





Abstract
Heterozygous pathogenic germline variants in CDC73 predispose to the development of primary hyperparathyroidism (pHPT) and, less frequently, ossifying fibroma of the jaw and renal and uterine tumors. Clinical information on CDC73-related disorders has so far been limited to small case series.
To assess the clinical manifestations and penetrance in CDC73-related disorders and to improve case detection in pHPT.
Nationwide retrospective Dutch cohort study.
Tertiary referral center.
We studied 89 patients with pHPT referred for germline CDC73 analysis and 43 subsequently tested relatives who proved to be mutation carriers.
Germline CDC73 mutation analysis.
CDC73 mutation detection yield, referral rate, and CDC73-related disease penetrance.
Pathogenic germline CDC73 variants were identified in 11 of the 89 referred pHPT patients (12.4%), with (suspected) hyperparathyroidism–jaw tumor (HPT-JT) syndrome (n = 3), familial isolated pHPT (n = 5), apparently sporadic parathyroid carcinoma (n = 2), and apparently sporadic parathyroid adenoma (n = 1). The estimated penetrance of CDC73-related disorders was 65% at age 50 years (95% confidence interval, 48% to 82%) in 43 nonindex mutation carriers.
Germline CDC73 analysis is recommended in individuals with (suspected) HPT-JT syndrome, familial isolated pHPT, atypical or malignant parathyroid histology, and young individuals with pHPT. These criteria would increase germline CDC73 mutation detection, enabling optimal clinical management of pHPT as well as genetic counseling and surveillance for family members at risk for developing CDC73-related disorders.
Primary hyperparathyroidism (pHPT) is a common endocrine disease with a prevalence of 1 to 4 per 1000 persons and with a peak incidence in the sixth decade of life (1). In the majority of cases, pHPT is caused by a single parathyroid adenoma (PA) and in <1% by a parathyroid carcinoma (PC) (2). A genetic predisposition for pHPT can be found in ∼10% of pHPT cases. This might be an underestimation because of unavailable, incomplete, or misdiagnosed family history; variable penetrance; or unknown genetic causes. To date, pathogenic variants in at least 11 genes have been found to be associated with hereditary pHPT. The most commonly identified hereditary syndromes associated with pHPT include multiple endocrine neoplasia type 1, 2a, or 4; and CaSR-, GCM2-, and CDC73-related disorders (3, 4). Inactivation of the CDC73 tumor suppressor gene (formerly known as HRPT2 and encoding parafibromin) predisposes heterozygous carriers to a spectrum of conditions: hyperparathyroidism–jaw tumor (HPT-JT) syndrome, familial isolated hyperparathyroidism (FIHP), and PC.
The penetrance of pHPT in CDC73-related disorders has been reported to be as high as 80% to 95% (5). The onset is typically in late adolescence or early adulthood, although patients younger than 10 years of age have also been reported (6, 7). PC may be found in >20% of patients with germline pathogenic CDC73 variants, which is higher than in other hereditary pHPT syndromes (5). Distinguishing between PA, atypical adenoma, and PC remains a challenge given the lack of specific differentiating clinical, biochemical, and histological features among these pathologies. However, the latter is of the utmost importance because it determines the extent and radical nature of initial surgery, which is in turn the major determinant of prognosis (5).
In addition to pHPT, patients with CDC73-related disorders are predisposed to developing ossifying fibromas of the mandible and/or maxilla, uterine tumors (e.g., adenofibromas, leiomyomas, adenomyosis, hyperplasia, adenosarcomas) and less frequently, a variety of malignant and nonmalignant renal lesions [e.g., Wilms tumor, clear cell renal carcinoma (RCC), papillary renal cell tumor, renal cysts] (5).
In total, about 100 index CDC73 mutation carriers have been reported to date, with no clearly identified phenotype-genotype relationship (5). The majority of germline (and somatic) pathogenic CDC73 variants are frameshift and nonsense variants, although missense variants as well as (small) deletions and insertions have been reported (7–9).
Limited data are available on the germline CDC73 mutation detection yield in patients with HPT-JT syndrome, FIHP, and PC. In this study, we performed a nationwide evaluation of germline CDC73 analyses undertaken in pHPT patients in the Netherlands and characterized the clinical manifestations and penetrance of 12 families with CDC73-related disorders.
Patients and Methods
Study population and design
All Dutch patients with an established diagnosis of pHPT referred for germline CDC73 analysis in the Netherlands from February 2004 through July 2016 were included in the study. There were no specific referral criteria for germline CDC73 analysis in the Netherlands during the study period. Data on sex, diagnosis, age at diagnosis, family history, and clinical manifestations were retrieved from DNA request forms.
Referred pHPT patients were grouped in four clinical subgroups based on their personal and/or family history: (1) (suspected) HPT-JT syndrome [pHPT and at least one HPT-JT syndrome-related feature or pHPT and a close relative with (suspected) HPT-JT syndrome], (2) FIHP (pHPT and at least one first- or second-degree relative with pHPT), (3) apparently sporadic PC (sPC), and (4) apparently sporadic PA (sPA). HPT-JT–related features included pHPT, ossifying fibroma of mandible and/or maxilla, renal lesions, and uterine tumors. According to the Dutch genetic testing strategy, before CDC73 analysis, germline MEN1 variants had to be excluded in patients with FIHP and sPAs diagnosed before age 35 years.
Index patients with pathogenic CDC73 variants or variants of uncertain significance were evaluated and counseled by a clinical geneticist in their regional university medical center. Written informed consent for collection of clinical, pathological, and molecular data was obtained from all index mutation carriers. Relatives were tested for the specific pathogenic CDC73 variant using cascade screening after counseling. All CDC73 mutation carriers were referred for surveillance aimed at detecting pHPT or jaw, renal, and/or uterine abnormalities. We also included in the study an extra family belonging to a Dutch index patient with CDC73-related disorder who underwent genetic testing abroad, whereas genetic testing via cascade screening of relatives was performed at our laboratory.
The study was approved by the local Ethical Committee of the Leiden University Medical Center (P15.016).
DNA sequencing and data analysis
Germline CDC73 mutation analysis was centralized in the Laboratory for Diagnostic Genome Analysis department of clinical genetics at the Leiden University Medical Center, the Netherlands, during the study period. Germline CDC73 mutation analysis was performed with Sanger sequencing. CDC73 deletion/duplication analysis was subsequently performed in 60 patients without pathogenic CDC73 variant using the MRC Holland P466-A1 kit (MRC Holland, Amsterdam, the Netherlands).
Coding variants were analyzed for their effect on function with Alamut software package v2.7 (Interactive Biosoftware, Rouen, France), which incorporates, for example, Align GVGD, SIFT, and PolyPhen2. Variants were annotated to the GenBank reference sequence NM_024529.4. The Leiden Open Variation Database (http://www.lovd.nl/CDC73) was consulted to find variants previously described and classified.
Histological and molecular analysis of parathyroid tumors
The overproducing parathyroid gland(s) were removed in all patients referred for germline CDC73 mutation analysis and all CDC73 mutation carriers diagnosed with pHPT as part of standard care. Available tumor tissue was reexamined by a referral pathologist in Leiden (H.M.). Parafibromin immunohistochemistry (IHC), somatic CDC73 analysis, and loss-of-heterozygosity analysis were performed on formalin-fixed, paraffin-embedded samples as previously described (10). IHC was scored positive (“normal”) if nuclear staining was detected in lesional cells and was only considered negative (“loss”) in the presence of positive internal controls.
Statistical analysis
To describe clinical characteristics, the mean ± standard deviation (SD) with range was calculated. Continuous variables were analyzed using an independent sample t test. Dichotomous variables were compared using the χ2 test. Age-related penetrance of pHPT was estimated using the Kaplan-Meier method. Statistical significance was set at P < 0.05; analyses were conducted using SPSS 23.0 (SPSS, Chicago, IL).
Results
CDC73-related disorders: case detection
Pathogenic germline CDC73 variants were identified in 11 of 89 (12.4%) clinically heterogeneous pHPT patients referred for mutation analysis. In total, seven different nonsense or frame shift pathogenic variants were identified; two families carried an exon 1 deletion and two families carried a large deletion spanning the entire CDC73 gene. The clinical characteristics of the study population (CDC73 vs non-CDC73) are listed in Table 1. Within the clinical subgroups, pathogenic germline CDC73 variants were identified in 3 of 18 patients with (suspected) HPT-JT (17%), in 5 of 19 patients with FIHP (26%), in 2 of 11 patients with sPC (18%), and in 1 of 41 patients with sPA (2%). The mean age (± SD) at diagnosis of pHPT was 32 ± 15 years (range, 13 to 54 years) in CDC73 mutation carriers and 42 ± 18 years (range, 10 to 81 years) in those without a detectable mutation (P = 0.068). Ten of the 11 CDC73 mutation carriers were male (91%), as opposed to 41% of nonmutation carriers (P ≤ 0.01). In total, 12 patients were diagnosed with PC (11 apparently sporadic and 1 in the context of FIHP). Family history was positive for pHPT in 73% of CDC73 mutation carriers, as opposed to only 24% in nonmutation carriers (P ≤ 0.01). A personal history of Wilms tumor was reported in one CDC73 mutation carrier and one patient carrying a variant of uncertain significance (see the following section). No other index CDC73 mutation carrier was diagnosed with renal abnormalities. In total, eight index nonmutation carriers had a personal history of renal abnormities (five with RCC and three with renal cysts).
Clinical Characteristics of 89 pHPT Patients Referred for Germline CDC73 Analysis
| Pathogenic CDC73 Variant n = 11 | No Pathogenic CDC73 Variant n = 78 | P Value | Yield, % 12.4 | |
|---|---|---|---|---|
| Age mean ± SD (y) | 32.3 ± 14.6 | 42.6 ± 18 | 0.068 | |
| Range (y) | 13–54 | 10–81 | ||
| Sex, male, n (%) | 10 (91) | 32 (41) | 0.002 | |
| (Suspected) HPT-JT syndrome, n (%) | 3 (27) | 15 (19) | 16.7 | |
| Familial isolated pHPT, n (%) | 5 (45)a | 14 (18) | 26.3 | |
| Sporadic parathyroid carcinoma, n (%) | 2 (18) | 9 (12) | 18.0 | |
| Sporadic parathyroid adenoma, n (%) | 1 (9) | 40 (51) | 2.4 | |
| Familiar pHPT, n (%) | 8 (73) | 19 (24) | 0.003 | |
| Recurrent pHPT or multiple PA | 0 | 12 | 0.162 | |
| Renal abnormalities | 1 | 9 | 0.810 | |
| Uterine abnormalities | 0 | 4 | 0.758 |
| Pathogenic CDC73 Variant n = 11 | No Pathogenic CDC73 Variant n = 78 | P Value | Yield, % 12.4 | |
|---|---|---|---|---|
| Age mean ± SD (y) | 32.3 ± 14.6 | 42.6 ± 18 | 0.068 | |
| Range (y) | 13–54 | 10–81 | ||
| Sex, male, n (%) | 10 (91) | 32 (41) | 0.002 | |
| (Suspected) HPT-JT syndrome, n (%) | 3 (27) | 15 (19) | 16.7 | |
| Familial isolated pHPT, n (%) | 5 (45)a | 14 (18) | 26.3 | |
| Sporadic parathyroid carcinoma, n (%) | 2 (18) | 9 (12) | 18.0 | |
| Sporadic parathyroid adenoma, n (%) | 1 (9) | 40 (51) | 2.4 | |
| Familiar pHPT, n (%) | 8 (73) | 19 (24) | 0.003 | |
| Recurrent pHPT or multiple PA | 0 | 12 | 0.162 | |
| Renal abnormalities | 1 | 9 | 0.810 | |
| Uterine abnormalities | 0 | 4 | 0.758 |
One of these patients was diagnosed with a PC. In total, 9 of 12 PCs were revised by a referral pathologist.
Clinical Characteristics of 89 pHPT Patients Referred for Germline CDC73 Analysis
| Pathogenic CDC73 Variant n = 11 | No Pathogenic CDC73 Variant n = 78 | P Value | Yield, % 12.4 | |
|---|---|---|---|---|
| Age mean ± SD (y) | 32.3 ± 14.6 | 42.6 ± 18 | 0.068 | |
| Range (y) | 13–54 | 10–81 | ||
| Sex, male, n (%) | 10 (91) | 32 (41) | 0.002 | |
| (Suspected) HPT-JT syndrome, n (%) | 3 (27) | 15 (19) | 16.7 | |
| Familial isolated pHPT, n (%) | 5 (45)a | 14 (18) | 26.3 | |
| Sporadic parathyroid carcinoma, n (%) | 2 (18) | 9 (12) | 18.0 | |
| Sporadic parathyroid adenoma, n (%) | 1 (9) | 40 (51) | 2.4 | |
| Familiar pHPT, n (%) | 8 (73) | 19 (24) | 0.003 | |
| Recurrent pHPT or multiple PA | 0 | 12 | 0.162 | |
| Renal abnormalities | 1 | 9 | 0.810 | |
| Uterine abnormalities | 0 | 4 | 0.758 |
| Pathogenic CDC73 Variant n = 11 | No Pathogenic CDC73 Variant n = 78 | P Value | Yield, % 12.4 | |
|---|---|---|---|---|
| Age mean ± SD (y) | 32.3 ± 14.6 | 42.6 ± 18 | 0.068 | |
| Range (y) | 13–54 | 10–81 | ||
| Sex, male, n (%) | 10 (91) | 32 (41) | 0.002 | |
| (Suspected) HPT-JT syndrome, n (%) | 3 (27) | 15 (19) | 16.7 | |
| Familial isolated pHPT, n (%) | 5 (45)a | 14 (18) | 26.3 | |
| Sporadic parathyroid carcinoma, n (%) | 2 (18) | 9 (12) | 18.0 | |
| Sporadic parathyroid adenoma, n (%) | 1 (9) | 40 (51) | 2.4 | |
| Familiar pHPT, n (%) | 8 (73) | 19 (24) | 0.003 | |
| Recurrent pHPT or multiple PA | 0 | 12 | 0.162 | |
| Renal abnormalities | 1 | 9 | 0.810 | |
| Uterine abnormalities | 0 | 4 | 0.758 |
One of these patients was diagnosed with a PC. In total, 9 of 12 PCs were revised by a referral pathologist.
CDC73 variant of uncertain significance
One CDC73 variant of uncertain significance [c.14T>G, p.(Leu5Arg)] was further identified in a female aged 37 years with pHPT and a history of a Wilms tumor at age 2 years. IHC showed global loss of parafibromin staining in her PA, and loss of heterozygosity of the wild-type CDC73 allele was also seen. The Wilms tumor sample was not available for further investigation. Family history showed a maternal cousin with pHPT age 30 years, whereas the mother and aunt were unaffected. Segregation analysis confirmed the presence of the variant in the affected cousin. However, IHC showed positive parafibromin staining in her PA and no pathogenic somatic CDC73 variants or loss of heterozygosity of the wild-type CDC73 allele. The c.14T>G variant has not been reported in the Single Nucleotide Polymorphism Database (dbSNP), Exome Sequencing Project (ESP), Exome Aggregation Consortium (ExAc), Genome of the Netherlands (GoNL), or ClinVar databases and affects an evolutionarily conserved amino acid. The substitution of the leucine residue by an arginine residue results in a relatively large difference in physical and chemical properties [Grantham score, 102 (range, 0 to 215)] (11). AGVGD, SIFT, and PolyPhen software predicted that this amino acid change will have a major effect on protein function. In silico RNA splice prediction software predicted no substantial change compared with the wild-type sequence.
Clinical manifestations in families with CDC73-related disorders
The characteristics of the index CDC73 mutation carriers and their tested relatives are shown in Table 2. Analysis of 77 relatives who were tested via cascade screening for their familial pathogenic CDC73 variant revealed 43 nonindex mutation carriers in 10 families. Detailed information on all CDC73 mutation carriers can be found in Supplemental Table 1 and in pedigrees A through K (Supplemental Fig. 1). The mean age (± SD) at DNA analysis was 42 ± 20 years (range, 10 to 80 years) in the nonindex CDC73 mutation carriers. In total, 24 of 43 (56%) nonindex mutation carriers were diagnosed with one or more CDC73-related disorder features, including pHPT (n = 20), ossifying fibroma of the jaw (n = 5), renal abnormalities (n = 8), and uterine fibroids (n = 1). In nonindex mutation carriers, pHPT was associated with a single PA, atypical adenoma, and PC in 17 (85%), 1 (5%), and 2 (10%) cases, respectively. In addition, at least eight family members from five different families (families A, C, D, J, and K) have been diagnosed with pHPT but have not (yet) been tested for the pathogenic CDC73 variant in their family. The age-related overall penetrance values for the 43 nonindex CDC73 mutation carriers were 11% at age 25 [95% confidence interval (CI), 2% to 20%], 65% at age 50 (95% CI, 48% to 82%), and 83% at age 70 (95% CI, 57% to 99%) (Fig. 1A). The mean age (± SD) at diagnosis of pHPT was 39 ± 14 years (range, 10 to 67 years) in the affected nonindex mutation carriers, compared with 33 ± 15 years (range, 13 to 54 years) in the index mutation carriers (P = 0.32). The age-related pHPT penetrance values for the 43 nonindex CDC73 mutation carriers were 8% at age 25 (95% CI, 0% to 16%), 53% at age 50 (95% CI, 33% to 74%), and 75% at age 70 (95% CI, 54% to 95%) (Fig. 1B).
Overview of the Clinical and Molecular Characteristics of 12 Index CDC73 Mutation Carriers and Their Tested Relatives
| ID | Sex | Tumors Observed (Age at Detection, y) | Family History | Phenotype | Germline CDC73 Variant | Tested Relatives | Nonindex Carriers (Symptomatic) | Not Tested Symptomatic Relatives |
|---|---|---|---|---|---|---|---|---|
| A | M | PA (54) | pHPT | FIHP | c.226C>T, p.(Arg76*) | 6 | 2 (1) | 2 |
| B | M | PC (54), RCC (57) | Negative | sPC | c.544dup, p.(Ile182Asnfs*11) | 3 | 1 (0) | 0 |
| C | F | PA (17) | pHPT | FIHP | c.358C>T, p.(Arg120*) | 1 | 1 (1) | 1 |
| Da | M | PA (34) | pHPT, renal cysts | Suspected HPT-JT syndrome | c.687_688dellAG, p.(Arg229Serfs*37) | 37 | 24 (14) | 2 |
| E | M | Jaw (15), PA (22) | pHPT, Wilms tumor | HPT-JT syndrome | c.3_15dup, p.(Ser6Glyfs*5) | 3 | 3 (3) | 0 |
| F | M | PA (13) | Negative | sPA | Whole gene deletion | 4 | 2 (0) | 0 |
| G | M | PC (45) | pHPT | FIHP | Whole gene deletion | 9 | 4 (2) | 0 |
| H | M | Wilms tumor (8), PA (33) | pHPT, uterine fibroids | FIHP | c.3_15dup, p.(Ser6Glyfs*5) | 3 | 1 (1) | 0 |
| I | M | PC (18) | Negative | sPC | Exon 1 deletion | 0 | 0 | |
| J | M | PA (40) | pHPT | FIHP | Exon 1 deletion | 2 | 1 (1) | 1 |
| K | M | PA (25) | pHPT | FIHP | c.685_688delAGAG, p.(Arg229Tyrfs*27) | 0 | 2 | |
| L | M | PA (40) | pHPT, jaw | HPT-JT syndrome | c.760C>T, p.(Gln254*) | 8 | 4 (1) | 0 |
| ID | Sex | Tumors Observed (Age at Detection, y) | Family History | Phenotype | Germline CDC73 Variant | Tested Relatives | Nonindex Carriers (Symptomatic) | Not Tested Symptomatic Relatives |
|---|---|---|---|---|---|---|---|---|
| A | M | PA (54) | pHPT | FIHP | c.226C>T, p.(Arg76*) | 6 | 2 (1) | 2 |
| B | M | PC (54), RCC (57) | Negative | sPC | c.544dup, p.(Ile182Asnfs*11) | 3 | 1 (0) | 0 |
| C | F | PA (17) | pHPT | FIHP | c.358C>T, p.(Arg120*) | 1 | 1 (1) | 1 |
| Da | M | PA (34) | pHPT, renal cysts | Suspected HPT-JT syndrome | c.687_688dellAG, p.(Arg229Serfs*37) | 37 | 24 (14) | 2 |
| E | M | Jaw (15), PA (22) | pHPT, Wilms tumor | HPT-JT syndrome | c.3_15dup, p.(Ser6Glyfs*5) | 3 | 3 (3) | 0 |
| F | M | PA (13) | Negative | sPA | Whole gene deletion | 4 | 2 (0) | 0 |
| G | M | PC (45) | pHPT | FIHP | Whole gene deletion | 9 | 4 (2) | 0 |
| H | M | Wilms tumor (8), PA (33) | pHPT, uterine fibroids | FIHP | c.3_15dup, p.(Ser6Glyfs*5) | 3 | 1 (1) | 0 |
| I | M | PC (18) | Negative | sPC | Exon 1 deletion | 0 | 0 | |
| J | M | PA (40) | pHPT | FIHP | Exon 1 deletion | 2 | 1 (1) | 1 |
| K | M | PA (25) | pHPT | FIHP | c.685_688delAGAG, p.(Arg229Tyrfs*27) | 0 | 2 | |
| L | M | PA (40) | pHPT, jaw | HPT-JT syndrome | c.760C>T, p.(Gln254*) | 8 | 4 (1) | 0 |
Abbreviations: F, female; jaw, ossifying fibroma jaw; M, male.
Published previously in Haven CJ, Wong FK, van Dam EW, van der Juijt R, van Asperen C, Jansen J, Rosenberg C, de Wit M, Roijers J, Hoppener J, Lips CJ, Larsson C, Teh BT, Morreau H. A genotypic and histopathological study of a large Dutch kindred with hyperparathyroidism–jaw tumor syndrome. J Clin Endocrinol Metab. 2000;85:1449–1454.
Overview of the Clinical and Molecular Characteristics of 12 Index CDC73 Mutation Carriers and Their Tested Relatives
| ID | Sex | Tumors Observed (Age at Detection, y) | Family History | Phenotype | Germline CDC73 Variant | Tested Relatives | Nonindex Carriers (Symptomatic) | Not Tested Symptomatic Relatives |
|---|---|---|---|---|---|---|---|---|
| A | M | PA (54) | pHPT | FIHP | c.226C>T, p.(Arg76*) | 6 | 2 (1) | 2 |
| B | M | PC (54), RCC (57) | Negative | sPC | c.544dup, p.(Ile182Asnfs*11) | 3 | 1 (0) | 0 |
| C | F | PA (17) | pHPT | FIHP | c.358C>T, p.(Arg120*) | 1 | 1 (1) | 1 |
| Da | M | PA (34) | pHPT, renal cysts | Suspected HPT-JT syndrome | c.687_688dellAG, p.(Arg229Serfs*37) | 37 | 24 (14) | 2 |
| E | M | Jaw (15), PA (22) | pHPT, Wilms tumor | HPT-JT syndrome | c.3_15dup, p.(Ser6Glyfs*5) | 3 | 3 (3) | 0 |
| F | M | PA (13) | Negative | sPA | Whole gene deletion | 4 | 2 (0) | 0 |
| G | M | PC (45) | pHPT | FIHP | Whole gene deletion | 9 | 4 (2) | 0 |
| H | M | Wilms tumor (8), PA (33) | pHPT, uterine fibroids | FIHP | c.3_15dup, p.(Ser6Glyfs*5) | 3 | 1 (1) | 0 |
| I | M | PC (18) | Negative | sPC | Exon 1 deletion | 0 | 0 | |
| J | M | PA (40) | pHPT | FIHP | Exon 1 deletion | 2 | 1 (1) | 1 |
| K | M | PA (25) | pHPT | FIHP | c.685_688delAGAG, p.(Arg229Tyrfs*27) | 0 | 2 | |
| L | M | PA (40) | pHPT, jaw | HPT-JT syndrome | c.760C>T, p.(Gln254*) | 8 | 4 (1) | 0 |
| ID | Sex | Tumors Observed (Age at Detection, y) | Family History | Phenotype | Germline CDC73 Variant | Tested Relatives | Nonindex Carriers (Symptomatic) | Not Tested Symptomatic Relatives |
|---|---|---|---|---|---|---|---|---|
| A | M | PA (54) | pHPT | FIHP | c.226C>T, p.(Arg76*) | 6 | 2 (1) | 2 |
| B | M | PC (54), RCC (57) | Negative | sPC | c.544dup, p.(Ile182Asnfs*11) | 3 | 1 (0) | 0 |
| C | F | PA (17) | pHPT | FIHP | c.358C>T, p.(Arg120*) | 1 | 1 (1) | 1 |
| Da | M | PA (34) | pHPT, renal cysts | Suspected HPT-JT syndrome | c.687_688dellAG, p.(Arg229Serfs*37) | 37 | 24 (14) | 2 |
| E | M | Jaw (15), PA (22) | pHPT, Wilms tumor | HPT-JT syndrome | c.3_15dup, p.(Ser6Glyfs*5) | 3 | 3 (3) | 0 |
| F | M | PA (13) | Negative | sPA | Whole gene deletion | 4 | 2 (0) | 0 |
| G | M | PC (45) | pHPT | FIHP | Whole gene deletion | 9 | 4 (2) | 0 |
| H | M | Wilms tumor (8), PA (33) | pHPT, uterine fibroids | FIHP | c.3_15dup, p.(Ser6Glyfs*5) | 3 | 1 (1) | 0 |
| I | M | PC (18) | Negative | sPC | Exon 1 deletion | 0 | 0 | |
| J | M | PA (40) | pHPT | FIHP | Exon 1 deletion | 2 | 1 (1) | 1 |
| K | M | PA (25) | pHPT | FIHP | c.685_688delAGAG, p.(Arg229Tyrfs*27) | 0 | 2 | |
| L | M | PA (40) | pHPT, jaw | HPT-JT syndrome | c.760C>T, p.(Gln254*) | 8 | 4 (1) | 0 |
Abbreviations: F, female; jaw, ossifying fibroma jaw; M, male.
Published previously in Haven CJ, Wong FK, van Dam EW, van der Juijt R, van Asperen C, Jansen J, Rosenberg C, de Wit M, Roijers J, Hoppener J, Lips CJ, Larsson C, Teh BT, Morreau H. A genotypic and histopathological study of a large Dutch kindred with hyperparathyroidism–jaw tumor syndrome. J Clin Endocrinol Metab. 2000;85:1449–1454.
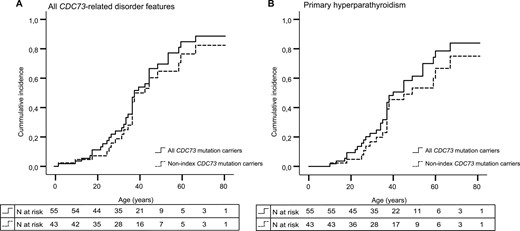
Age-related penetrance of CDC73-related disorder features in all CDC73 mutation carriers (n = 55) vs nonindex mutation carriers (n = 43). (A) Age-related penetrance of all CDC73-related disorder features for all CDC73 mutation carriers (black line) and only nonindex CDC73 mutation carriers (dotted line). (B) Age-related penetrance of pHPT for all CDC73 mutation carriers (black line) and only nonindex CDC73 mutation carriers (dotted line).
Discussion
Here, we report the results of a nationwide retrospective CDC73 survey to investigate CDC73 mutation detection yield and clinical phenotype in so-far genetically unexplained pHPT patients. We identified pathogenic germline CDC73 variants in 11 of 89 pHPT patients (12.4%). In our study population, mutation detection was associated with younger age, male sex, malignant disease, and a positive family history. The estimated penetrance of CDC73-related disorders was 83% at age 70 (95% CI, 57% to 99%) in 43 nonindex mutation carriers. Prospective studies in larger series of CDC73 mutation carriers, including genotype-phenotype relationships, genetic modifiers, and/or environmental factors, are required to determine the optimal age at which surveillance should be initiated and the monitoring intervals required to detect the different manifestations of CDC73-related disorders as they develop.
Improving future detection of CDC73-related disorder cases
In light of the relatively high incidence of pHPT and of the importance of genetic diagnosis, there is an unmet clinical need for development of guidelines for genetic testing. Based on data from our nationwide cohort analysis, we recommend germline CDC73 analysis in the four clinical subgroups of patients with pHPT listed next, a recommendation that is also in line with the 2015 Consensus Report on hereditary hyperparathyroidism of the European Society of Endocrine Surgeons (5).
All patients with HPT-JT syndrome
First, germline CDC73 analysis is recommended in individuals with (suspected) HPT-JT syndrome. Although the mutation detection yield (3/18, 17%) in our study population was lower than in a previous study (13/24, 54%), the high yield in the initial study might have been an overestimate due to ascertainment and selection bias (12).
All patients with familial pHPT (after exclusion of other gene abnormalities)
Second, germline CDC73 analysis is recommended in patients with FIHP after exclusion of pathogenic germline MEN1 variants. The mutation detection yield in our study population was 27% in patients with at least one first- or second-degree relative with pHPT. Different mutation detection yields ranging from 0% to 28% were found in previous, mostly small, studies (13–17).
All patients with PC or atypical histology of PA
The third subgroup of patients with pHPT in which germline CDC73 analysis is recommended includes individuals with apparently sporadic atypical or malignant parathyroid pathology. In our study population, the mutation detection yield in sPC was 17%. The detection yield observed in previous studies varies substantially per study population; ranging from 6%, 17% to 29%, 18%, 20%, and 31% to 38% in patients from Finland (18), Italy (19–21), France (7), United States/Japan (22), and China (23, 24), respectively. The study size and patient selection differed between studies and an unequivocal morphological diagnosis can be challenging. Referral to an experienced parathyroid surgeon and an expert pathologist should be considered in all patients with suspected PC. Subsequent parafibromin IHC and somatic CDC73 analysis could be considered for diagnostic and prognostic purposes (25). The frequency of pathogenic germline CDC73 variants in individuals with atypical adenoma has not been extensively studied and limited data are available on the contribution of IHC in cases with equivocal histology.
All patients with sporadic pHPT, younger than 35 years
The fourth subgroup of patients with pHPT in which germline CDC73 analysis is recommended includes young individuals with apparently sporadic benign pHPT, after exclusion of pathogenic germline MEN1 variants. In our study population, one patient with sPA (a 13-year-old boy) carried a pathogenic germline CDC73 variant. The yield of germline CDC73 testing in patients with sPA has barely been studied; therefore, no age-specific criteria can be identified. Dutch guidelines recommend germline MEN analysis in patients with pHPT diagnosed before age 35 years (26). For practical reasons, subsequent germline CDC73 analysis should also be considered in these patients.
Gene panel testing
To date, genetic testing for germline variants in genes predisposing to hereditary pHPT involved mainly sequential testing of single genes, prioritized according to clinical features. This type of testing protocol is expensive and time-consuming because at least 11 genes are associated with hereditary pHPT. The introduction of gene panel testing using next-generation sequencing would improve genetic testing for these rare disorders. However, complete analysis of CDC73 in next-generation sequencing panels will be challenging because of the presence GC-rich regions and frequent germline CDC73 deletions (4 of 12 in our study cohort).
Limitations and strengths of the study
The main strength of the current study is that all pHPT patients referred for germline CDC73 analysis in the Netherlands within a defined period (2004 through 2016) were included in the study. A further strength is that a total of 55 CDC73 mutation carriers from 12 families were clinically investigated in close collaboration with a number of Dutch university medical centers, representing one of the largest CDC73-related disorder series to date.
The study also has a number of limitations. The first is that the estimated mutation detection yield in this study was found in a retrospective diagnostic cohort, which despite being one of the largest CDC73-related cohorts published, might not be representative of the total patient population. Second, because we were not able to revise the histology of all parathyroid tumors from patients referred for germline CDC73 analysis, some patients may have been misclassified. And third, a possible explanation for the relatively low penetrance for jaw, uterine, and renal lesions could be inadequate surveillance and incomplete follow-up data. Alternatively, the high penetrance observed in prior studies (20% to 60%) (7, 9, 27, 28) is likely due to ascertainment and selection bias.
In conclusion, our data demonstrate that pathogenic germline CDC73 variants are frequently found in previously genetically unexplained pHPT patients. Our findings further suggest that genetic testing should be recommended in individuals with pHPT and HPT-JT syndrome-related features, familial isolated pHPT, atypical or malignant parathyroid histology, and in young individuals with pHPT. Gene panel testing or consecutive gene testing, including additional deletion and Sanger sequencing testing, should be considered, depending on the phenotype and available genetic testing options. Clinical use of these criteria will enhance the identification of individuals with CDC73-related disorders, thus improving both early detection of tumor development and genetic counseling.
Abbreviations:
- CI
confidence interval
- FIHP
familial isolated hyperparathyroidism
- HPT-JT
hyperparathyroidism–jaw tumor
- IHC
immunohistochemistry
- PA
parathyroid adenoma
- PC
parathyroid carcinoma
- pHPT
primary hyperparathyroidism
- RCC
clear cell renal carcinoma
- SD
standard deviation
- sPA
apparently sporadic parathyroid adenoma
- sPC
apparently sporadic parathyroid carcinoma.
Acknowledgments
The authors thank all patients who participated in the study.
Disclosure Summary: The authors have nothing to disclose.
References
Jackson MART, Hu MI, Perrier ND, Waguespack SG. CDC73-related disorders. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2017. Available at: https://www.ncbi.nlm.nih.gov/books/NBK3789/. Accessed 12 October 2017.
Author notes
These authors contributed equally to this study.

{kind=link}