (blue), functional but not feminizing ACC (red), and feminizing ACC (black). The symbols and error bars in the graph represent median and 2.5 and 97.5 percentile of all relative expression values. ZF, zona fasciculata; ZR, zona reticularis.
We then evaluated the immunostaining of steroidogenic tissue-specific enzymes (Fig. 2). compared with functional ACCs without feminization and nonfunctional ACCs, feminizing ACCs showed more intense membrane localization of CYP19A1, cytoplasmic AKR1C3 (HSD17B5) staining, and higher expression of LHCGR. However, no differences was observed between functional ACCs without feminization and nonfunctional ACCs. Cytoplasmic CYB5A staining showed no difference between the four groups (Fig. 2, Supplemental Table 4).
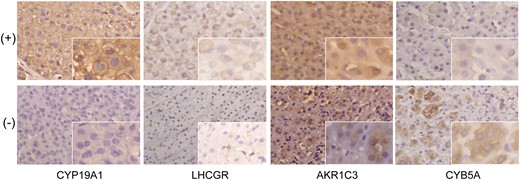
Immunohistochemical staining of steroidogenic tissue-specific enzymes in ACCs. Magnification ×200; insets ×400.
To further clarify, the elevated estrogen resulted from ACCs via elevated activity of estrogen synthesis. We profiled serum steroid hormone in one patient and found peripheral elevated 17-OH pregnenolone, deoxycorticosterone, 11-deoxycortisol, and E2, with decreased androstenedione and T compared with normal male controls. Interestingly, CYP19A1 activity indicated by E2/T ratios and CYP17A1 activity (17-OH progesterone/17-OH pregnenolone, androstenedione/17-OH progesterone) were both elevated (Supplemental Fig. 1).
Activity of Wnt/β-catenin pathway in functional ACCs
We found that β-catenin was significantly upregulated in the cytoplasm and nucleus of all feminizing ACCs (Supplemental Fig. 2A), although none of the feminizing ACCs harbored a somatic CTNNB1 mutation. Only 50% (2/4) of feminizing ACCs showed positive p53 staining. In two ACC cell lines (H295R and SW13), we found that steroidogenesis-related genes were undetectable in SW13 cells and highly expressed in H295R cells (Supplemental Fig. 2B). Steroid hormone profiling found high levels of cortisol and sex hormones in the supernatants of H295R cells but not in SW13 cells (Supplemental Fig. 2C). Note that H295R cells harbored a pathogenic p.S45P CTNNB1 variation.
Effect of Wnt pathway antagonists on steroidogenesis and cell differentiation in NCI-H295R cells
We treated H295R cells with niclosamide and PNU-74654 to inhibit Wnt signaling (Fig. 3A). As previously reported, niclosamide and PNU-74654 inhibited proliferation in a time- and dose-dependent manner (Fig. 3B). We collected the supernatants to profile steroid hormones. Niclosamide significantly suppressed almost all steroid hormones including estrogens, and cortisol decreased in a dose-dependent manner (P < 0.05). Cortisol and dehydroepiandrosterone were undetectable after 48 hours with 0.25 to 1 μM niclosamide (Fig. 3C). PNU-74654 significantly suppressed 11-deoxycortisol, androstenedione, and T (P < 0.05) (Fig. 3C).
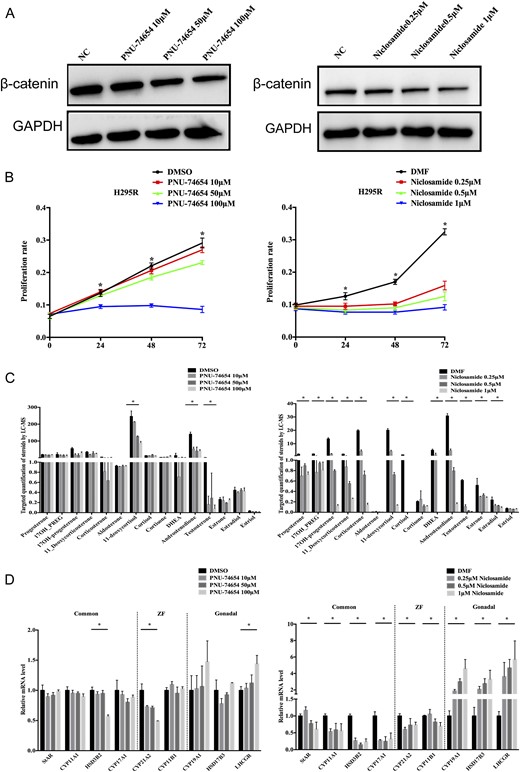
The effects of PNU-74654 and niclosamide on H295R cell proliferation, steroidogenesis, and differentiation. (A) β-catenin expression was determined by Western blot. (B) PNU-74654 and niclosamide inhibit cell proliferation in a time- and dose-dependent manner. (C) Steroid hormone secretion analysis was performed via liquid chromatography–tandem mass spectrometry after treatment with niclosamide and PNU-74654 in H295R cells. (D) The expression pattern of steroid tissue differentiation-related genes was analyzed by quantitative reverse transcription PCR with different dosages of niclosamide and PNU-74654. Error bars represent ±SD. *P < 0.01. DMF, N,N-dimethylformamide; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NC, negative control; ZF, zona fasciculata.
Accordingly, niclosamide significantly decreased HSD3B2, CYP17A1, and CYP11B1 mRNA 48 hours after treatment (P < 0.01; Fig. 3D). PNU-74654 decreased HSD3B2 and CYP21A2 by 50% (P < 0.01; Fig. 3D) 72 hours after treatment. However, gonadal-specific gene expression was elevated after inhibition of Wnt signaling. Niclosamide markedly increased CYP19A1 (1.88, 3.00, and 4.58-fold, P < 0.01), HSD17B3 (2.08, 2.77, and 3.30-fold, P < 0.01), and LHCGR (3.61, 4.67, and 5.70-fold, P < 0.01) mRNA (Fig. 3D), whereas PNU-74654 increased CYP19A1, HSD17B3, and LHCGR mRNA at concentrations >50 μM (Fig. 3D).
Discussion
In this study we described four cases of feminizing ACC. We clarified that excess estrogen production was associated with the elevated CYP19A1 activity of ACC, and dedifferentiation toward common adrenal-gonadal precursor cells of ACC cells may be the explanation.
Cushing syndrome with or without virilization due to excessive androgens is common in functional ACC. However, feminizing ACC is quite rare. In our cohort, we diagnosed four feminizing cases among 46 patients with ACC. All four patients presented with clinical features of gynecomastia or postmenopausal bleeding and high serum estrogens, which were normalized after surgery. The resected tumors showed histological features of ACC and high levels of CYP19A1. Most patients with feminizing ACC presented with gynecomastia and estrogen overproduction (5–10). Subclinical feminization in patients with only elevated E2 level but no clinical symptoms are rarely reported. In our cohort, no patients showed this subclinical feminization feature.
Adrenal precursor cells and bipotential gonadal precursor cells arise from a common ancestor. Previous studies have revealed combined steroidogenic characters of fetal adrenal and Leydig cells in both androgen-producing ACC and testicular adrenal rest tumor tissues (11, 22). Androgen-producing ACC tissues showed elevated expression of Leydig cell markers (CYP11A1, CYP17A1, HSD17B3, and INSL3), whereas high expression of adrenal cell markers (CYP11B1, CYP11B2, and MC2R) was demonstrated in testicular adrenal rest tumor tissues. Here we found that feminizing ACC tumors had gonadal cell features, including high expression of CYP19A1, AKR1C3, and LHCGR. We also found remarkably reduced CYB5A expression in feminizing ACC, indicative of fetal adrenal character (23). Consistent with immunohistochemical staining results, liquid chromatography–tandem mass spectrometry of adrenal vein samples showed elevated E2/T ratios, indicating increased activity of CYP19A1, which converted androgen to estrogen (7–10, 24). It is possible that ACCs acquire pluripotential steroidogenic function by dedifferentiation of the adrenocortical cells toward their common adrenal-gonadal precursor (25), thus contributing to the production of sex hormones in ACCs.
Wnt signaling pathways play key roles in the development, differentiation, and homeostasis of the adrenal gland (26–28). Wnt pathways have been associated with functional ACCs. Data from the Cancer Genome Atlas show a higher rate of Wnt-related mutations in functional ACCs compared with nonfunctional ACCs (48.9% vs 23.1%) (13). However, in adrenocortical adenomas the result is the opposite: 83% of the nonfunctioning adenomas showed abnormal β-catenin immunostaining vs 25% of functional adenomas (15). Activating CTNNB1 mutations and impaired β-catenin degradation have also been found in aldosterone-producing adenomas (29). In our study, high expression of β-catenin nuclear staining was observed in four feminizing ACC tumors and cell lines. Furthermore, we found that niclosamide and PNU-74654 not only inhibited cell proliferation but also decreased sex hormone or glucocorticoid production, thus demonstrating the steroidogenesis-inhibiting capacity of Wnt inhibitors. Intriguingly, we found that niclosamide significantly increased CYP19A1, HSD17B3, and LHCGR expression in a dose-dependent manner in H295R cells, in accordance with a previous study demonstrating that Wnt pathway activators suppressed ovarian follicular markers (30). However, the significance of Wnt inhibitors increasing gonadal markers expression warrants further study.
Limitations of the current study include the limited patient numbers and lack of in vivo data. In conclusion, our study identified the source of estrogen production and the possible molecular mechanism of feminizing ACC. Our findings suggest that Wnt inhibitors may be used to treat functional ACC, blocking both carcinogenesis and steroidogenesis. However, long-term adverse effects, such as sex hormone dysregulation, are worth investigating.
Abbreviations:
- ACC
adrenocortical carcinoma
- CYP19A1
aromatase cytochrome P450
- DHEAS
dehydroepiandrosterone sulfate
- E2
estradiol
- ENSAT
European Network for the Study of Adrenal Tumors
- T
testosterone
Acknowledgments
Financial Support: This study was supported by the National Natural Science Foundation of China (grants 81400779 to W.Z., 81500609 to Y.J., and 81600594 to C.Z.), the Chinese Society of Endocrinology (grant 13050890474 to W.W.), the Shanghai Science and Technology Committee (grant 15411966600 to L.J.), the Doctoral Innovation Fund Projects from Shanghai Jiao Tong University School of Medicine (grant BXJ201713 to L.W.), the National International Science Cooperation Foundation (grant 2015DFA30560 to W.W.), and the National Key Research and Development Program of China (grant 2016YFC0901500 to L.Y.).
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}